Discovery of Novel Angiotensin-Converting Enzyme Inhibitory Peptides from Todarodes pacificus and Their Inhibitory Mechanism: In Silico and In Vitro Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. In Silico Hydrolysis of the Myosin Heavy Chain of Todarodes Pacificus
2.2. In Silico Prediction of ACE Inhibitory Peptides from Myosin Heavy Chain of Todarodes Pacificus
2.3. In Silico Screening of ACE Inhibitory Peptides from Myosin Heavy Chain of Todarodes pacificus
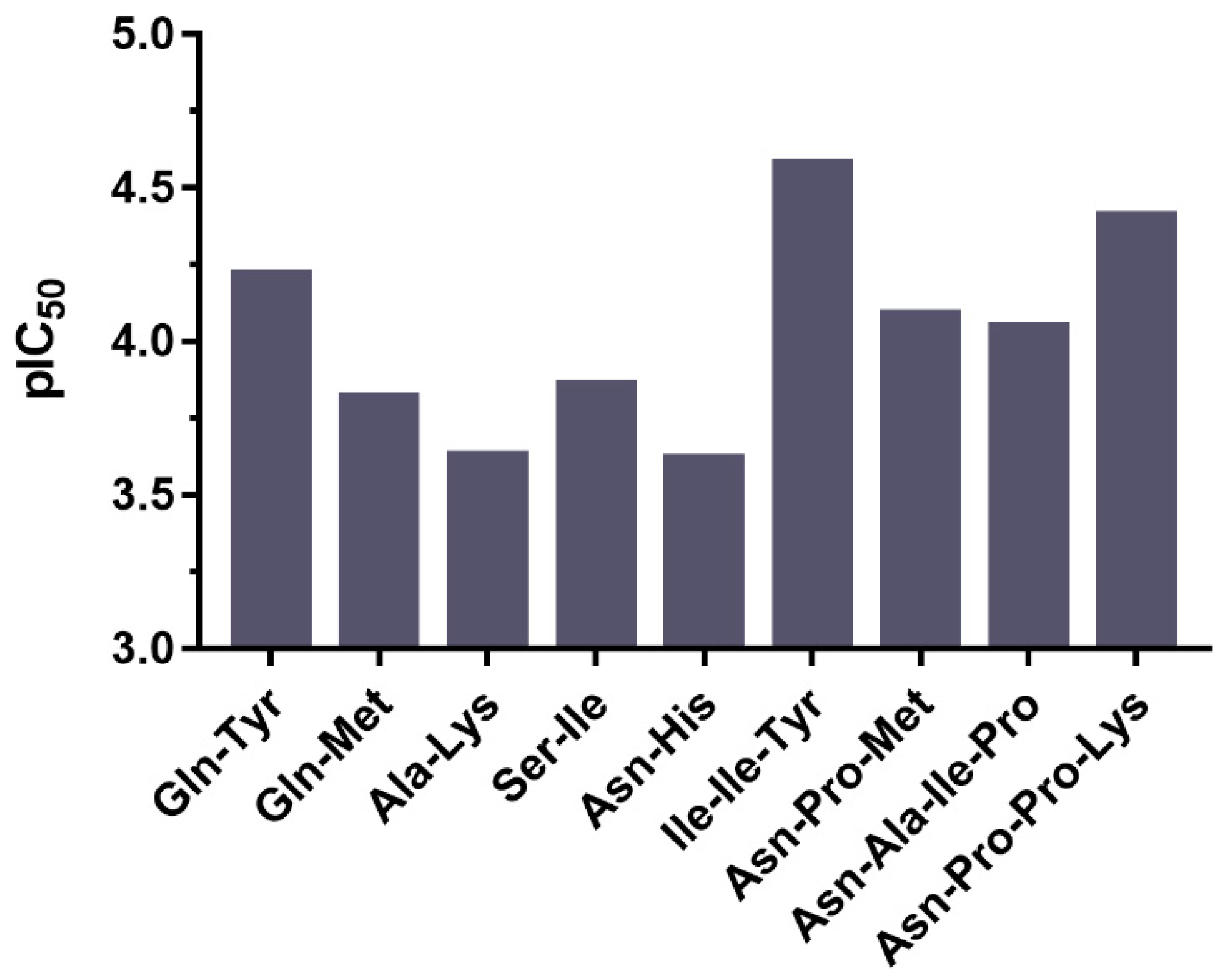
2.4. ACE Inhibitory Activity of Peptides Derived from Todarodes Pacificus
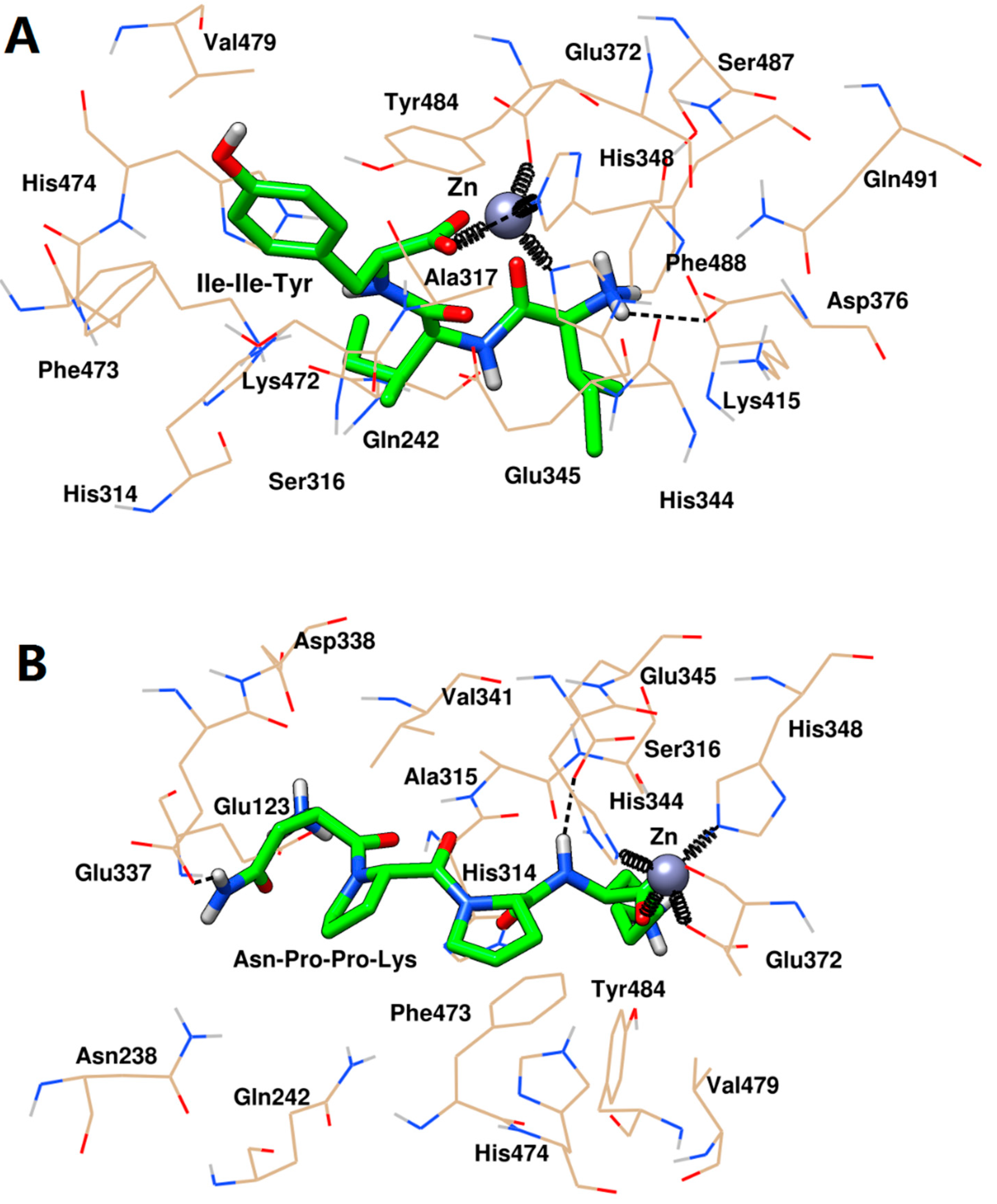
2.5. Inhibition Mechanisms of ACE by Ile-Ile-Tyr and Asn-Pro-Pro-Lys
2.5.1. Molecular Docking
2.5.2. MD Simulation
3. Materials and Methods
3.1. Reagents
3.2. In Silico Hydrolysis
3.3. Prediction of ACE Inhibitory Peptides
3.4. In Silico Prediction of Physicochemical Properties of Peptides
3.5. In Vitro ACE Inhibitory Activity
3.6. Molecular Docking of ACE with Peptides
3.6.1. Selection of Docking Parameters
3.6.2. Docking of ACE with Inhibitory Peptides
3.7. MD of ACE with Inhibitory Peptides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Acharya, K.R.; Sturrock, E.D.; Riordan, J.F.; Ehlers, M.R. Ace revisited: A new target for structure-based drug design. Nat. Rev. Drug Discov. 2003, 2, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.A.; FitzGerald, R.J. Angiotensin converting enzyme inhibitory peptides derived from food proteins: Biochemistry, bioactivity and production. Curr. Pharm. Design 2007, 13, 773–791. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Kowey, P.R.; Whelton, P.K.; Prisant, L.M. New guidelines for potassium replacement in clinical practice: A contemporary review by the National Council on Potassium in Clinical Practice. Arch. Intern. Med. 2000, 160, 2429–2436. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, L.J.; Morris, A.A.; Chapman, S.A. Angiotensin-converting enzyme inhibitor induced angioedema: Predictors of mechanical ventilation and treatment approaches. Intens. Care. Med. 2015, 41, 2233–2234. [Google Scholar] [CrossRef] [PubMed]
- Hicks, B.M.; Filion, K.B.; Yin, H.; Sakr, L.; Udell, J.A.; Azoulay, L. Angiotensin converting enzyme inhibitors and risk of lung cancer: Population based cohort study. BMJ Brit. Med. J. 2018, 363, k4209. [Google Scholar] [CrossRef] [PubMed]
- Miralles, B.; Amigo, L.; Recio, I. Critical review and perspectives on food-derived antihypertensive peptides. J. Agric. Food Chem. 2018, 66, 9384–9390. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, H.; Pihlanto, A. Bioactive peptides: Production and functionality. Int. Dairy J. 2006, 16, 945–960. [Google Scholar] [CrossRef]
- Xu, J.Y.; Qin, L.Q.; Wang, P.Y.; Li, W.; Chang, C. Effect of milk tripeptides on blood pressure: A meta-analysis of randomized controlled trials. Nutrition 2008, 24, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Sipola, M.; Finckenberg, P.; Santisteban, J.; Korpela, R.; Vapaatalo, H.; Nurminen, M.L. Long-term intake of milk peptides attenuates development of hypertension in spontaneously hypertensive rats. J. Physiol. Pharmacol. 2001, 52, 745–754. [Google Scholar] [PubMed]
- Byun, H.G.; Kim, S.K. Purification and characterization of angiotensin I converting enzyme (ACE) inhibitory peptides from Alaska pollack (Theragra chalcogramma) skin. Process Biochem. 2001, 36, 1155–1162. [Google Scholar] [CrossRef]
- Ahn, C.B.; Jeon, Y.J.; Kim, Y.T.; Je, J.Y. Angiotensin I converting enzyme (ACE) inhibitory peptides from salmon byproduct protein hydrolysate by Alcalase hydrolysis. Process Biochem. 2012, 47, 2240–2245. [Google Scholar] [CrossRef]
- Suetsuna, K.; Maekawa, K.; Chen, J.R. Antihypertensive effects of Undaria pinnatifida (wakame) peptide on blood pressure in spontaneously hypertensive rats. J. Nutr. Biochem. 2004, 15, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Udenigwe, C.C. Bioinformatics approaches, prospects and challenges of food bioactive peptide research. Trends Food Sci. Tech. 2014, 36, 137–143. [Google Scholar] [CrossRef]
- Lacroix, I.M.; Li-Chan, E.C. Evaluation of the potential of dietary proteins as precursors of dipeptidyl peptidase (DPP)-IV inhibitors by an in silico approach. J. Funct. Foods 2012, 4, 403–422. [Google Scholar] [CrossRef]
- Garg, S.; Apostolopoulos, V.; Nurgali, K.; Mishra, V.K. Evaluation of in silico approach for prediction of presence of opioid peptides in wheat. J. Funct. Foods 2018, 41, 34–40. [Google Scholar] [CrossRef]
- Yu, Z.; Wu, S.; Zhao, W.; Ding, L.; Shiuan, D.; Chen, F.; Li, J.; Liu, J. Identification and the molecular mechanism of a novel myosin-derived ACE inhibitory peptide. Food Funct. 2018, 9, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Lafarga, T.; O’Connor, P.; Hayes, M. Identification of novel dipeptidyl peptidase-IV and angiotensin-I-converting enzyme inhibitory peptides from meat proteins using in silico analysis. Peptides 2014, 59, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Bleakley, S.; Hayes, M.; O’Shea, N.; Gallagher, E.; Lafarga, T. Predicted release and analysis of novel ACE-I, renin, and DPP-IV inhibitory peptides from common oat (Avena sativa) protein hydrolysates using in silico analysis. Foods 2017, 6, 108. [Google Scholar] [CrossRef]
- Choi, S.; Puligundla, P.; Mok, C. Impact of corona discharge plasma treatment on microbial load and physicochemical and sensory characteristics of semi-dried squid (Todarodes pacificus). Food Sci. Biotechnol. 2017, 26, 1137–1144. [Google Scholar] [CrossRef]
- Nam, K.A.; You, S.G.; Kim, S.M. Molecular and physical characteristics of squid (Todarodes pacificus) skin collagens and biological properties of their enzymatic hydrolysates. J. Food Sci. 2008, 73, C249–C255. [Google Scholar] [CrossRef]
- Lin, L.; Lv, S.; Li, B. Angiotensin-I-converting enzyme (ACE)-inhibitory and antihypertensive properties of squid skin gelatin hydrolysates. Food Chem. 2012, 131, 225–230. [Google Scholar] [CrossRef]
- Alemán, A.; Pérez-Santín, E.; Bordenave-Juchereau, S.; Arnaudin, I.; Gómez-Guillén, M.C.; Montero, P. Squid gelatin hydrolysates with antihypertensive, anticancer and antioxidant activity. Food Res. Int. 2011, 44, 1044–1051. [Google Scholar] [CrossRef]
- Alemán, A.; Gómez-Guillén, M.C.; Montero, P. Identification of ace-inhibitory peptides from squid skin collagen after in vitro gastrointestinal digestion. Food Res. Int. 2013, 54, 790–795. [Google Scholar] [CrossRef]
- Toldrá, F.; Reig, M.; Aristoy, M.C.; Mora, L. Generation of bioactive peptides during food processing. Food Chem. 2018, 267, 395–404. [Google Scholar] [CrossRef]
- Norris, R.; O’Keeffe, M.B.; Poyarkov, A.; FitzGerald, R.J. Peptide identification and angiotensin converting enzyme (ACE) inhibitory activity in prolyl endoproteinase digests of bovine αs-casein. Food Chem. 2015, 188, 210–217. [Google Scholar] [CrossRef]
- Norris, R.; Poyarkov, A.; O’Keeffe, M.B.; FitzGerald, R.J. Characterisation of the hydrolytic specificity of Aspergillus niger derived prolyl endoproteinase on bovine β-casein and determination of ACE inhibitory activity. Food Chem. 2014, 156, 29–36. [Google Scholar] [CrossRef]
- Iwaniak, A.; Minkiewicz, P.; Darewicz, M.; Sieniawski, K.; Starowicz, P. BIOPEP database of sensory peptides and amino acids. Food Res. Int. 2016, 85, 155–161. [Google Scholar] [CrossRef]
- Gangopadhyay, N.; Wynne, K.; O’Connor, P.; Gallagher, E.; Brunton, N.P.; Rai, D.K.; Hayes, M. In silico and in vitro analyses of the angiotensin-I converting enzyme inhibitory activity of hydrolysates generated from crude barley (Hordeum vulgare) protein concentrates. Food Chem. 2016, 203, 367–374. [Google Scholar] [CrossRef]
- Lafarga, T.; Rai, D.K.; O’Connor, P.; Hayes, M. A bovine fibrinogen-enriched fraction as a source of peptides with in vitro renin and angiotensin-I-converting enzyme inhibitory activities. J. Agric. Food Chem. 2015, 63, 8676–8684. [Google Scholar] [CrossRef]
- Kumar, R.; Chaudhary, K.; Chauhan, J.S.; Nagpal, G.; Kumar, R.; Sharma, M.; Raghava, G.P. An in silico platform for predicting, screening and designing of antihypertensive peptides. Sci. Rep-UK 2015, 5, 12512. [Google Scholar] [CrossRef]
- Kumar, R.; Chaudhary, K.; Sharma, M.; Nagpal, G.; Chauhan, J.S.; Singh, S.; Gautam, A.; Raghava, G.P. AHTPDB: A comprehensive platform for analysis and presentation of antihypertensive peptides. Nucleic Acids Res. 2014, 43, D956–D962. [Google Scholar] [CrossRef]
- Ketnawa, S.; Rawdkuen, S. Purification and characterization of ACE inhibitory peptide from aquatic resources: A review. Int. J. Pl. An and Env. Sci. 2013, 3, 220–233. [Google Scholar]
- Khedr, S.; Deussen, A.; Kopaliani, I.; Zatschler, B.; Martin, M. Effects of tryptophan-containing peptides on angiotensin-converting enzyme activity and vessel tone ex vivo and in vivo. Eur. J. Nutr. 2018, 57, 907–915. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.; Raghava, P.S.; Open Source Drug Discovery Consortium. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliver. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Wang, Y.K.; He, H.L.; Chen, X.L.; Sun, C.Y.; Zhang, Y.Z.; Zhou, B.C. Production of novel angiotensin I-converting enzyme inhibitory peptides by fermentation of marine shrimp Acetes chinensis with Lactobacillus fermentum SM 605. Appl. Microbiol. Biot. 2008, 79, 785–791. [Google Scholar] [CrossRef]
- Yang, Y.; Tao, G.; Liu, P.; Liu, J. Peptide with angiotensin I-converting enzyme inhibitory activity from hydrolyzed corn gluten meal. J. Agric. Food Chem. 2007, 55, 7891–7895. [Google Scholar] [CrossRef]
- Arihara, K.; Nakashima, Y.; Mukai, T.; Ishikawa, S.; Itoh, M. Peptide inhibitors for angiotensin I-converting enzyme from enzymatic hydrolysates of porcine skeletal muscle proteins. Meat Sci. 2001, 57, 319–324. [Google Scholar] [CrossRef]
- Natesh, R.; Schwager, S.L.; Sturrock, E.D.; Acharya, K.R. Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature 2003, 421, 551. [Google Scholar] [CrossRef]
- Zhu, J.; Li, H.; Xu, Y.; Wang, D. Construction of fucoxanthin vector based on binding of whey protein isolate and its subsequent complex coacervation with lysozyme. J. Agric. Food Chem. 2019, 67, 2980–2990. [Google Scholar] [CrossRef]
- Ning, X.; Zhang, Y.; Yuan, T.; Li, Q.; Tian, J.; Guan, W.; Liu, B.; Zhang, W.; Xu, X.; Zhang, Y. Enhanced thermostability of glucose oxidase through computer-aided molecular design. Int. J. Mol. Sci. 2018, 19, 425. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Chen, G. Influence of secondary-structure folding on the mutually exclusive folding process of GL5/I27 protein: Evidence from molecular dynamics simulations. Int. J. Mol. Sci. 2016, 17, 1962. [Google Scholar] [CrossRef]
- Ali, S.; Khan, F.I.; Mohammad, T.; Lan, D.; Hassan, M.; Wang, Y. Identification and evaluation of inhibitors of lipase from Malassezia restricta using virtual high-throughput screening and molecular dynamics studies. Int. J. Mol. Sci. 2019, 20, 884. [Google Scholar] [CrossRef]
- Fülöp, V.; Böcskei, Z.; Polgár, L. Prolyl oligopeptidase: An unusual β-propeller domain regulates proteolysis. Cell 1998, 94, 161–170. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of octanol−water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Case, D.; Ben-Shalom, I.; Brozell, B.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M., Jr. MCPB.py: A python based metal center parameter builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, D.; Wang, C.; Song, Y.; Zhu, J.; Zhang, X. Discovery of Novel Angiotensin-Converting Enzyme Inhibitory Peptides from Todarodes pacificus and Their Inhibitory Mechanism: In Silico and In Vitro Studies. Int. J. Mol. Sci. 2019, 20, 4159. https://doi.org/10.3390/ijms20174159
Yu D, Wang C, Song Y, Zhu J, Zhang X. Discovery of Novel Angiotensin-Converting Enzyme Inhibitory Peptides from Todarodes pacificus and Their Inhibitory Mechanism: In Silico and In Vitro Studies. International Journal of Molecular Sciences. 2019; 20(17):4159. https://doi.org/10.3390/ijms20174159
Chicago/Turabian StyleYu, Dingyi, Cong Wang, Yufeng Song, Junxiang Zhu, and Xiaojun Zhang. 2019. "Discovery of Novel Angiotensin-Converting Enzyme Inhibitory Peptides from Todarodes pacificus and Their Inhibitory Mechanism: In Silico and In Vitro Studies" International Journal of Molecular Sciences 20, no. 17: 4159. https://doi.org/10.3390/ijms20174159
APA StyleYu, D., Wang, C., Song, Y., Zhu, J., & Zhang, X. (2019). Discovery of Novel Angiotensin-Converting Enzyme Inhibitory Peptides from Todarodes pacificus and Their Inhibitory Mechanism: In Silico and In Vitro Studies. International Journal of Molecular Sciences, 20(17), 4159. https://doi.org/10.3390/ijms20174159