p53, miR-34a and EMP1—Newly Identified Targets of TFF3 Signaling in Y79 Retinoblastoma Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
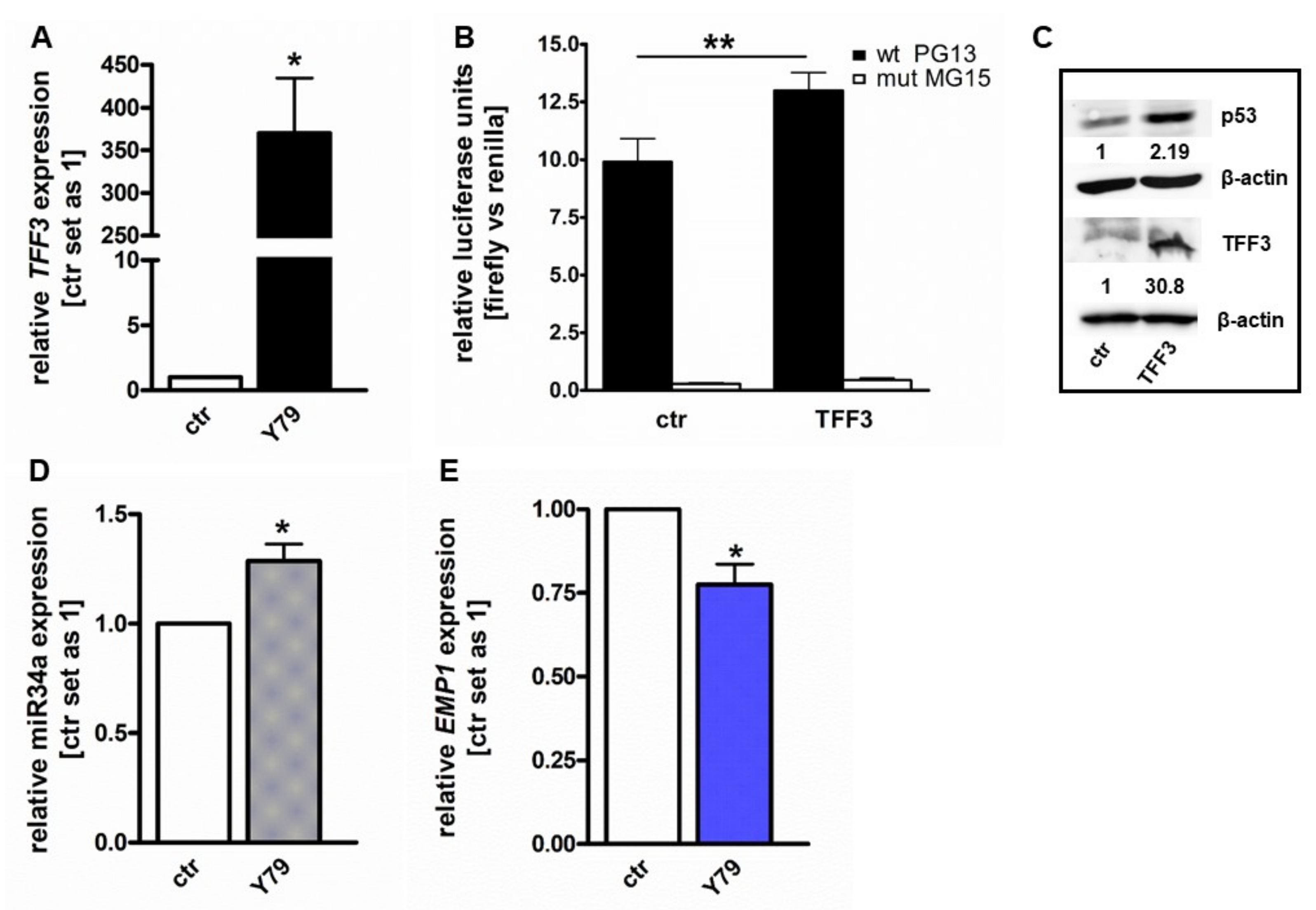
2.1. TFF3 Overexpression Activates p53 and miR-34a and Inhibits EMP1 Expression
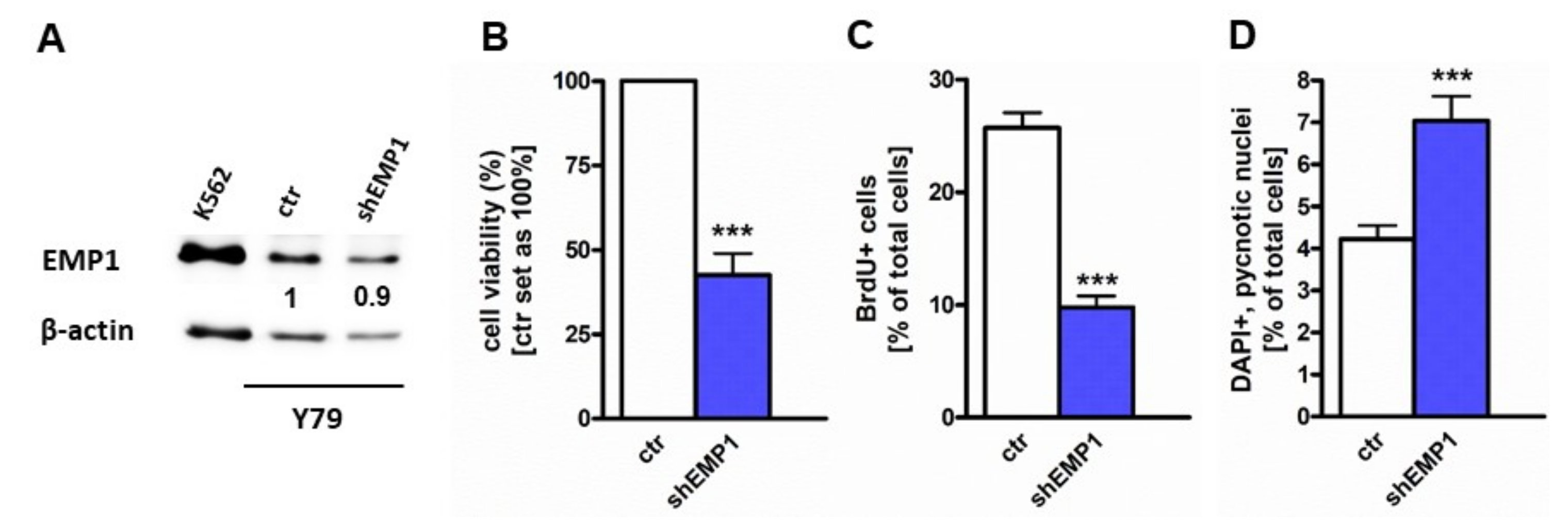
2.2. EMP1 Knockdown Inhibits Growth and Induces Apoptosis in Y79 Cells
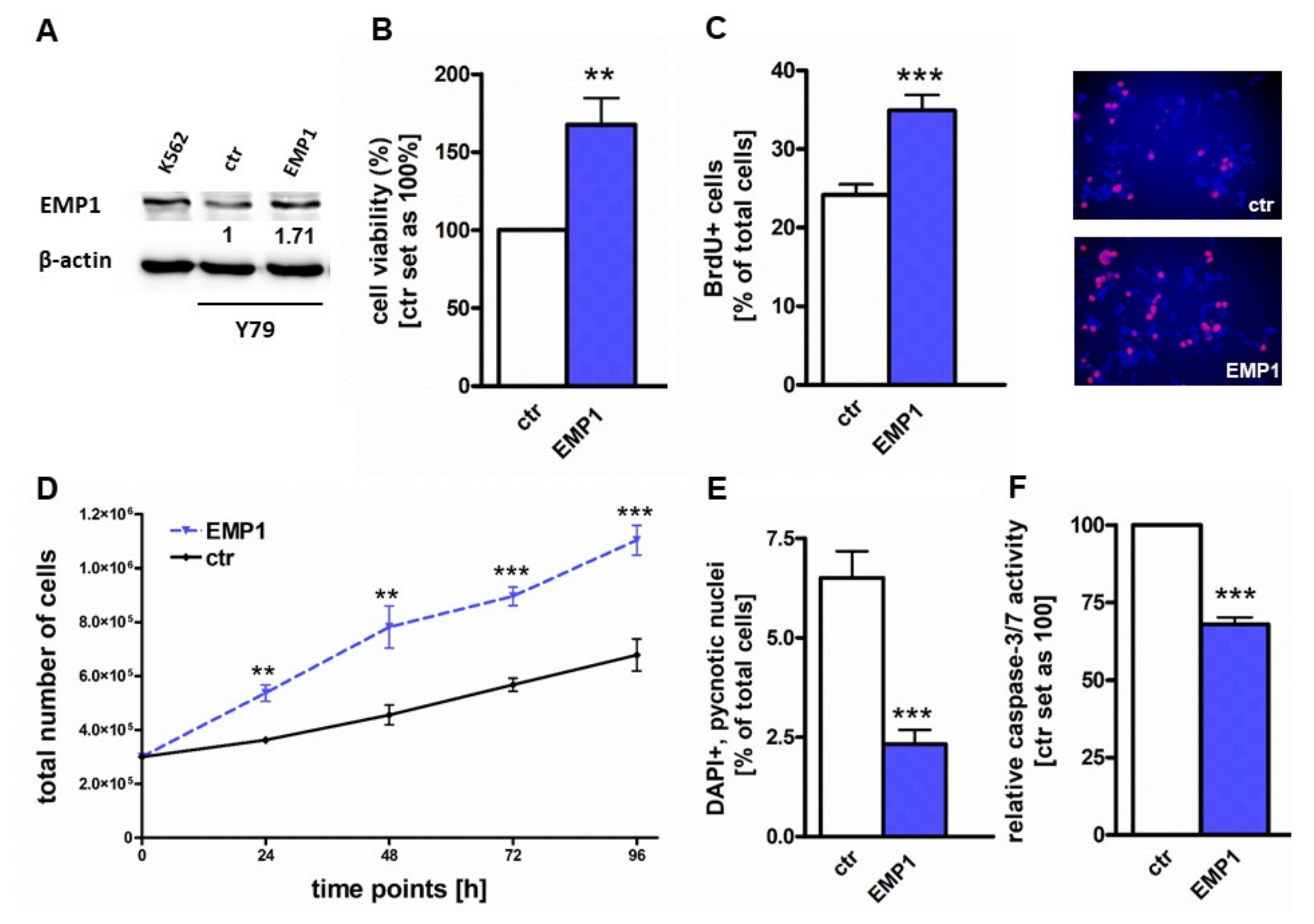
2.3. EMP1 Overexpression Induces Growth and Inhibits Apoptosis in Y79 Cells
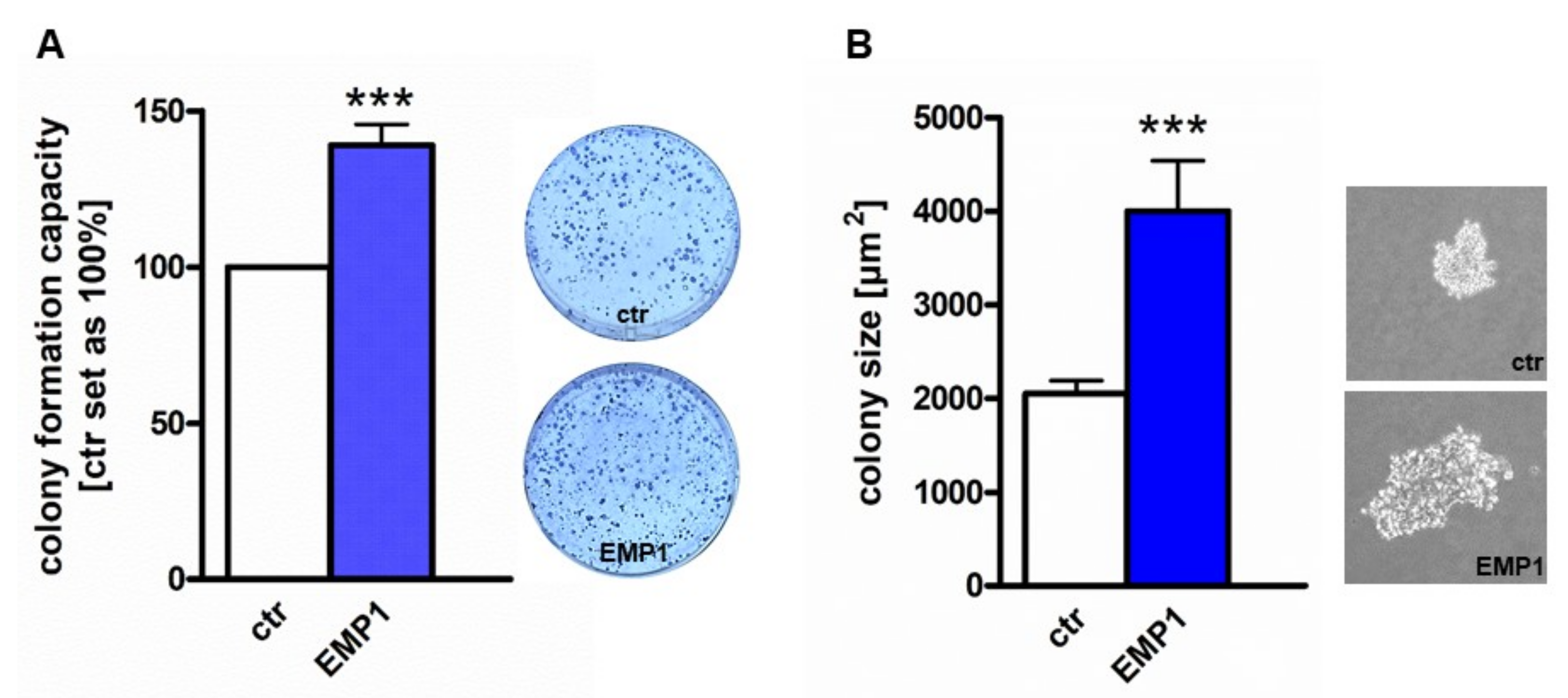
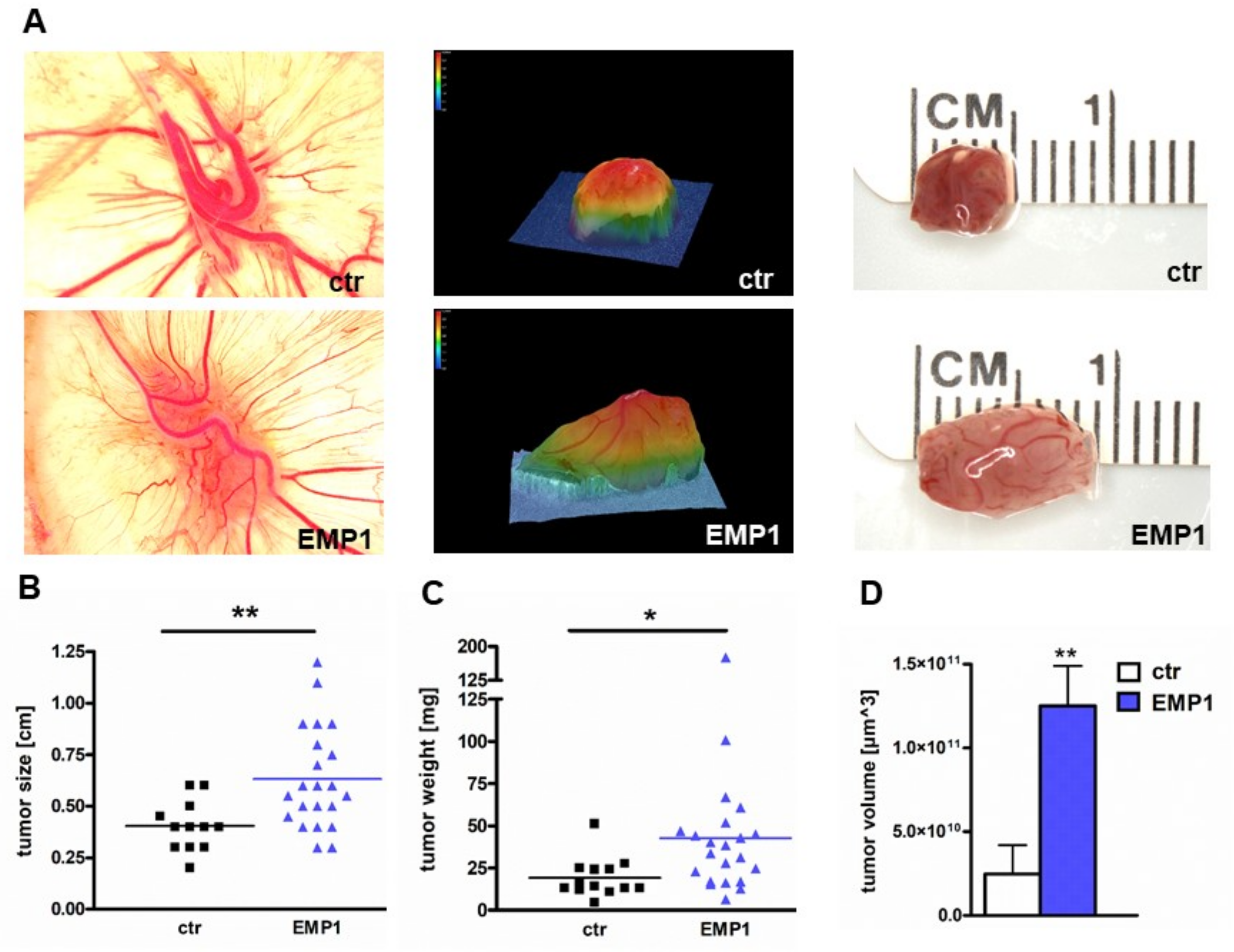
2.4. EMP1 Overexpression Increases Tumorigenicity of Y79 Cells In Vitro and In Vivo
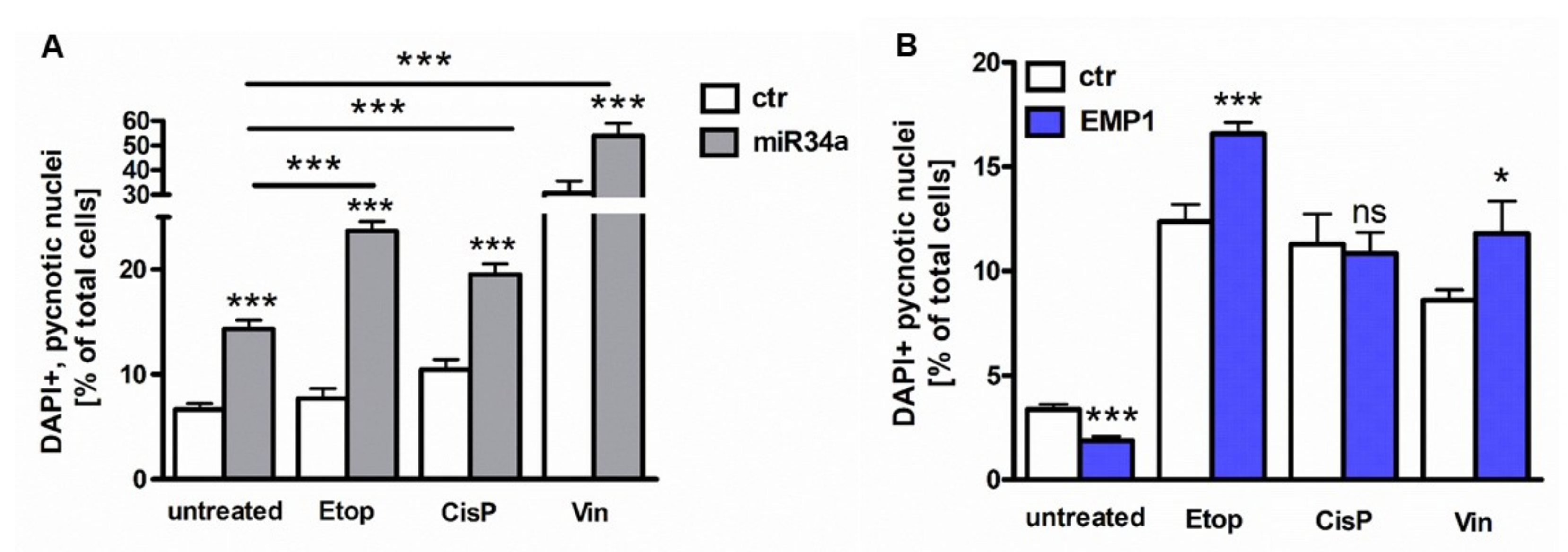
2.5. MiR-34a and EMP1 Overexpression Enhances Chemosensitivity of Y79 RB Cells
3. Discussion
4. Material and Methods
4.1. Human Retina and Retinoblastoma Samples
4.2. Cell Culture
4.3. Treatment with Chemotherapeutics
4.4. Plasmids and Lentiviral Expression Vectors
4.5. TFF3 and EMP1 Overexpression and EMP1 Knockdown in Y79 Retinoblastoma Cells
4.6. RNA Extraction and Quantitative Real-Time PCR
4.7. Western Blotting
4.8. Cell Viability
4.9. Growth Kinetic
4.10. Cell Proliferation and Apoptosis Detection
4.11. Caspase-Glo 3/7 Assay
4.12. Colony Formation and Soft Agarose Assay
4.13. CAM Assay
4.14. Luciferase Assay
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dimaras, H.; Kimani, K.; Dimba, E.A.O.; Gronsdahl, P.; White, A.; Chan, H.S.L.; Gallie, B.L. Retinoblastoma. Lancet 2012, 379, 1436–1446. [Google Scholar] [CrossRef] [Green Version]
- Dimaras, H.; Corson, T.W. Retinoblastoma, the visible CNS tumor: A review. J. Neurosci. Res. 2018, 97, 29–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corson, T.W.; Gallie, B.L. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosom. Cancer 2007, 46, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Dimaras, H.; Khetan, V.; Halliday, W.; Orlic-Milacic, M.; Prigoda, N.L.; Piovesan, B.; Marrano, P.; Corson, T.W.; Eagle, R.C.; Squire, J.A.; et al. Loss of RB1 induces non-proliferative retinoma: Increasing genomic instability correlates with progression to retinoblastoma. Hum. Mol. Genet. 2008, 17, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Rushlow, D.E.; Mol, B.M.; Kennett, J.Y.; Yee, S.; Pajovic, S.; Thériault, B.L.; Prigoda-Lee, N.L.; Spencer, C.; Dimaras, H.; Corson, T.W.; et al. Characterisation of retinoblastomas without RB1 mutations: Genomic, gene expression, and clinical studies. Lancet Oncol. 2013, 14, 327–334. [Google Scholar] [CrossRef]
- Singh, U.; Malik, M.A.; Goswami, S.; Shukla, S.; Kaur, J. Epigenetic regulation of human retinoblastoma. Tumor Boil. 2016, 37, 14427–14441. [Google Scholar] [CrossRef]
- Yousef, Y.A.; Soliman, S.E.; Astudillo, P.P.P.; Durairaj, P.; Dimaras, H.; Chan, H.S.L.; Héon, E.; Gallie, B.L.; Shaikh, F. Intra-arterial Chemotherapy for Retinoblastoma. JAMA Ophthalmol. 2016, 134, 1202. [Google Scholar] [CrossRef]
- Munier, F.L.; Gaillard, M.-C.; Balmer, A.; Soliman, S.; Podilsky, G.; Moulin, A.P.; Beck-Popovic, M. Intravitreal chemotherapy for vitreous disease in retinoblastoma revisited: From prohibition to conditional indications. Br. J. Ophthalmol. 2012, 96, 1078–1083. [Google Scholar] [CrossRef]
- Hoffmann, W.; Hauser, F. The P-domain or trefoil motif: A role in renewal and pathology of mucous epithelia? Trends Biochem. Sci. 1993, 18, 239–243. [Google Scholar] [CrossRef]
- Hoffmann, W. Trefoil factors TFF (trefoil factor family) peptide-triggered signals promoting mucosal restitution. Cell. Mol. Life Sci. CMLS 2005, 62, 2932–2938. [Google Scholar] [CrossRef]
- Thim, L.; May, F.E.B. Structure of mammalian trefoil factors and functional insights. Cell. Mol. Life Sci. CMLS 2005, 62, 2956–2973. [Google Scholar] [CrossRef] [PubMed]
- Kjellev, S. The trefoil factor family—Small peptides with multiple functionalities. Cell. Mol. Life Sci. CMLS 2009, 66, 1350–1369. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.; Dünker, N. Trefoil factor family peptides—Friends or foes? Biomol. Concepts 2015, 6, 343–359. [Google Scholar] [CrossRef] [PubMed]
- McFall, R.C.; Sery, T.W.; Makadon, M. Characterization of a new continuous cell line derived from a human retinoblastoma. Cancer Res. 1977, 37, 1003–1010. [Google Scholar] [PubMed]
- Weise, A.; Dünker, N. High trefoil factor 1 (TFF1) expression in human retinoblastoma cells correlates with low growth kinetics, increased cyclin-dependent kinase (CDK) inhibitor levels and a selective down-regulation of CDK6. Histochem. Cell Biol. 2013, 139, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Khoury, T.; Chadha, K.; Javle, M.; Donohue, K.; LeVea, C.; Iyer, R.; Okada, H.; Nagase, H.; Tan, D. Expression of Intestinal Trefoil Factor (TFF-3) in Hepatocellular Carcinoma. Int. J. Pancreatol. 2005, 35, 171–178. [Google Scholar] [CrossRef]
- Yamachika, T.; Werther, J.L.; Bodian, C.; Babyatsky, M.; Tatematsu, M.; Yamamura, Y.; Chen, A.; Itzkowitz, S. Intestinal trefoil factor: A marker of poor prognosis in gastric carcinoma. Clin. Cancer Res. 2002, 8, 1092–1099. [Google Scholar] [PubMed]
- Philippeit, C.; Busch, M.; Dünker, N. Epigenetic Control of Trefoil Factor Family (TFF) Peptide Expression in Human Retinoblastoma Cell Lines. Cell. Physiol. Biochem. 2014, 34, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Große-Kreul, J.; Busch, M.; Winter, C.; Pikos, S.; Stephan, H.; Dünker, N. Forced Trefoil Factor Family Peptide 3 (TFF3) Expression Reduces Growth, Viability, and Tumorigenicity of Human Retinoblastoma Cell Lines. PLoS ONE 2016, 11, e0163025. [Google Scholar] [CrossRef] [PubMed]
- Soutto, M.; Chen, Z.; Saleh, M.A.; Katsha, A.; Zhu, S.; Zaika, A.; Belkhiri, A.; El-Rifai, W. TFF1 activates p53 through down-regulation of miR-504 in gastric cancer. Oncotarget 2014, 5, 5663–5673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, M.; Große-Kreul, J.; Wirtz, J.J.; Beier, M.; Stephan, H.; Royer-Pokora, B.; Metz, K.; Dünker, N.; Große-Kreul, J.; Royer-Pokora, B. Reduction of the tumorigenic potential of human retinoblastoma cell lines by TFF1 overexpression involves p53/caspase signaling and miR-18a regulation. Int. J. Cancer 2017, 141, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Lowenstein, C.J. MiR-34, SIRT1, and p53: The feedback loop. Cell Cycle 2009, 8, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Huang, J.; Xie, M.; Yu, Y.; Zhu, S.; Kang, R.; Cao, L.; Tang, D.; Duan, X. MIR34A regulates autophagy and apoptosis by targeting HMGB1 in the retinoblastoma cell. Autophagy 2014, 10, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Dalgard, C.L.; Gonzalez, M.; Deniro, J.E.; O’Brien, J.M. Differential MicroRNA-34a Expression and Tumor Suppressor Function in Retinoblastoma Cells. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4542–4551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-W.; Cheng, H.-L.; Ding, Y.-R.; Chou, L.-H.; Chow, N.-H. EMP1, EMP 2, and EMP3 as novel therapeutic targets in human cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2017, 1868, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.K.; Kannan, N.; Grandison, P.M.; Mitchell, M.D.; Lobie, P.E. Are trefoil factors oncogenic? Trends Endocrinol. Metab. TEM 2008, 19, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Emami, S.; Rodrigues, S.; Rodrigue, C.M.; Le Floch, N.; Rivat, C.; Attoub, S.; Bruyneel, E.; Gespach, C. Trefoil factor family (TFF) peptides and cancer progression. Mol. Med. TFF Pept. 2004, 25, 885–898. [Google Scholar]
- Levak, M.T.; Mihalj, M.; Koprivčić, I.; Lovrić, I.; Novak, S.; Bijelić, N.; Lončar, M.B.; Belovari, T.; Kralik, K.; Pauzar, B. Differential expression of tff genes and proteins in breast tumors. Acta Clin. Croat. 2018, 57, 264–277. [Google Scholar]
- Buache, E.; Etique, N.; Alpy, F.; Stoll, I.; Muckensturm, M.; Reina-San-Martin, B.; Chenard, M.P.; Tomasetto, C.; Rio, M.C. Deficiency in trefoil factor 1 (TFF1) increases tumorigenicity of human breast cancer cells and mammary tumor development in TFF1-knockout mice. Oncogene 2011, 30, 3261–3273. [Google Scholar] [CrossRef] [Green Version]
- Abols, A.; Ducena, K.; Andrejeva, D.; Sadovska, L.; Zandberga, E.; Vilmanis, J.; Narbuts, Z.; Tars, J.; Eglitis, J.; Pirags, V.; et al. Trefoil factor 3 is required for differentiation of thyroid follicular cells and acts as a context-dependent tumor suppressor. Neoplasma 2015, 62, 914–924. [Google Scholar] [CrossRef] [Green Version]
- Misso, G.; Di Martino, M.T.; De Rosa, G.; Farooqi, A.A.; Lombardi, A.; Campani, V.; Zarone, M.R.; Gullà, A.; Tagliaferri, P.; Tassone, P.; et al. Mir-34: A new weapon against cancer? Molecular therapy. Nucleic Acids 2014, 3, e194. [Google Scholar] [PubMed]
- Dimopoulos, K.; Gimsing, P.; Grønbaek, K.; Grønbæk, K. Aberrant microRNA expression in multiple myeloma. Eur. J. Haematol. 2013, 91, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.M.; Li, C.W.; Li, Y.Y.; Li, T.Y.; Pan, X.L.; Liu, J.; Bin Lin, Z.; Zhao, L.; Shi, L.; Wang, D.Y. Down-regulation of EMP1 is associated with epithelial hyperplasia and metaplasia in nasal polyps. Histopathology 2013, 63, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Taylor, V.; Welcher, A.A.; Amgen, E.P.; Suter, U. Epithelial Membrane Protein-1, Peripheral Myelin Protein 22, and Lens Membrane Protein 20 Define a Novel Gene Family. J. Boil. Chem. 1995, 270, 28824–28833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredel, M. Functional Network Analysis Reveals Extended Gliomagenesis Pathway Maps and Three Novel MYC-Interacting Genes in Human Gliomas. Cancer Res. 2005, 65, 8679–8689. [Google Scholar] [CrossRef]
- Johnson, A.H.; Frierson, H.F.; Zaika, A.; Powell, S.M.; Roche, J.; Crowe, S.; Moskaluk, C.A.; El-Rifai, W. Expression of Tight-Junction Protein Claudin-7 Is an Early Event in Gastric Tumorigenesis. Am. J. Pathol. 2005, 167, 577–584. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-T.; Kong, J.-P.; Ding, F.; Wang, X.-Q.; Wang, M.-R.; Liu, L.-X.; Wu, M.; Liu, Z.-H. Analysis of gene expression profile induced by EMP-1 in esophageal cancer cells using cDNA Microarray. World J. Gastroenterol. 2003, 9, 392–398. [Google Scholar] [CrossRef]
- Kornberg, L.J.; Villaret, D.; Popp, M.; Lui, L.; McLaren, R.; Brown, H.; Cohen, D.; Yun, J.; McFadden, M. Gene Expression Profiling in Squamous Cell Carcinoma of the Oral Cavity Shows Abnormalities in Several Signaling Pathways. Laryngoscope 2005, 115, 690–698. [Google Scholar] [CrossRef]
- Sun, G.G.; Lu, Y.F.; Fu, Z.Z.; Cheng, Y.J.; Hu, W.N. EMP1 inhibits nasopharyngeal cancer cell growth and metastasis through induction apoptosis and angiogenesis. Tumour Biol. 2014, 35, 3185–3193. [Google Scholar] [CrossRef]
- Ariës, I.M.; Jerchel, I.S.; Dungen, R.E.S.R.V.D.; Berk, L.C.J.V.D.; Boer, J.M.; Horstmann, M.A.; Escherich, G.; Pieters, R.; Boer, M.L.D. EMP1, a novel poor prognostic factor in pediatric leukemia regulates prednisolone resistance, cell proliferation, migration and adhesion. Leukemia 2014, 28, 1828–1837. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Xiong, S.-H.; Hu, M.; Zhang, C.-S. Knockdown Epithelial Membrane Protein 1 Suppresses Human Degenerative Intervertebral Disc-Derived Nucleus Pulposus Cell Proliferation. Cartilage 2011, 2, 300–306. [Google Scholar] [CrossRef]
- Wang, X.; Dong, K.; Gao, P.; Long, M.; Lin, F.; Weng, Y.; Ouyang, Y.; Ren, J.; Zhang, H. microRNA-34a Sensitizes Lung Cancer Cell Lines to DDP Treatment Independent of p53 Status. Cancer Biother. Radiopharm. 2013, 28, 45–50. [Google Scholar] [CrossRef]
- Vinall, R.L.; Ripoll, A.Z.; Wang, S.; Pan, C.-X.; deVere White, R.W. MiR-34a chemosensitizes bladder cancer cells to cisplatin treatment regardless of p53-Rb pathway status. Int. J. Cancer 2012, 130, 2526–2538. [Google Scholar] [CrossRef]
- Reid, T.W.; Rabson, A.S.; Tralka, T.S.; Wilcox, J.L.; Albert, D.M.; Russell, P.; Craft, J.; Chu, E.W. Characteristics of an Established Cell Line of Retinoblastoma. J. Natl. Cancer Inst. 1974, 53, 347–360. [Google Scholar] [CrossRef]
- Busch, M.; Philippeit, C.; Weise, A.; Dünker, N. Re-characterization of established human retinoblastoma cell lines. Histochem. Cell Biol. 2015, 143, 325–338. [Google Scholar] [CrossRef]
- Stegmeier, F.; Hu, G.; Rickles, R.J.; Hannon, G.J.; Elledge, S.J. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc. Natl. Acad. Sci. USA 2005, 102, 13212–13217. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, L.; Neveling, K.; Borkens, S.; Schneider, H.; Freund, M.; Grassman, E.; Theiss, S.; Wawer, A.; Burdach, S.; Auerbach, A.D.; et al. Correct mRNA Processing at a Mutant TT Splice Donor in FANCC Ameliorates the Clinical Phenotype in Patients and Is Enhanced by Delivery of Suppressor U1 snRNAs. Am. J. Hum. Genet. 2010, 87, 480–493. [Google Scholar] [CrossRef] [Green Version]
- Haubold, M. Bone morphogenetic protein 4 (BMP4) signaling in retinoblastoma cells. Int. J. Boil. Sci. 2010, 6, 700–715. [Google Scholar] [CrossRef]
- Zijlstra, A.; Mellor, R.; Panzarella, G.; Aimes, R.T.; Hooper, J.D.; Marchenko, N.D.; Quigley, J.P. A quantitative analysis of rate-limiting steps in the metastatic cascade using human-specific real-time polymerase chain reaction. Cancer Res. 2002, 62, 7083–7092. [Google Scholar]
- Palmer, T.D.; Lewis, J.; Zijlstra, A. Quantitative Analysis of Cancer Metastasis using an Avian Embryo Model. J. Vis. Exp. 2011. [Google Scholar] [CrossRef]
- El-Deiry, W. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Elso, C.M.; Roberts, L.J.; Smyth, G.K.; Thomson, R.J.; Baldwin, T.M.; Foote, S.J.; Handman, E. Leishmaniasis host response loci (lmr1-3) modify disease severity through a Th1/Th2-independent pathway. Genes Immun. 2004, 5, 93–100. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Busch, M.; Klein, S.; Große-Kreul, J.; Scheiner, O.; Metz, K.; Stephan, H.; Dünker, N. p53, miR-34a and EMP1—Newly Identified Targets of TFF3 Signaling in Y79 Retinoblastoma Cells. Int. J. Mol. Sci. 2019, 20, 4129. https://doi.org/10.3390/ijms20174129
Busch M, Klein S, Große-Kreul J, Scheiner O, Metz K, Stephan H, Dünker N. p53, miR-34a and EMP1—Newly Identified Targets of TFF3 Signaling in Y79 Retinoblastoma Cells. International Journal of Molecular Sciences. 2019; 20(17):4129. https://doi.org/10.3390/ijms20174129
Chicago/Turabian StyleBusch, Maike, Stefan Klein, Jan Große-Kreul, Oliver Scheiner, Klaus Metz, Harald Stephan, and Nicole Dünker. 2019. "p53, miR-34a and EMP1—Newly Identified Targets of TFF3 Signaling in Y79 Retinoblastoma Cells" International Journal of Molecular Sciences 20, no. 17: 4129. https://doi.org/10.3390/ijms20174129
APA StyleBusch, M., Klein, S., Große-Kreul, J., Scheiner, O., Metz, K., Stephan, H., & Dünker, N. (2019). p53, miR-34a and EMP1—Newly Identified Targets of TFF3 Signaling in Y79 Retinoblastoma Cells. International Journal of Molecular Sciences, 20(17), 4129. https://doi.org/10.3390/ijms20174129