Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function
by
Julián Piñeiro Fernández
1,†, 
Kimberly A. Luddy
1,2,*,†,
Cathal Harmon
3 and
Cliona O’Farrelly
1,4,* 1
School of Biochemistry and Immunology, Trinity College Dublin, D02 PN40 Dublin, Ireland
2
Department of Cancer Physiology, H. Lee Moffitt Cancer Center, Tampa, FL 33626, USA
3
Brigham and Women’s Hospital, Harvard Institutes of Medicine, Harvard Medical School, Boston, MA 02138, USA
4
School of Medicine, Trinity College Dublin, D02 PN40 Dublin, Ireland
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2019, 20(17), 4131; https://doi.org/10.3390/ijms20174131
Submission received: 2 August 2019
/
Revised: 16 August 2019
/
Accepted: 20 August 2019
/
Published: 24 August 2019
(This article belongs to the Special Issue Natural Killer and NKT Cells)
Abstract
:The liver is a complex organ with critical physiological functions including metabolism, glucose storage, and drug detoxification. Its unique immune profile with large numbers of cytotoxic CD8+ T cells and significant innate lymphoid population, including natural killer cells, T cells, MAIT cells, and iNKTcells, suggests an important anti-tumor surveillance role. Despite significant immune surveillance in the liver, in particular large NK cell populations, hepatic cell carcinoma (HCC) is a relatively common outcome of chronic liver infection or inflammation. The liver is also the second most common site of metastatic disease. This discordance suggests immune suppression by the environments of primary and secondary liver cancers. Classic tumor microenvironments (TME) are poorly perfused, leading to accumulation of tumor cell metabolites, diminished O2, and decreased nutrient levels, all of which impact immune cell phenotype and function. Here, we focus on changes in the liver microenvironment associated with tumor presence and how they affect NK function and phenotype.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
“The myth of Prometheus means that all the sorrows of the world have their seat in the liver. But it needs a brave man to face so humble a truth.”—François Mauriac, in Le Nœud de vipères (1932)
From ancient Greeks to modern biologists, the liver has long fascinated humans. Its large, yet unassuming exterior veils the profound functions and unique behavior of this vital organ. The liver is the only internal organ with the capacity to self-regenerate. Once thought to harbor all human emotions, the liver is now known to be the birthplace of fetal blood cells, the key site for detoxification, and the primary location of energy generation and storage. It controls our appetites, mediates inflammation, and modulates immune activity [1,2,3]. Unlike other organs, the liver receives blood from two separate systems: 30% of its supply arrives from the circulation, while the remaining 70% comes from the hepatic portal system that drains from the GI tract, gallbladder, pancreas, and spleen, carrying a multitude of harmless dietary and microbial antigens. Although classified as a non-lymphoid organ, the immunological composition of the liver is strikingly unique, containing large populations of innate lymphocytes with strong anti-tumor potential, including natural killer cells (NK cells), natural killer T cells (NKT cells), mucosal-associated invariant T cells (MAIT cells), and gamma delta T cells ( T cells) [4,5,6,7]. These fast-responding cytotoxic cells are charged with protecting the liver and hence the rest of the body from ingested pathogens and transformed hepatocytes, as well as disseminated tumor cells arriving in the hepatic vein.
NK cells, which make up to 50% of the liver lymphocyte population, are cytotoxic cells with anti-tumor functions that are mediated through the release of cytotoxic granules, TRAIL and FasL [5]. Unlike their adaptive counterparts, CD8 T cells, NK cells do not rely on antigen presentation; instead, they are activated through a cascade of various activating and inactivating receptors (Figure 1). This allows NK cells to target stressed and damaged self cells. Liver NK populations include high proportions of CD56bright cells and also a population of liver-resident NK cells, which are characterized by higher expression of CXCR6 and CD69, altered expression of the transcription factors Eomes and Tbet, and exhibit a strong cytotoxic function [2,5,8]. Despite being enriched with large numbers of NK cells, malignant cells can embed and thrive in some livers.
Hepatocellular carcinoma (HCC) accounts for the vast majority of primary liver cancers, which originate from hepatocytes that become transformed. Chronic inflammation caused by infection, autoimmunity, obesity, excess alcohol, and iron accumulation [9,10,11,12,13,14] are primary drivers of hepatocyte transformation. Liver metastases or secondary cancers are typically from primaries of the GI tract, but can also include breast, lung, and melanoma, which often arrive through the portal vein [15]. The liver provides quite a different microenvironment to other organs, perhaps explaining why classic checkpoint inhibition has hitherto failed in patients with liver metastases [16,17]. Changes in nutrient flow, vasculature, and immune profiles create selection pressures for arriving cancer cells. Only those most fit for the new environment will survive and repopulate the new site, changing the local microenvironment in the process. Here, we discuss the conditions of the tumor microenvironment in the liver and its effects on the phenotype and functions of hepatic NK cells.
2. Hepatic Tumor Microenvironment
“Prometheus gave us fire and we use it to light cigarettes.”—Marty Rubin
The tumor microenvironment (TME) is formed when cancer cells begin to invade and change the surrounding tissue architecture. Composed of tumor cells, immune cells, and stromal cells, the TME is a unique ecosystem providing a protective niche for tumor cells to thrive [6,18]. Common abiotic features of the TME include low oxygen concentrations, altered metabolite accumulations, acidic pH, as well as a collection of immunosuppressive cytokines and growth factors (Figure 2). These harsh conditions not only shape the tumor cell repertoire, but also strongly impact the health and function of resident and tumor-infiltrating immune cell populations. The TME can:
NK cells are no exception. The various features of the TME can suppress NK cell function, skew their differentiation, change their phenotype, halt their proliferation, and even induce apoptosis.
2.1. Hypoxia
The healthy liver is marked by regions of high and low oxygen levels, fitting the needs of hepatocytes tasked with different metabolic roles [24,25]. While much of the normal liver is highly vascularized, poor vessel perfusion and rapid uptake of O2 by densely-packed tumor cells often results in hypoxic environments within the tumor. With a median oxygen level of 0.8%, hepatic cellular carcinoma is one of the most hypoxic tumor types [26]. Furthermore, the perivenous portions of the liver, hypothesized to receive the majority of disseminated tumor cells, have low oxygen tension [15]. Cancer cells arriving from primary sites in the GI tract, the breast, and the lung must undergo metabolic changes through the regulation of hypoxia-induced transcription factors (e.g., HIF-1 and c-Myc), which will help develop low-oxygen tolerance to survive this hypoxic environment [27]. Highly-glycolytic cancer cells express HIF-1, which under normal conditions, is ubiquitinated and degraded in the cytoplasm [28]. However, when oxygen is low, HIF-1 re-enters the nucleus and binds Hif-1, stabilizing it as a protein and exerting its function as a transcription factor [29,30]. In the liver, hypoxia-derived HIF-1 induces changes in surface and soluble MHC class I polypeptide-related sequence A (MICA), thus impairing NK cell’s ability to recognize the tumor [23,31]. In some cases, Hif-1, along with other hypoxia-related pathways such as unfolded protein response, also drive tumor cell autophagy [32]. Reversal of autophagy by silencing important genes involved in this process, such as Beclin1, increases NK cell infiltration into the tumor [33].
Hypoxia can have diverse direct effects on NK cells and may even be beneficial for their function under short-term exposure [34,35]. However, following long-term oxygen deprivation, NK cells themselves upregulate HIF-1 [36], resulting in an altered transcriptional profile [34]. Hif-1 downregulates the expression of natural cytotoxicity receptors, NKp30, NKp44, NKp46, and the natural killer group 2D (NKG2D) receptor, activators of NK cells [36]. HIF-1 regulates important genes related to metabolism, cell proliferation, and apoptosis. Metabolic effects of Hif-1 on NK cells include the altered expression of glycolytic enzymes (e.g., PMK2 and PGK1) [37], metabolite transporters, (e.g., GLUT1 and 3, SLC1A5, and MCT4) [37], and enzymes involved in biosynthesis (e.g., FAS and 6PGDH) [38]. Hypoxia inactivates mammalian target of rapamycin (mTOR) in NK cells [39], a protein complex that senses nutrient deficits and controls NK cell growth, maturation, and differentiation [40]. The mechanism is not entirely defined, but it is clear that HIF-1 activation leads to DNA damage and replication arrest, which inhibits mTOR through regulation of DNA damage response 1 (REDD1) [41]. It may also promote degradation of granzyme B through autophagy, as occurs during starvation [42]. Inhibition of mTOR signaling in hepatic NK cells by inactivating or blocking the mTORC1 pathway (gene knockout) also results in the reduction of mature NK cells (lower numbers of CD11b+ cells) and loss of IFN production downstream of NKG2D activation and impaired OXPHOS metabolism [43], showing the importance of this pathway in hypoxia-related processes. Hypoxic conditions also reduce intracellular granzyme B and perforin [44].
The acquisition of new blood vessels alleviates the hypoxic burden on tumor cells, allowing for uncontrolled growth. While NK cells are the primary effector cells of the innate immune system, there are subsets of NK cells with differing phenotypes. Decidual NK cells are highly angiogenic cells with a pivotal role in pregnancy [45,46]. Diminished oxygen levels and increased TGF in the TME can polarize NK cell differentiation into a proangiogenic phenotype [46,47,48]. Proangiogenic genes, vascular endothelial growth factor (VEGF) and TGF-, are upregulated in immune cells, usually in a Hif-1-dependent manner [37]. Particularly, tumor-infiltrating NK cells are more likely to develop a CD56 bright phenotype [49] upon interaction with PD-L1 in hypoxia [49]. These CD56 bright NK cells express lower levels of STAT5, necessary for NK cell immunosurveillance [50]. The lack of STAT5A and STAT5B promotes higher levels of the immunosuppressive cytokine TGF1, which upregulates VEGF in healthy NK cells (along with others like P1GF) [45]. VEGF binds VEGF receptor 2 (VEGFR2) on the surface of endothelial cells, inducing the formation of new blood vessels. In liver cancers, agrin expression induces endothelial cell recruitment and adhesion to the tumor site, thus promoting angiogenesis and upregulating and stabilizing VEGFR2 [51].
2.2. Tumor Metabolism
Cell metabolism undergoes drastic changes during transformation (Figure 3). Under normal conditions, epithelial cells, as well as hepatocytes generate ATP from glucose slowly and efficiently by oxidative phosphorylation [52]. However, cancer cells switch from oxidative phosphorylation (OXPHOS) to aerobic glycolysis as a primary means of glucose metabolism, to meet the increased energy and biomolecular needs of transformation [53,54,55,56,57,58,59,60]. Production of ATP is far less efficient during glycolysis, with just two molecules of ATP generated for each molecule of glucose in contrast to the 37–39 produced via OXPHOS. However, aerobic glycolysis skips the OXPHOS pathway and quickly generates ATP along with a wealth of biomolecules involved in the anabolism and biosynthesis of new structures for cell growth and proliferation [61,62,63,64,65]. One of the most important of these is the pentose-phosphate pathway (PPP), which transforms glucose-6-phosphate, an intermediary metabolite from glycolysis, into ribulose-5-phosphate, which will eventually become de novo-synthesized nucleotides. NADPH generated in the process will be used in lipid synthesis [64,65]. As previously reviewed by Gillies et al., the glycolytic shift in cancer cells involves a wide variety of pathways including hypoxia-inducible factor, myc, PI3K/Akt/mTOR, p53, and Ras [66]. The dramatic increase in glucose metabolism leads to an equally dramatic increase in metabolic end products, which accumulate in the tumor microenvironment and elicits immunoregulatory functions on intratumoral NK cells.
2.2.1. Lactate
Glycolysis generates several intermediate products, which contribute to biosynthesis pathways or NADH production (see [67] for more details). One key intermediate, pyruvate, the conjugate base of pyruvic acid, can be transported into the mitochondria for use in the citric acid cycle, converted to fatty acids, or carbohydrates through gluconeogenesis. In oxygen-poor environments and transformed cells, a large portion of pyruvate is converted to lactate by lactate dehydrogenase [67]. When the intracellular levels of lactate become too high, the proton-linked monocarboxylate transporters (MCT) pump lactate outside of the cell.
The healthy liver is the primary site of lactate absorption via gluconeogenesis [68]. However, in advanced liver disease and cancer, lactate production is increased, and lactate clearance is decreased. This may lead to a build up of lactate in the liver microenvironment [69,70]. Previous work from our group demonstrated that conditioned media derived from ex vivo cultures of colorectal cancer liver metastasis tissues had elevated levels of lactate (>10 mmol/L) compared to non-cancer liver tissue controls (<3 mmol/L) [69]. High tumor lactate levels are a poor prognostic indicator in many cancers including liver cancers [69]. Hepatic cytotoxic lymphocytes, such as NK cells and CD8 T cells, become inactivated when levels of intracellular lactate are high, either by downregulating the expression of NKp46, CD107a, and granzyme B [71] or simply by undergoing apoptosis due to the mitochondrial damage induced by reactive oxygen species, particularly in the hepatic CD56bright subpopulations [69].
2.2.2. Adenosine
Adenosine is a purine metabolite that suppresses NK cell proliferation, maturation [72], and effector function through the mTOR pathway [73]. Adenosine is generated by tumor cell cleavage of ATP into AMP and then AMP into free adenosine (the responsible enzymes for these processes are the ectonucleotidases CD39 and CD73, respectively) [74]. Under low oxygen concentrations within the tumor, neoplastic cells and some Tregs [75] express higher levels of CD39 and CD73 in an HIF-1/mTOR-dependent manner [76]. Additionally, NK cells can synthesize and secrete adenosine themselves. CD56bright NK cells produce free adenosine via CD38 and CD203a, acting as an immunosuppressor cell by regulating other lymphocytes, mainly CD4+ T cells, while CD56dim express lower levels of CD73 and CD39 [77].
This metabolite not only suppresses NK cell function, but many other immune cells including T cells, and recruits and activates other immunosuppressive cells such as regulatory T cells [78] and myeloid-derived suppressor cells (MDSCs). MDSC inhibit CD4 helper T cells, a primary source of NK cell-supportive cytokines. All of this decreases the activity and the killing potential of NK cells, but it does not directly affect the antibody-dependent cell cytotoxicity (ADCC) of NK cells [23], although antibody secretion is impaired due to the effects of the tumor on antibody-producing plasma cells [79,80]. Blocking adenosine signaling reverses the immunosuppressive effects and may be a potential therapeutic option in combination with other NK cell-based immunotherapies, including adoptive transfer and checkpoint blockade [39,81,82].
2.2.3. Tryptophan Catabolism
Tryptophan is an essential amino acid catabolized through either the serotonin or kynurenine pathways. In the former, tryptophan is used by the nervous system to produce serotonin and is required for the synthesis of melatonin and vitamin B3 [83]. The kynurenine pathway is mediated by intracellular enzymes, indoleamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase 2 (TDO). Kynurenine is further processed to quinolinic acid and picolinic acid or released from the cell via CD98 [84]. IDO is an IFN--inducible gene expressed in the thymus, GI tract, lung, maternal–fetal interface, and importantly, by regulatory myeloid cells and antigen-presenting cells [85]. TDO expression is induced by glucocorticoids in hepatic cells and is responsible for maintaining systemic tryptophan levels [85,86,87]. Both enzymes are overexpressed in various cancer types. Constitutive and inducible IDO is expressed at high levels in colorectal cancer and is significantly correlated with liver metastasis [88]. TDO is constitutively expressed in glioma, bladder cancer, melanoma, and HCC [86].
The tryptophan/kynurenine pathway is associated with immune suppression in cancer. Overexpression of IDO and TDO by cancer cells, tumor-infiltrating myeloid cells, and aberrant hepatocytes causes a depletion of tryptophan in the surrounding environment. Munn et al. and others hypothesized that tryptophan depletion is responsible for the immunosuppressive effects of tumoral IDO [85]. They showed that without adequate tryptophan levels, lymphocytes are unable to proliferate [89]. Others hypothesized that downstream metabolites including l-kynurenine, 3-hydroxykynurenine, 3-hydroxyanthranilic acid, and picolinic acid are toxic to lymphocytes. At high levels, these metabolites inhibit NK cell proliferation by inducing cell cycle arrest [90]. l-kynurenine blocks cytokine upregulation of NK cell-activating receptors NKp46 and NKG2D at both the transcript and protein level [91]. Furthermore, l-kynurenine inhibited the cytotoxic effects of activated NK cells. These effects are reversible upon removal of the metabolite [91]. In hepatocellular carcinoma, tumor-associated fibroblasts suppress hepatic NK cell activity through IDO and prostaglandin E2 (PGE2, an immunosuppressive lipid product of arachidonic acid metabolism) secretion [92] and can be reversed or palliated by anti-IDO [93]. The mechanisms driving the inhibition of NK cells by either tryptophan depletion or metabolite accumulation are unknown, but may be guided by nutrient sensing.
2.3. Acidic pH
Tumor cells consume and breakdown large amounts of glucose through aerobic and anaerobic processes. The outcome is the secretion of acids in the form of lactate and CO2 [66]. Carbonic anhydrases (CA) convert CO2 into bicarbonate and protons, resulting in acidification of the local environment. CA-IX is a membrane-bound and soluble CA. It is expressed in response to hypoxia and acidosis and correlates with poor prognosis in cirrhosis and HCC [94,95]. Lactate, the secreted end product of glycolysis, further acidifies the environment when hydrogens are removed by ion transporters [66]. In the TME, this build-up of protons can induce a drop in the extracellular pH to 6.6, causing dramatic effects on infiltrating immune cells [96].
The acidification of the tumor microenvironment has different effects on immune cells. On the one hand, it has been shown to increase the maturation of dendritic cells, as well as the activation and recruitment of MDSCs and Treg cells [97], while driving M2 differentiation [98,99,100]. On the other hand, low pH inhibits cytotoxic functions of lymphocytes and blocks secretion of inflammatory cytokines such as IFN [101] and TNF [96]. This effect is amplified in the spleen compared to liver-resident NK cells in rat models, suggesting some acid adaptation of liver-resident NK cells, as they exhibit lower rates of cell death, better morphology, and higher accumulation of granules compared to splenic NK cells in the same acidic environment [102]. Systemic buffering in murine models restored IFN expression by NK cells [101] and inhibited the formation of hepatic metastasis [103].
Interestingly, when tumor-related acidosis is extrapolated to other diseases and other microenvironments, such as the microenvironment generated by cryptococcoma (Cryptococcus gattii and Cryptococcus neoformans infections), the acidic pH in the center of the mass (analogous to the tumor mass) produces increased NK cell degranulation and perforin-mediated killing compared to pH 7.4 [104]. This suggests that acidic pH on its own may not be the sole inducer of the diminished anti-tumor activity of NK cells. Rather, it may be that low pH boosts the immunosuppressive effects of other components of the TME.
2.4. Cytokines and Growth Factors
The healthy liver is rich in a plethora of cytokines (e.g., IL-1, IL-1, TNF, and IL-6) and growth factors (e.g., IGF-I, IGF-II, EGF, and TGF-) that stimulate hepatocyte growth and induce activation upon the correct stimuli [105]. Following tissue injury, Kupffer cells, liver-specific macrophages, secrete pro-inflammatory cytokines such as IL-1, IL-6, and TNF- [106,107]. TNF- plays a key role in maintaining liver homeostasis by promoting tissue regeneration along with IL-6 in an STAT3/NFB-dependent manner [108]. As the wound is repaired, Kupffer cells undergo a shift in phenotype and secrete anti-inflammatory cytokines such as IL-4 and IL-10 [109]. Persistent liver injury and chronic liver disease create a state of perpetual inflammation that can support malignant transformation [110]. Pro-inflammatory cytokine levels are particularly high at early stages of tumor development, indicating that during tumor formation, a pro-inflammatory environment plays a role in tumor development [111]. High levels of pro-inflammatory cytokines such as IL-6, IL-8, and TNF- are present in the tissue from patients undergoing resection for CRLM [112] and are associated with poor prognosis and tissue damage [113].
While the cytokines and growth factors associated with inflammation may support tumor growth at these early stages, they also activate cytotoxic immune cells, strengthening a cycle of immune-mediated tumor cell death and birth. The term “immunoediting” is often used to describe an evolutionary process on tumor development driven by the immune system [114,115,116]. For example, IL-21, secreted mainly by helper follicular T cells, NKT cells, and Th17 cells, increases the cytotoxicity and proliferation of NK cells and other immune cells [117,118]. It has been shown that IL-21 and IL-2 have synergistic effects, and when combined, the expression of CD25, NKG2A, and CD86 on CD56bright NK cells is upregulated [119]. NKp44 and NKG2D are only upregulated in the presence of IL-2, but with IL-21 costimulation, NKG2D is slightly downregulated, suggesting a negative effect of IL-21 on NKG2D expression [120]. IL-15 promotes NK cell activation, proliferation, and survival [121] via PI3K, PDPK1, and AKT [39], and its expression is tissue-specific. IL-15 is secreted by monocytes, tissue-resident Kupffer cells, activated macrophages, and epithelial cells [122,123] and plays a role in antiviral responses in the liver. IL-15, which shares a similar structure with IL-2, also plays a key role in overcoming NK cell dysfunction and exhaustion by restoring the cytotoxic activity in CRLM and HCC [124]. Other cytokines such as IL-13 (and less importantly, IL-33 and IL-5), Th2 type cytokines, are correlated with fibrosis progression in chronic liver inflammation diseases in mice [113], activating pro-fibrotic genes through the STAT6-ZEB1 pathway [125,126].
As the tumor progresses, the cytokine pattern shifts towards an immune-evasive anti-inflammatory response, resulting in diminished activation of the immune system. Upregulated anti-inflammatory cytokines within the TME include IL-10 and TGF- [127], similar to those observed in chronic inflammation processes [128]. The reasons behind these changes over time are not completely understood, but the physicochemical properties of the tumors are thought to be the main reason [128]. IL-10 induces dysregulation of NK surface receptor expression, including NKG2A. Hepatic NK cells have been shown to upregulate NKG2A and downregulate Ly49 (an MHC I-binding receptor) in the presence of IL-10, resulting in an exhaustion-like phenotype and an inability to respond appropriately to stimuli [129,130]. TGF- inhibits NK cell function by inhibiting cytokine-mediated activation of mTOR [131]. Furthermore, inhibition of TGF- signaling restores NK cell functionality in murine models of colorectal cancer liver metastasis [132].
Hepatocellular carcinoma often results from chronic viral infections including, hepatitis C virus, and hepatitis B virus (HCV and HBV). The presence of virus in the tumor can further complicate the immune response [133]. Tumoral HCV and HBV infections have differential effects on the resident NK cell population. High viral loads in patients with established HCC correlate with tumor progression and poor outcomes [134]. Virally-infected cells stimulate monocytes to express IL-18, which activates NK cells, as well as immunosuppressive cytokines, TGF, and IL-10 [135]. During the course of viral-induced HCC, hepatic NK cells are exposed to both transformed cells and infected cells. Infected hepatocytes activate NK cells, which can lead to viral clearance. However, chronic infections are often accompanied by diminished NK cell function and IFN production [135]. Qa-1, expressed by infected hepatocytes, binds to NKG2A on NK cells and induces exhaustion [136]. Chronic HCV promotes CD56 neg NK cells with diminished cytotoxicity [137].
3. Conclusions
NK cell activation is a complex process that is sensitive to the heterogeneous microenvironments within the tumor (Figure 4). While here, we review the common abiotic components of the liver TME (hypoxia, metabolites, acidosis, cytokines, and growth factors) and their effects on hepatic NK cells, there are many factors beyond the scope of this review that also contribute to reduced intratumoral NK cell function, including:
Immunotherapies, particularly checkpoint inhibitors, have revolutionized the way we treat cancers. The immune system now sits at the forefront of drug development and trial design in oncology. However, the liver remains the least responsive organ to current checkpoint immunotherapies, perhaps due to additional checkpoints expressed by NK cells yet to be targeted, as well as the strong immunosuppression of the metastatic liver environment. While T cells are currently a prime target of cancer immunologists, many are beginning to see the potential of NK cells. Their lack of dependence on antigen expression and rapid activation make them an attractive addition to T cell-based immunotherapies, particularly in the prevention of metastatic disease. To further the development of NK cell-based immunotherapy, we must understand the complex and often apparently redundant pathways of activation and inactivation of these highly-cytotoxic cells. The high frequency of liver-resident NK cells in healthy liver suggests an essential role for targeting and preventing liver metastasis.
Organ-specific immune responses, especially tumor surveillance mechanisms, are as diverse as the tissues in which they are housed. This is particularly true in common sites of metastasis: the lymph nodes, liver, and bone marrow. Disseminating cancer cells are faced with novel immunological challenges upon entering new tissue that select only for those tumor cells able to thrive in the face of these new predators. Perhaps the resulting immunologic bottlenecks can be better exploited with organ-specific immunotherapies designed to target the tissue housing the current lesion rather than that of the primary site.
Funding
This work was supported by grants from the Health Research Board of Ireland (HRB, HRA—POR/2013/424), the Science Foundation Ireland Investigator Award (12/IA/1667), and Trinity St. James’s Cancer Institute—Cancer Immunology Stimulus Award.
Conflicts of Interest
The authors declare that this review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| MDPI | Multidisciplinary Digital Publishing Institute |
| CRLM | Colorectal liver metastasis |
| HCC | Hepatocellular carcinoma |
| TME | Tumor microenvironment |
| GI | Gastrointestinal |
| NK cell | Natural killer cell |
| NKT cell | Natural killer T cell |
| MAIT cell | Mucosal-associated invariant T cell |
| Treg | Regulatory T cell |
| MDSC | Myeloid-derived suppressor cell |
| CD | Cluster of differentiation |
| IFN | Interferon gamma |
| TNF | Tumor necrosis factor alpha |
| TGF- | Tumor growth factor beta |
| IL | Interleukin |
| NKG2 | Natural killer group 2 receptor |
| OxPhos | Oxidative phosphorylation |
| PPP | Pentose-phosphate pathway |
| mTOR | Mammalian target of Rapamycin |
| ATP | Adenosine triphosphate |
| cAMP | Cyclic adenosine monophosphate |
| LADH | Lactate dehydrogenase |
| IDO | Indoleamine 2,3-dioxygenase |
| TDO | Tryptophan 2,3-dioxygenase |
| NAD | Nicotinamide adenine dinucleotide |
| MCT | Monocarboxylase transporter |
| ADCC | Antibody-dependent cell cytotoxicity |
| PGE2 | Prostaglandin E2 |
| CA | Carbonic anhydrase |
| ROS | Reactive oxygen species |
| FAS | Fatty acid synthase |
| REDD1 | Regulation of DNA damage response 1 |
| HIF-1 | Hypoxia inducible factor 1 |
| MICA | MHC class I polypeptide-related sequence A |
| TRAIL | TNF-related apoptosis-inducing ligand |
| 6PGDH | 6-Phosphogluconate dehydrogenase |
| PI3K | Phosphoinositide 3-kinase |
| PKB | Protein kinase B |
| PDPK1 | 3-phosphoinositide-dependent protein kinase-1 |
References
- Knell, A.J. Liver function and failure: The evolution of liver physiology. J. R. Coll. Physicians Lond. 1980, 14, 205–208. [Google Scholar] [PubMed]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Hua, J. Immune cells in liver regeneration. Oncotarget 2017, 8, 3628–3639. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. Immune tolerance in liver disease. Hepatology 2014, 60, 2109–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundstrom, P.; Szeponik, L.; Ahlmanner, F.; Sundquist, M.; Wong, J.S.B.; Lindskog, E.B.; Gustafsson, B.; Quiding-Jarbrink, M. Tumor-infiltrating mucosal-associated invariant T (MAIT) cells retain expression of cytotoxic effector molecules. Oncotarget 2019, 10, 2810–2823. [Google Scholar] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Wisse, E.; Tian, Z. Liver natural killer cells: Subsets and roles in liver immunity. Cell. Mol. Immunol. 2016, 13, 328–336. [Google Scholar] [CrossRef]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar]
- Xie, Y. Hepatitis B Virus-Associated Hepatocellular Carcinoma. Adv. Exp. Med. Biol. 2017, 1018, 11–21. [Google Scholar]
- Shiani, A.; Narayanan, S.; Pena, L.; Friedman, M. The Role of Diagnosis and Treatment of Underlying Liver Disease for the Prognosis of Primary Liver Cancer. Cancer Control 2017, 24, 1073274817729240. [Google Scholar] [CrossRef] [PubMed]
- Pinero, F.; Pages, J.; Marciano, S.; Fernandez, N.; Silva, J.; Anders, M.; Zerega, A.; Ridruejo, E.; Ameigeiras, B.; D’Amico, C.; et al. Fatty liver disease, an emerging etiology of hepatocellular carcinoma in Argentina. World J. Hepatol. 2018, 10, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306. [Google Scholar] [CrossRef]
- Kim, D.W.; Talati, C.; Kim, R. Hepatocellular carcinoma (HCC): Beyond sorafenib-chemotherapy. J. Gastrointest. Oncol. 2017, 8, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Nam, J.M.; Giaccia, A.J. Hypoxia: Signaling the Metastatic Cascade. Trends Cancer 2016, 2, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Qin, S. Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Opportunities and Challenges. Oncologist 2019, 24, S3–S10. [Google Scholar] [PubMed] [Green Version]
- Ray, K. Colorectal cancer: Evading the immune response in metastasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 191. [Google Scholar] [CrossRef]
- Yang, K.R.; Mooney, S.M.; Zarif, J.C.; Coffey, D.S.; Taichman, R.S.; Pienta, K.J. Niche inheritance: A cooperative pathway to enhance cancer cell fitness through ecosystem engineering. J. Cell. Biochem. 2014, 115, 1478–1485. [Google Scholar] [CrossRef]
- Terry, S.; Buart, S.; Chouaib, S. Hypoxic Stress-Induced Tumor and Immune Plasticity, Suppression, and Impact on Tumor Heterogeneity. Front. Immunol. 2017, 8, 1625. [Google Scholar] [CrossRef]
- Giese, M.A.; Hind, L.E.; Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 2019, 133, 2159–2167. [Google Scholar] [CrossRef]
- Gonzalez-Gugel, E.; Saxena, M.; Bhardwaj, N. Modulation of innate immunity in the tumor microenvironment. Cancer Immunol. Immunother. 2016, 65, 1261–1268. [Google Scholar] [CrossRef]
- Davoodzadeh Gholami, M.; Kardar, G.A.; Saeedi, Y.; Heydari, S.; Garssen, J.; Falak, R. Exhaustion of T lymphocytes in the tumor microenvironment: Significance and effective mechanisms. Cell. Immunol. 2017, 322, 1–14. [Google Scholar] [CrossRef]
- Hasmim, M.; Messai, Y.; Ziani, L.; Thiery, J.; Bouhris, J.H.; Noman, M.Z.; Chouaib, S. Critical Role of Tumor Microenvironment in Shaping NK Cell Functions: Implication of Hypoxic Stress. Front. Immunol. 2015, 6, 482. [Google Scholar] [CrossRef]
- Lee-Montiel, F.T.; George, S.M.; Gough, A.H.; Sharma, A.D.; Wu, J.; DeBiasio, R.; Vernetti, L.A.; Taylor, D.L. Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp. Biol. Med. (Maywood) 2017, 242, 1617–1632. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Xu, Q.Y.; Wu, H.M.; Hua, J. Effect of lipid-induced macrophage M1/M2 polarization on lipid metabolism in hepatocytes. Zhonghua Gan Zang Bing Za Zhi 2018, 26, 276–281. [Google Scholar]
- Chen, C.; Lou, T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget 2017, 8, 46691–46703. [Google Scholar] [CrossRef]
- Loo, J.M.; Scherl, A.; Nguyen, A.; Man, F.Y.; Weinberg, E.; Zeng, Z.; Saltz, L.; Paty, P.B.; Tavazoie, S.F. Extracellular metabolic energetics can promote cancer progression. Cell 2015, 160, 393–406. [Google Scholar] [CrossRef]
- Groulx, I.; Lee, S. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol. Cell. Biol. 2002, 22, 5319–5336. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Gassmann, M.; Chilov, D.; Wenger, R.H. Regulation of the hypoxia-inducible factor-1 alpha. ARNT is not necessary for hypoxic induction of HIF-1 alpha in the nucleus. Adv. Exp. Med. Biol. 2000, 475, 87–99. [Google Scholar]
- Sheppard, S.; Ferry, A.; Guedes, J.; Guerra, N. The Paradoxical Role of NKG2D in Cancer Immunity. Front. Immunol. 2018, 9, 1808. [Google Scholar] [CrossRef]
- Daskalaki, I.; Gkikas, I.; Tavernarakis, N. Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front. Cell Dev. Biol. 2018, 6, 104. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Paggetti, J.; Moussay, E.; Berchem, G.; Janji, B. Driving Natural Killer cells toward the melanoma tumor battlefield: Autophagy as a valuable therapeutic target. Oncoimmunology 2018, 7, e1452583. [Google Scholar] [CrossRef] [Green Version]
- Parodi, M.; Raggi, F.; Cangelosi, D.; Manzini, C.; Balsamo, M.; Blengio, F.; Eva, A.; Varesio, L.; Pietra, G.; Moretta, L.; et al. Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front. Immunol. 2018, 9, 2358. [Google Scholar] [CrossRef] [Green Version]
- Velasquez, S.Y.; Killian, D.; Schulte, J.; Sticht, C.; Thiel, M.; Lindner, H.A. Short Term Hypoxia Synergizes with Interleukin 15 Priming in Driving Glycolytic Gene Transcription and Supports Human Natural Killer Cell Activities. J. Biol. Chem. 2016, 291, 12960–12977. [Google Scholar] [CrossRef] [Green Version]
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef]
- Chambers, A.M.; Lupo, K.B.; Matosevic, S. Tumor Microenvironment-Induced Immunometabolic Reprogramming of Natural Killer Cells. Front. Immunol. 2018, 9, 2517. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef]
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Tsaih, S.W.; Lemke, A.; Flister, M.J.; Thakar, M.S.; Malarkannan, S. mTORC1 and mTORC2 differentially promote natural killer cell development. eLife 2018, 7, e35619. [Google Scholar] [CrossRef]
- Sarkar, S.; Germeraad, W.T.; Rouschop, K.M.; Steeghs, E.M.; van Gelder, M.; Bos, G.M.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef]
- Bruno, A.; Focaccetti, C.; Pagani, A.; Imperatori, A.S.; Spagnoletti, M.; Rotolo, N.; Cantelmo, A.R.; Franzi, F.; Capella, C.; Ferlazzo, G.; et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia 2013, 15, 133–142. [Google Scholar] [CrossRef]
- Cerdeira, A.S.; Rajakumar, A.; Royle, C.M.; Lo, A.; Husain, Z.; Thadhani, R.I.; Sukhatme, V.P.; Karumanchi, S.A.; Kopcow, H.D. Conversion of peripheral blood NK cells to a decidual NK-like phenotype by a cocktail of defined factors. J. Immunol. 2013, 190, 3939–3948. [Google Scholar] [CrossRef]
- Keskin, D.B.; Allan, D.S.; Rybalov, B.; Andzelm, M.M.; Stern, J.N.; Kopcow, H.D.; Koopman, L.A.; Strominger, J.L. TGFbeta promotes conversion of CD16+ peripheral blood NK cells into CD16- NK cells with similarities to decidual NK cells. Proc. Natl. Acad. Sci. USA 2007, 104, 3378–3383. [Google Scholar] [CrossRef]
- Allan, D.S.; Rybalov, B.; Awong, G.; Zuniga-Pflucker, J.C.; Kopcow, H.D.; Carlyle, J.R.; Strominger, J.L. TGF-Î2 affects development and differentiation of human natural killer cell subsets. Eur. J. Immunol. 2010, 40, 2289–2295. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Njah, K.; Chakraborty, S.; Qiu, B.; Arumugam, S.; Raju, A.; Pobbati, A.V.; Lakshmanan, M.; Tergaonkar, V.; Thibault, G.; Wang, X.; et al. A Role of Agrin in Maintaining the Stability of Vascular Endothelial Growth Factor Receptor-2 during Tumor Angiogenesis. Cell Rep. 2019, 28, 949–965. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar]
- Vazquez, A.; Kamphorst, J.J.; Markert, E.K.; Schug, Z.T.; Tardito, S.; Gottlieb, E. Cancer metabolism at a glance. J. Cell. Sci. 2016, 129, 3367–3373. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Ko, H.; Yun, M. Cancer Metabolism as a Mechanism of Treatment Resistance and Potential Therapeutic Target in Hepatocellular Carcinoma. Yonsei Med. J. 2018, 59, 1143–1149. [Google Scholar] [CrossRef]
- Jin, F.; Wang, Y.; Zhu, Y.; Li, S.; Liu, Y.; Chen, C.; Wang, X.; Zen, K.; Li, L. The miR-125a/HK2 axis regulates cancer cell energy metabolism reprogramming in hepatocellular carcinoma. Sci. Rep. 2017, 7, 3089. [Google Scholar] [CrossRef]
- Jiao, L.; Zhang, H.L.; Li, D.D.; Yang, K.L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.Y.; Ravichandran, S.; et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy 2018, 14, 671–684. [Google Scholar] [CrossRef]
- Hu, H.; Zhu, W.; Qin, J.; Chen, M.; Gong, L.; Li, L.; Liu, X.; Tao, Y.; Yin, H.; Zhou, H.; et al. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology 2017, 65, 515–528. [Google Scholar] [CrossRef]
- Zarrinpar, A. Metabolic Pathway Inhibition in Liver Cancer. SLAS Technol. 2017, 22, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Kowalik, M.A.; Columbano, A.; Perra, A. Emerging Role of the Pentose Phosphate Pathway in Hepatocellular Carcinoma. Front. Oncol. 2017, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Emerging roles of lipid metabolism in cancer progression. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 254–260. [Google Scholar] [CrossRef]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 76. [Google Scholar] [CrossRef]
- Gillies, R.J.; Robey, I.; Gatenby, R.A. Causes and consequences of increased glucose metabolism of cancers. J. Nucl. Med. 2008, 49 (Suppl. 2), 24S–42S. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Kaloyianni, M.; Freedland, R.A. Contribution of several amino acids and lactate to gluconeogenesis in hepatocytes isolated from rats fed various diets. J. Nutr. 1990, 120, 116–122. [Google Scholar] [CrossRef]
- Harmon, C.; Robinson, M.W.; Hand, F.; Almuaili, D.; Mentor, K.; Houlihan, D.D.; Hoti, E.; Lynch, L.; Geoghegan, J.; O’Farrelly, C. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol. Res. 2019, 7, 335–346. [Google Scholar] [CrossRef]
- Teilhet, C.; Morvan, D.; Joubert-Zakeyh, J.; Biesse, A.S.; Pereira, B.; Massoulier, S.; Dechelotte, P.; Pezet, D.; Buc, E.; Lamblin, G.; et al. Specificities of Human Hepatocellular Carcinoma Developed on Non-Alcoholic Fatty Liver Disease in Absence of Cirrhosis Revealed by Tissue Extracts 1H-NMR Spectroscopy. Metabolites 2017, 7, 49. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef]
- Chambers, A.M.; Wang, J.; Lupo, K.B.; Yu, H.; Atallah Lanman, N.M.; Matosevic, S. Adenosinergic Signaling Alters Natural Killer Cell Functional Responses. Front. Immunol. 2018, 9, 2533. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.Y.; Ni, X.C.; Yi, Y.; He, H.W.; Wang, J.X.; Fu, Y.P.; Sun, J.; Zhou, J.; Cheng, Y.F.; Jin, J.J.; et al. Overexpression of CD39 in hepatocellular carcinoma is an independent indicator of poor outcome after radical resection. Medicine (Baltimore) 2016, 95, e4989. [Google Scholar] [CrossRef]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Chiesa, S.; Imperatori, A.; Zanellato, S.; Mortara, L.; Gattorno, M.; Pistoia, V.; et al. CD56brightCD16- NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4+ T Cell Proliferation. J. Immunol. 2015, 195, 965–972. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-Î2-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef]
- Kaku, H.; Cheng, K.F.; Al-Abed, Y.; Rothstein, T.L. A novel mechanism of B cell-mediated immune suppression through CD73 expression and adenosine production. J. Immunol. 2014, 193, 5904–5913. [Google Scholar] [CrossRef]
- Sakowicz-Burkiewicz, M.; Kocbuch, K.; Grden, M.; Maciejewska, I.; Szutowicz, A.; Pawelczyk, T. Impact of adenosine receptors on immunoglobulin production by human peripheral blood B lymphocytes. J. Physiol. Pharmacol. 2012, 63, 661–668. [Google Scholar]
- Wang, J.; Matosevic, S. Adenosinergic signaling as a target for natural killer cell immunotherapy. J. Mol. Med. 2018, 96, 903–913. [Google Scholar] [CrossRef]
- Barrow, A.D.; Colonna, M. Tailoring Natural Killer cell immunotherapy to the tumour microenvironment. Semin. Immunol. 2017, 31, 30–36. [Google Scholar] [CrossRef]
- Strasser, B.; Gostner, J.M.; Fuchs, D. Mood, food, and cognition: Role of tryptophan and serotonin. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 55–61. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Lee, J.R.; Dalton, R.R.; Messina, J.L.; Sharma, M.D.; Smith, D.M.; Burgess, R.E.; Mazzella, F.; Antonia, S.J.; Mellor, A.L.; Munn, D.H. Pattern of recruitment of immunoregulatory antigen-presenting cells in malignant melanoma. Lab. Investig. 2003, 83, 1457–1466. [Google Scholar] [CrossRef]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef]
- Badawy, A.A. Targeting tryptophan availability to tumors: The answer to immune escape? Immunol. Cell Biol. 2018, 96, 1026–1034. [Google Scholar] [CrossRef]
- Sucher, R.; Fischler, K.; Oberhuber, R.; Kronberger, I.; Margreiter, C.; Ollinger, R.; Schneeberger, S.; Fuchs, D.; Werner, E.R.; Watschinger, K.; et al. IDO and regulatory T cell support are critical for cytotoxic T lymphocyte-associated Ag-4 Ig-mediated long-term solid organ allograft survival. J. Immunol. 2012, 188, 37–46. [Google Scholar] [CrossRef]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef]
- Finkelmeier, F.; Canli, O.; Peiffer, K.H.; Walter, D.; Tal, A.; Koch, C.; Pession, U.; Vermehren, J.; Trojan, J.; Zeuzem, S.; et al. Circulating hypoxia marker carbonic anhydrase IX (CA9) in patients with hepatocellular carcinoma and patients with cirrhosis. PLoS ONE 2018, 13, e0200855. [Google Scholar] [CrossRef]
- Huang, W.J.; Jeng, Y.M.; Lai, H.S.; Fong, I.U.; Sheu, F.Y.; Lai, P.L.; Yuan, R.H. Expression of hypoxic marker carbonic anhydrase IX predicts poor prognosis in resectable hepatocellular carcinoma. PLoS ONE 2015, 10, e0119181. [Google Scholar] [CrossRef]
- Fischer, B.; Muller, B.; Fischer, K.G.; Baur, N.; Kreutz, W. Acidic pH inhibits non-MHC-restricted killer cell functions. Clin. Immunol. 2000, 96, 252–263. [Google Scholar] [CrossRef]
- Motallebnezhad, M.; Jadidi-Niaragh, F.; Qamsari, E.S.; Bagheri, S.; Gharibi, T.; Yousefi, M. The immunobiology of myeloid-derived suppressor cells in cancer. Tumor Biol. 2016, 37, 1387–1406. [Google Scholar] [CrossRef]
- Selleri, S.; Bifsha, P.; Civini, S.; Pacelli, C.; Dieng, M.M.; Lemieux, W.; Jin, P.; Bazin, R.; Patey, N.; Marincola, F.M.; et al. Human mesenchymal stromal cell-secreted lactate induces M2-macrophage differentiation by metabolic reprogramming. Oncotarget 2016, 7, 30193–30210. [Google Scholar] [CrossRef]
- El-Kenawi, A.; Gatenbee, C.; Robertson-Tessi, M.; Bravo, R.; Dhillon, J.; Balagurunathan, Y.; Berglund, A.; Visvakarma, N.; Ibrahim-Hashim, A.; Choi, J.; et al. Acidity promotes tumor progression by altering macrophage phenotype in prostate cancer. bioRxiv 2018. [Google Scholar] [CrossRef]
- Martinez, D.; Vermeulen, M.; von Euw, E.; Sabatte, J.; Maggini, J.; Ceballos, A.; Trevani, A.; Nahmod, K.; Salamone, G.; Barrio, M.; et al. Extracellular acidosis triggers the maturation of human dendritic cells and the production of IL-12. J. Immunol. 2007, 179, 1950–1959. [Google Scholar] [CrossRef]
- Potzl, J.; Roser, D.; Bankel, L.; Homberg, N.; Geishauser, A.; Brenner, C.D.; Weigand, M.; Rocken, M.; Mocikat, R. Reversal of tumor acidosis by systemic buffering reactivates NK cells to express IFN-Î3 and induces NK cell-dependent lymphoma control without other immunotherapies. Int. J. Cancer 2017, 140, 2125–2133. [Google Scholar] [CrossRef]
- Lv, L.H.; Yu, J.D.; Li, G.L.; Long, T.Z.; Zhang, W.; Chen, Y.J.; Min, J.; Wan, Y.L. Functional distinction of rat liver natural killer cells from spleen natural killer cells under normal and acidic conditions in vitro. Hepatobiliary Pancreat. Dis. Int. 2012, 11, 285–293. [Google Scholar] [CrossRef]
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A.; et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009, 69, 2260–2268. [Google Scholar] [CrossRef]
- Islam, A.; Li, S.S.; Oykhman, P.; Timm-McCann, M.; Huston, S.M.; Stack, D.; Xiang, R.F.; Kelly, M.M.; Mody, C.H. An acidic microenvironment increases NK cell killing of Cryptococcus neoformans and Cryptococcus gattii by enhancing perforin degranulation. PLoS Pathog. 2013, 9, e1003439. [Google Scholar] [CrossRef]
- Ramadori, G.; Armbrust, T. Cytokines in the liver. Eur. J. Gastroenterol. Hepatol. 2001, 13, 777–784. [Google Scholar] [CrossRef]
- Li, P.; He, K.; Li, J.; Liu, Z.; Gong, J. The role of Kupffer cells in hepatic diseases. Mol. Immunol. 2017, 85, 222–229. [Google Scholar] [CrossRef]
- Tsutsui, H.; Nishiguchi, S. Importance of Kupffer cells in the development of acute liver injuries in mice. Int. J. Mol. Sci. 2014, 15, 7711–7730. [Google Scholar] [CrossRef]
- Sudo, K.; Yamada, Y.; Saito, K.; Shimizu, S.; Ohashi, H.; Kato, T.; Moriwaki, H.; Ito, H.; Seishima, M. TNF-alpha and IL-6 signals from the bone marrow derived cells are necessary for normal murine liver regeneration. Biochim. Biophys. Acta 2008, 1782, 671–679. [Google Scholar] [CrossRef]
- Knolle, P.; Schlaak, J.; Uhrig, A.; Kempf, P.; Meyer zum Buschenfelde, K.H.; Gerken, G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J. Hepatol. 1995, 22, 226–229. [Google Scholar] [CrossRef]
- Bishayee, A. The role of inflammation and liver cancer. Adv. Exp. Med. Biol. 2014, 816, 401–435. [Google Scholar]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef]
- Ueda, T.; Shimada, E.; Urakawa, T. Serum levels of cytokines in patients with colorectal cancer: Possible involvement of interleukin-6 and interleukin-8 in hematogenous metastasis. J. Gastroenterol. 1994, 29, 423–429. [Google Scholar] [CrossRef]
- Hammerich, L.; Tacke, F. Interleukins in chronic liver disease: Lessons learned from experimental mouse models. Clin. Exp. Gastroenterol. 2014, 7, 297–306. [Google Scholar]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef]
- Santegoets, S.J.; Turksma, A.W.; Suhoski, M.M.; Stam, A.G.; Albelda, S.M.; Hooijberg, E.; Scheper, R.J.; van den Eertwegh, A.J.; Gerritsen, W.R.; Powell, D.J.; et al. IL-21 promotes the expansion of CD27+ CD28+ tumor infiltrating lymphocytes with high cytotoxic potential and low collateral expansion of regulatory T cells. J. Transl. Med. 2013, 11, 37. [Google Scholar] [CrossRef]
- Skak, K.; Frederiksen, K.S.; Lundsgaard, D. Interleukin-21 activates human natural killer cells and modulates their surface receptor expression. Immunology 2008, 123, 575–583. [Google Scholar] [CrossRef]
- Burgess, S.J.; Marusina, A.I.; Pathmanathan, I.; Borrego, F.; Coligan, J.E. IL-21 down-regulates NKG2D/DAP10 expression on human NK and CD8+ T cells. J. Immunol. 2006, 176, 1490–1497. [Google Scholar] [CrossRef]
- Carson, W.E.; Ross, M.E.; Baiocchi, R.A.; Marien, M.J.; Boiani, N.; Grabstein, K.; Caligiuri, M.A. Endogenous production of interleukin 15 by activated human monocytes is critical for optimal production of interferon-gamma by natural killer cells in vitro. J. Clin. Investig. 1995, 96, 2578–2582. [Google Scholar] [CrossRef]
- Yoshikai, Y.; Nishimura, H. The role of interleukin 15 in mounting an immune response against microbial infections. Microbes Infect. 2000, 2, 381–389. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Kelly, A.M.; Doherty, D.G.; Traynor, O.; McEntee, G.; Kelly, J.; Hegarty, J.E.; O’Farrelly, C. Hepatic interleuklin 15 (IL-15) expression: Implications for local NK/NKT cell homeostasis and development. Clin. Exp. Immunol. 2004, 138, 94–101. [Google Scholar] [CrossRef]
- Easom, N.J.W.; Stegmann, K.A.; Swadling, L.; Pallett, L.J.; Burton, A.R.; Odera, D.; Schmidt, N.; Huang, W.C.; Fusai, G.; Davidson, B.; et al. IL-15 Overcomes Hepatocellular Carcinoma-Induced NK Cell Dysfunction. Front. Immunol. 2018, 9, 1009. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Zhang, J.; Liu, H.; Wan, L.; Zhang, H.; Huang, Q.; Xu, E.; Lai, M. IL-13/STAT6 signaling plays a critical role in the epithelial-mesenchymal transition of colorectal cancer cells. Oncotarget 2016, 7, 61183–61198. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Munker, S.; Mullenbach, R.; Weng, H.L. IL-13 Signaling in Liver Fibrogenesis. Front. Immunol. 2012, 3, 116. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Gabrilovich, D.I. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cancer Metastasis Rev. 2006, 25, 323–331. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Lassen, M.G.; Lukens, J.R.; Dolina, J.S.; Brown, M.G.; Hahn, Y.S. Intrahepatic IL-10 maintains NKG2A+Ly49- liver NK cells in a functionally hyporesponsive state. J. Immunol. 2010, 184, 2693–2701. [Google Scholar] [CrossRef]
- Sun, C.; Xu, J.; Huang, Q.; Huang, M.; Wen, H.; Zhang, C.; Wang, J.; Song, J.; Zheng, M.; Sun, H.; et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology 2017, 6, e1264562. [Google Scholar] [CrossRef]
- Viel, S.; Marcais, A.; Guimaraes, F.S.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-Î2 inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal 2016, 9, ra19. [Google Scholar] [CrossRef]
- Otegbeye, F.; Ojo, E.; Moreton, S.; Mackowski, N.; Lee, D.A.; de Lima, M.; Wald, D.N. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE 2018, 13, e0191358. [Google Scholar]
- Sun, C.; Sun, H.Y.; Xiao, W.H.; Zhang, C.; Tian, Z.G. Natural killer cell dysfunction in hepatocellular carcinoma and NK cell-based immunotherapy. Acta Pharmacol. Sin. 2015, 36, 1191–1199. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.J.; Kim, Y.J. Hepatitis B viral load affects prognosis of hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 12039–12044. [Google Scholar] [CrossRef]
- Yoon, J.C.; Yang, C.M.; Song, Y.; Lee, J.M. Natural killer cells in hepatitis C: Current progress. World J. Gastroenterol. 2016, 22, 1449–1460. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, X.M.; Li, S.R.; Twelkmeyer, T.; Wang, W.H.; Zhang, S.Y.; Wang, S.F.; Chen, J.Z.; Jin, X.; Wu, Y.Z.; et al. NKG2A is a NK cell exhaustion checkpoint for HCV persistence. Nat. Commun. 2019, 10, 1507. [Google Scholar] [CrossRef]
- Strunz, B.; Hengst, J.; Deterding, K.; Manns, M.P.; Cornberg, M.; Ljunggren, H.G.; Wedemeyer, H.; Bjorkstrom, N.K. Chronic hepatitis C virus infection irreversibly impacts human natural killer cell repertoire diversity. Nat. Commun. 2018, 9, 2275. [Google Scholar] [CrossRef]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef]
- Bernardini, G.; Antonangeli, F.; Bonanni, V.; Santoni, A. Dysregulation of Chemokine/Chemokine Receptor Axes and NK Cell Tissue Localization during Diseases. Front. Immunol. 2016, 7, 402. [Google Scholar] [CrossRef]
- Sungur, C.M.; Murphy, W.J. Positive and negative regulation by NK cells in cancer. Crit. Rev. Oncog. 2014, 19, 57–66. [Google Scholar] [CrossRef]
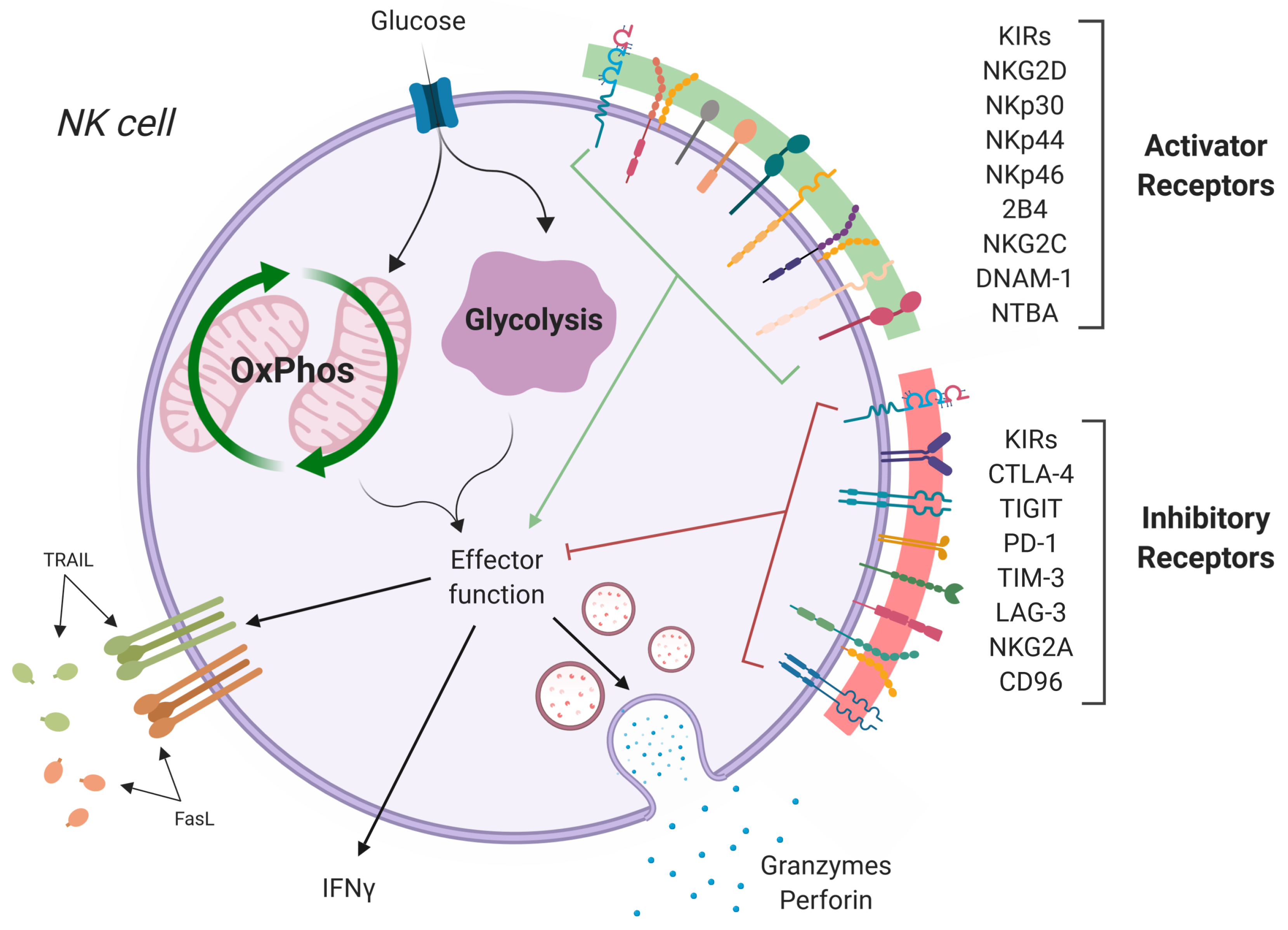
Figure 1.
NK cell activation/inhibition. NK cells become activated through a complex network of activating receptors (green) and inhibitory receptors (red). Loss of inhibition or amplification of activating signals trigger NK cell activation, inducing metabolic changes and driving effector functions, including release of cytotoxic granules, pro-inflammatory cytokines (IFN), and death receptor signaling (TRAIL, FasL). Figure created with BioRender.com.
Figure 1.
NK cell activation/inhibition. NK cells become activated through a complex network of activating receptors (green) and inhibitory receptors (red). Loss of inhibition or amplification of activating signals trigger NK cell activation, inducing metabolic changes and driving effector functions, including release of cytotoxic granules, pro-inflammatory cytokines (IFN), and death receptor signaling (TRAIL, FasL). Figure created with BioRender.com.

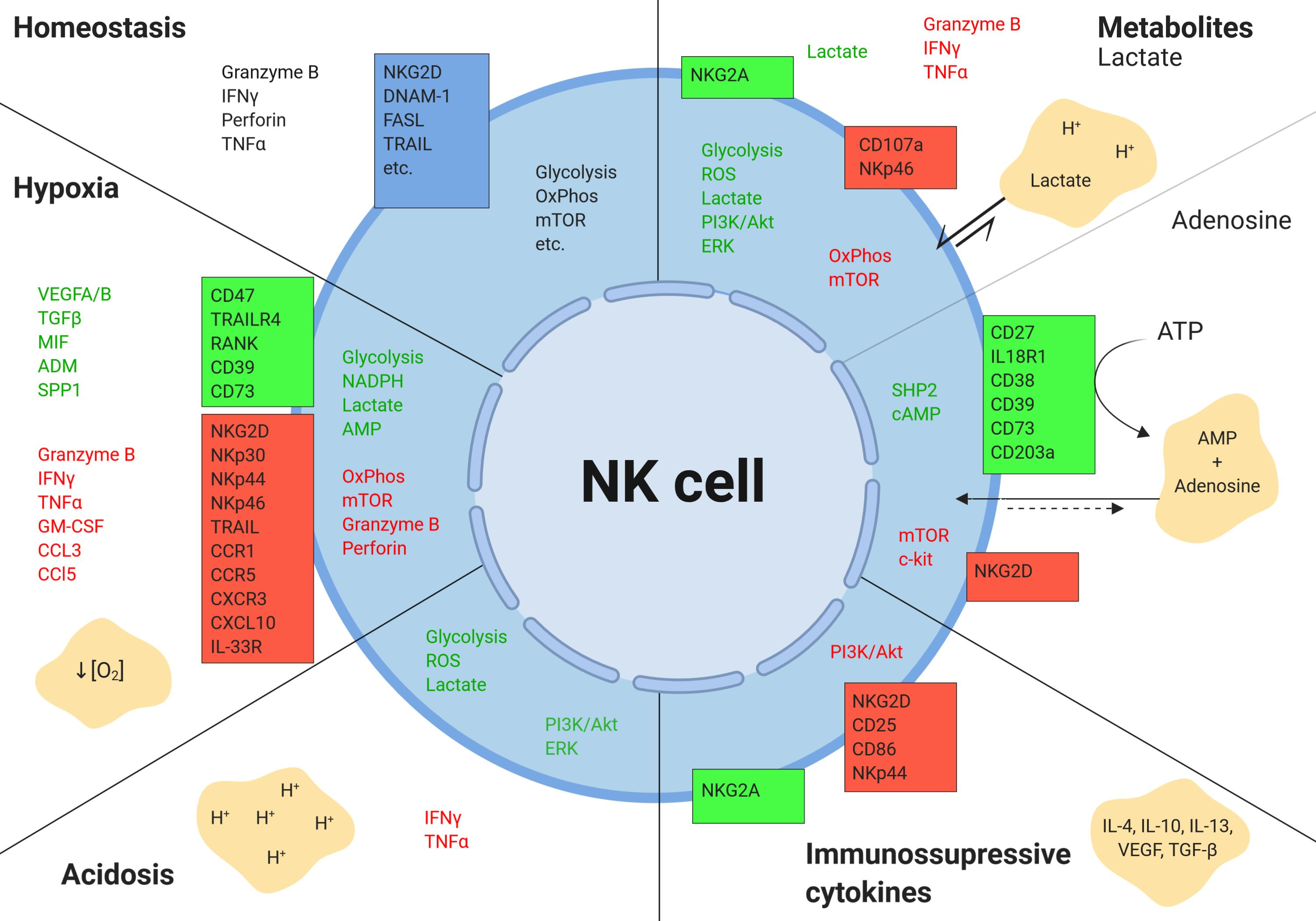
Figure 2.
Components of the tumor microenvironment. The tumor microenvironment is a complex ecosystem of heterogeneous tumor cells, stromal cells, and a variety of immune cells residing in a network of dysregulated vasculature and collagen. Poor perfusion and densely-packed glycolytic tumor cells create pockets of diminished oxygen levels, acidic pH, poor nutrient loads, anti-inflammatory cytokines, chemokines, and accumulated metabolic by-products, such as lactate. Tumor-infiltrating immune cells of both the myeloid and lymphoid lineages are found within the TME. Figure created with BioRender.com.
Figure 2.
Components of the tumor microenvironment. The tumor microenvironment is a complex ecosystem of heterogeneous tumor cells, stromal cells, and a variety of immune cells residing in a network of dysregulated vasculature and collagen. Poor perfusion and densely-packed glycolytic tumor cells create pockets of diminished oxygen levels, acidic pH, poor nutrient loads, anti-inflammatory cytokines, chemokines, and accumulated metabolic by-products, such as lactate. Tumor-infiltrating immune cells of both the myeloid and lymphoid lineages are found within the TME. Figure created with BioRender.com.

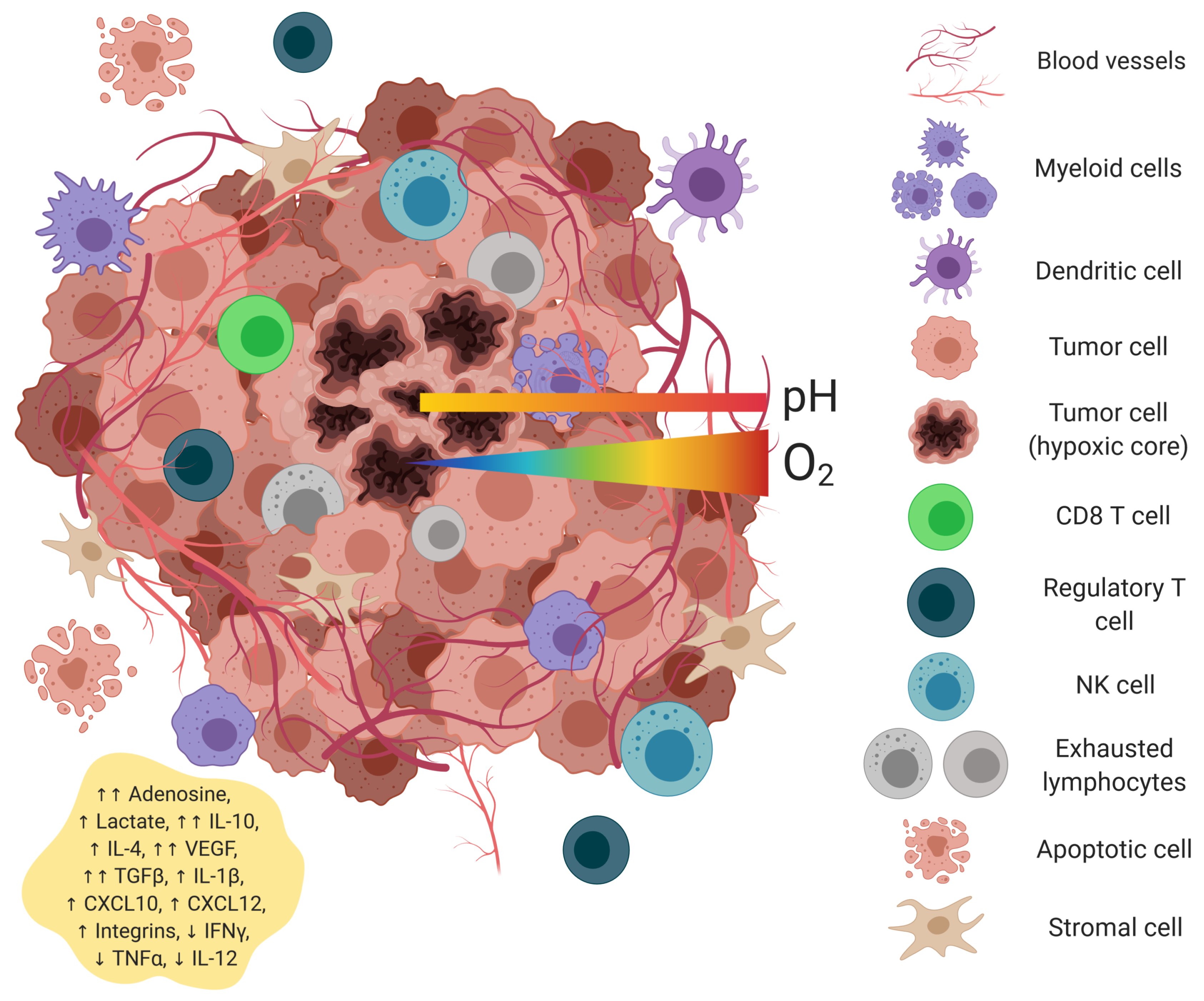
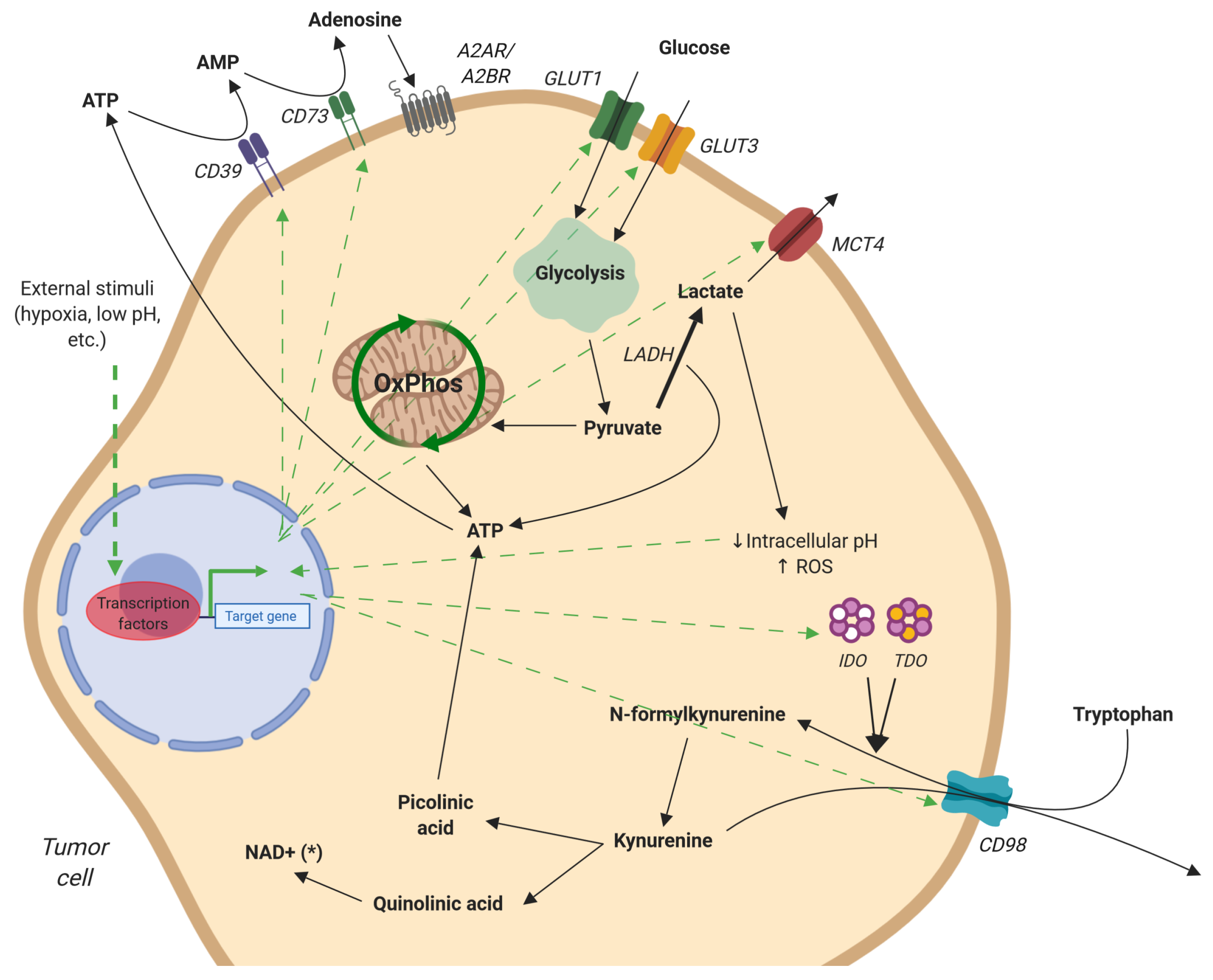
Figure 3.
Metabolism of adenosine, glucose, and tryptophan by tumor cells. Free adenosine is produced by the cleavage of extracellular ATP by CD39 and CD73. Signaling through A2AR, adenosine initiates an anti-inflammatory response. Glucose uptake is increased, via GLUT1/3, after which it is converted to pyruvate via glycolysis. Excess pyruvate is transformed into lactate and either exported by MCT4 or remains in the cytoplasm, decreasing cellular pH, resulting in ROS production. Tryptophan, transported by CD98, is transformed to kynurenine by IDO and TDO. Kynurenine is either released from the cell via CD98 or further processed to quinolinic acid and picolinic acid. These intermediates can be used for the generation of NAD+ and ATP, respectively. Green arrows represent stimulation or upregulation. Figure created with BioRender.com.
Figure 3.
Metabolism of adenosine, glucose, and tryptophan by tumor cells. Free adenosine is produced by the cleavage of extracellular ATP by CD39 and CD73. Signaling through A2AR, adenosine initiates an anti-inflammatory response. Glucose uptake is increased, via GLUT1/3, after which it is converted to pyruvate via glycolysis. Excess pyruvate is transformed into lactate and either exported by MCT4 or remains in the cytoplasm, decreasing cellular pH, resulting in ROS production. Tryptophan, transported by CD98, is transformed to kynurenine by IDO and TDO. Kynurenine is either released from the cell via CD98 or further processed to quinolinic acid and picolinic acid. These intermediates can be used for the generation of NAD+ and ATP, respectively. Green arrows represent stimulation or upregulation. Figure created with BioRender.com.

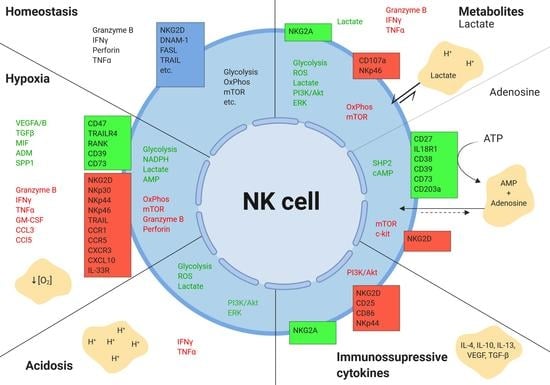
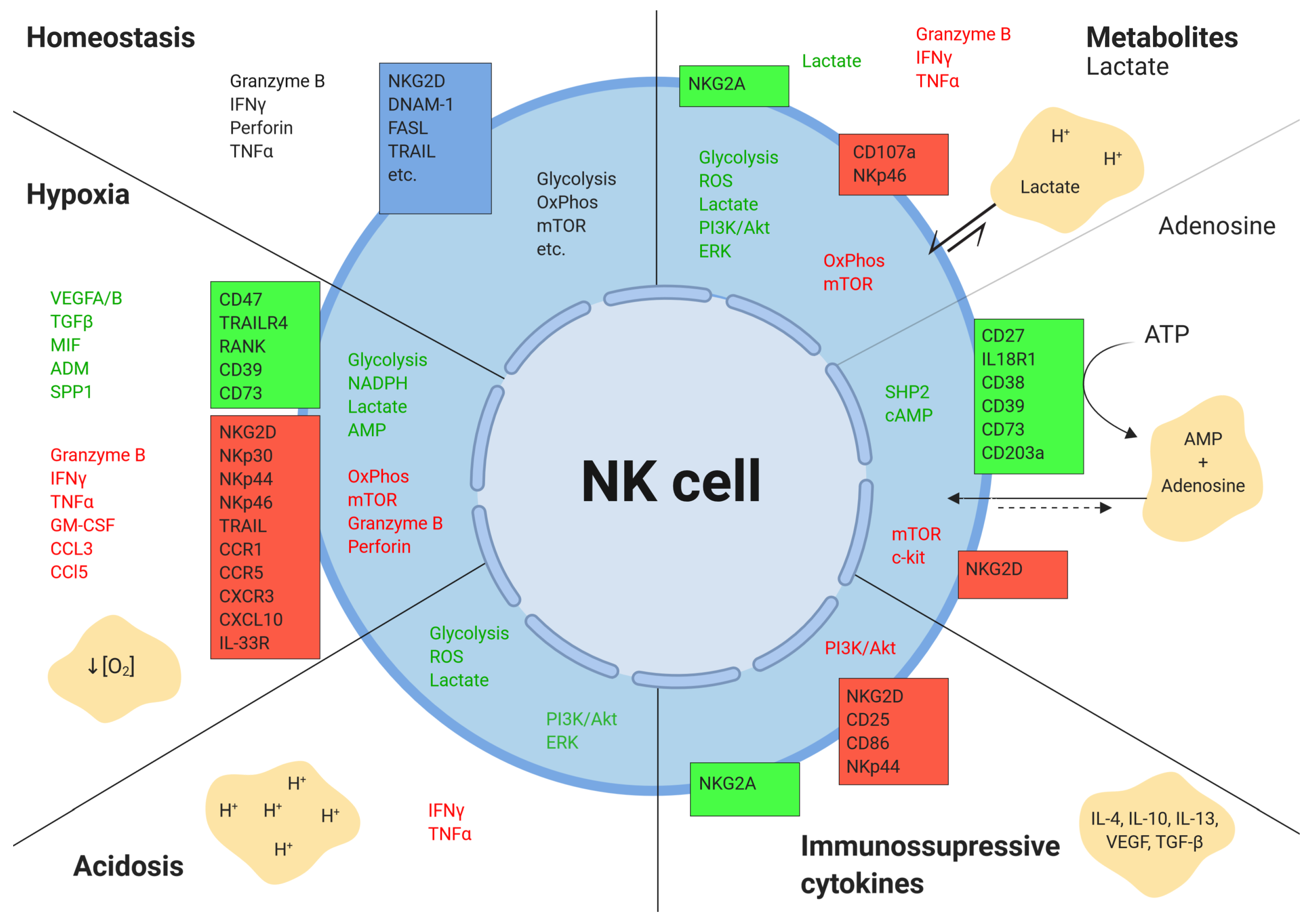
Figure 4.
Effects of the tumor microenvironment on NK cells.The tumor microenvironment alters the expression of various secreted, surface, and intracellular molecules in NK cells. Boxes represent surface receptors. Green indicates upregulation, and red depicts downregulation. Figure created with BioRender.com.
Figure 4.
Effects of the tumor microenvironment on NK cells.The tumor microenvironment alters the expression of various secreted, surface, and intracellular molecules in NK cells. Boxes represent surface receptors. Green indicates upregulation, and red depicts downregulation. Figure created with BioRender.com.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Piñeiro Fernández, J.; Luddy, K.A.; Harmon, C.; O’Farrelly, C. Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function. Int. J. Mol. Sci. 2019, 20, 4131. https://doi.org/10.3390/ijms20174131
AMA Style
Piñeiro Fernández J, Luddy KA, Harmon C, O’Farrelly C. Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function. International Journal of Molecular Sciences. 2019; 20(17):4131. https://doi.org/10.3390/ijms20174131
Chicago/Turabian StylePiñeiro Fernández, Julián, Kimberly A. Luddy, Cathal Harmon, and Cliona O’Farrelly. 2019. "Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function" International Journal of Molecular Sciences 20, no. 17: 4131. https://doi.org/10.3390/ijms20174131
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.