Perineuronal Nets and Their Role in Synaptic Homeostasis


 and
and 
Abstract
1. Introduction
2. The Role of Extracellular Matrix in the Functioning of the Blood-Brain Barrier
3. Structure of Perineuronal Nets
4. Characteristics of Perineuronal Nets Components
4.1. Hyaluronan
4.2. Proteoglycans
4.2.1. Glycosaminoglycans
4.2.2. Core Proteins
4.2.3. Chondroitin Sulphate Proteoglycans
4.2.4. Tenascins
4.2.5. Hapln Proteins
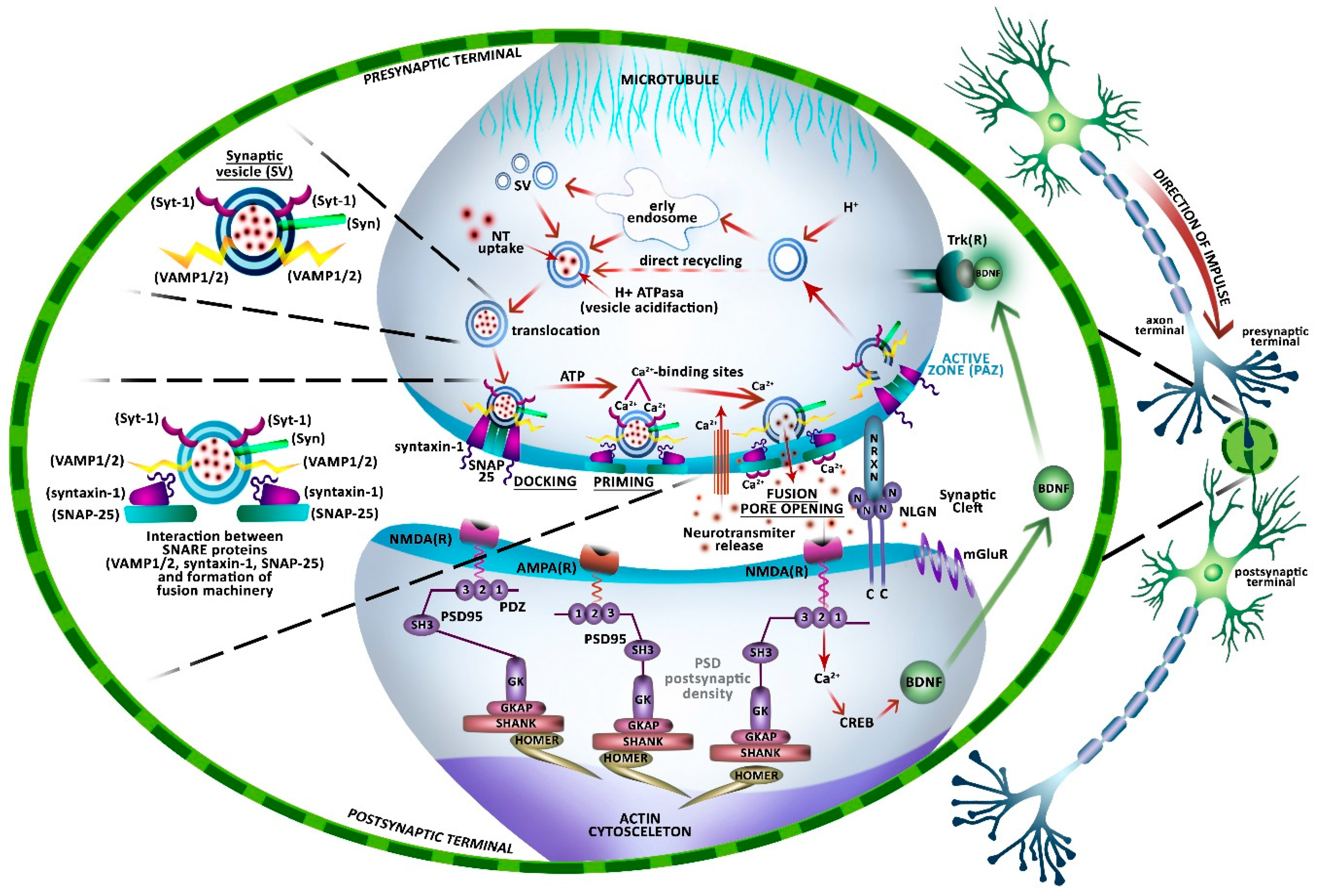
5. Neuronal Communication
6. Participation of Perineuronal Nets in Synaptic Plasticity
7. Perineuronal Nets - Clinical Aspects
8. Perspectives for Further Research
9. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPARs | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors |
| AD | Alzheimer’s disease |
| SHANK | SH3 and ankyrin repeat domains protein |
| ASD | Autism spectrum disorders |
| AQPs | Aquaporins |
| BBB | Blood brain barrier |
| BMECs | Brain microvascular endothelial cells |
| BDNF | Brain-derived neurotrophic factor |
| Bral1 | Brain-specific hyaluronan-binding protein 1 |
| Bral2 | Brain-specific hyaluronan-binding protein 2 |
| Crtl1 | Cartilage-linking protein 1 |
| VAMP3 | Cellubrevin |
| CS | Chondroitin sulphate |
| CSPGs | Chondroitin sulphate proteoglycans |
| ChABC | Chondroitinase ABC |
| DAI | Diffuse axonal injury |
| GAG-GAG | Disaccharide polymers glucosaminoglycan- glucosaminoglycan |
| DLG4 | Discs Large MAGUK scaffold protein 4 |
| VAMP8 | Endobrevin |
| ECM | Extracellular matrix |
| Gal | Galactose |
| GalNAc | Galactose-N-acetylogalactosamine |
| GABA | Gamma-aminobutyric acid |
| GCS | Glasgow Coma Scale |
| GPI | Glycosylphosphatidylinositol |
| GlcA | Glucosamine |
| GAGs | Glucosaminoglycans |
| GlcNAc | Glucose N-acetyloglucosamine |
| GlcA-GlcNAc | Glucuronic acid–acetylogalactosamine |
| GlcA | Glucuronic acid |
| GKAP | Guanylate kinase-associated protein |
| GK | Guanylate kinase-like domain |
| HOMER | Homer protein |
| HAPLN-1, -3 and -4 | Hyaluronan and proteoglycan binding link protein |
| HAS | Hyaluronan synthase |
| HA | Hyaluronic acid (hyaluronan, |
| IdoA | Iduronic acid |
| MAGUK | Membrane-associated guanylate guanyl kinases |
| mGluR | Metabotropic glutamate receptor |
| MMPs | Metalloproteinases |
| VAMP5 | Myobrevin |
| NRXN1 | Neurexin 1 |
| NRXN2 | Neurexin 2 |
| NRXN3 | Neurexin 3 |
| NLGN1 | Neuroligin 1 |
| NLGN3 | Neuroligin 3 |
| NLGN4X, | Neuroligin 4, X-linked precursor |
| NLGN4Y | Neuroligin 4, Y-linked precursor |
| NLGN | Neuroligin |
| NMDA(R) | N-methyl-D-aspartate receptors |
| PD | Parkinson’s disease |
| PNNs | Perineuronal nets |
| PSD | Postsynaptic densities |
| PSD95/DLG4 | Postsynaptic density protein-95 |
| PAZ | Presynaptic active zone |
| NRXN1, NRXN2, NRXN3 | Neurexin 1,2,3 |
| PGs | Proteoglycans |
| SNAP | Soluble N-ethylmaleimide sensitive factor attachment protein |
| SNARE | Soluble N-ethylmaleimide sensitive factor attachment protein receptor |
| SVs | Synaptic vesicles |
| VAMP1/VAMP2; VAMP1/2 | Synaptobrevin ½ |
| Syp | Synaptophysin |
| SNAP-25 | Synaptosomal nerve-associated protein 25 |
| v-SNAREs | Vesicle-associated SNARE proteins |
| Syt-1 | Synaptotagmin 1 |
| t-SNAREs | Target-localized SNARE proteins |
| Tn-C | Tenascin-C |
| Tn-R | Tenascin-R |
| TJs | Tight junctions |
| Ti-VAMP | Tetanus-Insensitive VAMP |
| TIMP | Tissue inhibitor of metalloproteinase |
| TBI | Traumatic brain injury |
| Trk(R) | Tropomyosin receptor kinase B |
| VAMP4 | Vesicle-associated membrane protein 4 |
| VAMP | Vesicle-associated membrane protein |
| VAMP7 | Vesicle-associated membrane protein 7 |
References
- Cragg, B. Brain extracellular space fixed for electron microscopy. Neurosci. Lett. 1979, 15, 301–306. [Google Scholar] [CrossRef]
- Testa, D.; Prochiantz, A.; Di Nardo, A.A. Perineuronal nets in brain physiology and disease. Semin. Cell Dev. Biol. 2019, 89, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Zadka, L. The importance of metalloproteinases and their tissue inhibitors in the neoplastic process. Neuroskop 2008, 10, 106–109. [Google Scholar]
- Przybyłowska, K.; Błasiak, J. Matrix metalloproteinases and their role in cancer progression. Postępy Biochem. 2001, 47, 212–222. [Google Scholar] [PubMed]
- Ffrench-Constant, C.; Colognato, H. Integrins: Versatile integrators of extracellular signals. Trends Cell Biol. 2004, 14, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Dityatev, A.; Schachner, M. The extracellular matrix and synapses. Cell Tissue Res. 2006, 326, 647–654. [Google Scholar] [CrossRef]
- Ziemiańska, K.; Konopka, A.; Wilczyński, G.M. The role of extracellular proteolysis in synaptic plasticity of the central nervous system. Postep. Hig. Med. Dosw. 2012, 66, 959–975. [Google Scholar] [CrossRef]
- Matthews, R.T.; Kelly, G.M.; Zerillo, C.A.; Gray, G.; Tiemeyer, M.; Hockfield, S. Aggrecan glycoforms contribute to the molecular heterogeneity of perineuronal nets. J. Neurosci. 2002, 22, 7536–7547. [Google Scholar] [CrossRef]
- Spreafico, R.; De Biasi, S.; Vitellaro-Zuccarello, L. The Perineuronal Net: A Weapon for a challenge. J. Hist. Neurosci. 1999, 8, 179–185. [Google Scholar] [CrossRef]
- Miyata, S.; Nadanaka, S.; Igarashi, M.; Kitagawa, H. Structural variation of chondroitin sulfate chains contributes to the molecular heterogeneity of perineuronal nets. Front. Integr. Neurosci. 2018, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Sorg, B.A.; Berretta, S.; Blacktop, J.M.; Fawcett, J.W.; Kitagawa, H.; Kwok, J.C.F.; Miquel, M. Casting a Wide Net: Role of Perineuronal Nets in Neural Plasticity. J. Neurosci. 2016, 36, 11459–11468. [Google Scholar] [CrossRef] [PubMed]
- Bozzelli, P.L.; Alaiyed, S.; Kim, E.; Villapol, S.; Conant, K. Proteolytic remodeling of perineuronal nets: Effects on synaptic plasticity and neuronal population dynamics. Neural Plast. 2018, 2018, 5735789. [Google Scholar] [PubMed]
- Reicheft, A.C.; Hare, D.J.; Bussey, T.J.; Saksida, L.M. Perineuronal nets: Plasticity, protection, and therapeutic potential. Trends Neurosci. 2019, 42, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Prager, O.; Kamintsky, L.; Hasam-Henderson, L.A.; Schoknecht, K.; Wuntke, V.; Papageorgiou, I.; Swolinsky, J.; Muoio, V.; Bar-Klein, G.; Vazana, U.; et al. Seizure-induced microvascular injury is associated with impaired neurovascular coupling and blood-brain barrier dysfunction. Epilepsia 2019, 60, 322–336. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Toriumi, H.; Yamada, S.; Hoshino, H.; Suzuki, N. Astrocytes and pericytes cooperatively maintain a capillary-like structure composed of endothelial cells on gel matrix. Brain Res. Neth. 2011, 1406, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Bauer, H.C.; Krizbai, I.A.; Bauer, H.; Traweger, A. You Shall Not Pass”-tight junctions of the blood brain barrier. Front. Neurosci. 2014, 8, 392. [Google Scholar] [CrossRef] [PubMed]
- Redzic, Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: Similarities and differences. Fluids Barriers CNS 2011, 8, 3. [Google Scholar] [CrossRef]
- Bendayan, R.; Lee, G.; Bendayan, M. Functional expression and localization of P-glycoprotein at the blood brain barrier. Microsc. Res. Tech. 2002, 57, 365–380. [Google Scholar] [CrossRef]
- Rhodes, K.E.; Fawcett, J.W. Chondroitin sulphate proteoglycans: Preventing plasticity or protecting the CNS? J. Anat. 2004, 204, 33–48. [Google Scholar] [CrossRef]
- Itano, N.; Sawai, T.; Yoshida, M.; Lenas, P.; Yamada, Y.; Imagawa, M.; Shinomura, T.; Hamaguchi, M.; Yoshida, Y.; Ohnuki, Y.; et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J. Biol. Chem. 1999, 274, 25085–25092. [Google Scholar] [CrossRef] [PubMed]
- Carulli, D.; Rhodes, K.E.; Brown, D.J.; Bonnert, T.P.; Pollack, S.J.; Oliver, K.; Strata, P.; Fawcett, J.W. Composition of perineuronal nets in the adult rat cerebellum and the cellular origin of their components. J. Comp. Neurol. 2006, 494, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Z.X.; Jin, T.; Wang, Z.Y.; Zhao, P. Tau pathology promotes the reorganization of the extracellular matrix and inhibits the formation of perineuronal nets by regulating the expression and the distribution of hyaluronic acid synthases. J. Alzheimers Dis. 2017, 57, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.C.F.; Carulli, D.; Fawcett, J.W. In vitro modeling of perineuronal nets: Hyaluronan synthase and link protein are necessary for their formation and integrity. J. Neurochem. 2010, 114, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Mikami, T.; Kitagawa, H. Biosynthesis and function of chondroitin sulfate. Biochim. Biophys. Acta. 2013, 1830, 4719–4733. [Google Scholar] [CrossRef]
- Schwartz, N.B. Regulation of chondroitin sulfate synthesis. Effect of beta-xylosides on synthesis of chondroitin sulfate proteoglycan, chondroitin sulfate chains, and core protein. J. Biol. Chem. 1977, 252, 6316–6321. [Google Scholar] [PubMed]
- Oohashi, T.; Edamatsu, M.; Bekku, Y.; Carulli, D. The hyaluronan and proteoglycan link proteins: Organizers of the brain extracellular matrix and key molecules for neuronal function and plasticity. Exp. Neurol. 2015, 274, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y. Lecticans: Organizers of the brain extracellular matrix. Cell Mol. Life Sci. 2000, 57, 276–289. [Google Scholar] [CrossRef]
- Day, J.M.; Olin, A.I.; Murdoch, A.D.; Canfield, A.; Sasaki, T.; Timpl, R.; Hardingham, T.E.; Aspberg, A. Alternative splicing in the aggrecan G3 domain influences binding interactions with Tenascin-C and other extracellular matrix proteins. J. Biol. Chem. 2004, 279, 12511–12518. [Google Scholar] [CrossRef]
- Rauch, U.; Clement, A.; Retzler, C.; Fröhlich, L.; Fässler, R.; Göhring, W.; Faissner, A. Mapping of a defined neurocan binding site to distinct domains of tenascin-C. J. Biol. Chem. 1997, 272, 26905–26912. [Google Scholar] [CrossRef]
- Oohira, A.; Matsui, F.; Tokita, Y.; Yamauchi, S.; Aono, S. Molecular interactions of neural chondroitin sulfate proteoglycans in the brain development. Arch. Biochem. Biophys. 2000, 374, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Bandtlow, C.E.; Zimmermann, D.R. Proteoglycans in the developing brain: New conceptual insights for old proteins. Physiol. Rev. 2000, 80, 1267–1290. [Google Scholar] [CrossRef] [PubMed]
- Asher, R.A.; Morgenstern, D.A.; Fidler, P.S.; Adcock, K.H.; Oohira, A.; Braistead, J.E.; Levine, J.M.; Margolis, R.U.; Rogers, J.H.; Fawcett, J.W. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J. Neurosci. 2000, 20, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Asher, R.A.; Morgenstern, D.A.; Shearer, M.C.; Adcock, K.H.; Pesheva, P.; Fawcett, J.W. Versican is upregulated in CNS injury and is a product of oligodendrocyte lineage cells. J. Neurosci. 2002, 22, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Thon, N.; Haas, C.A.; Rauch, U.; Merten, T.; Fassler, R.; Frotscher, M.; Deller, T. The chondroitin sulphate proteoglycan brevican is upregulated by astrocytes after entorhinal cortex lesions in adult rats. Eur. J. Neurosci. 2000, 12, 2547–2558. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, E.J.; Moon, L.D.F.; Popat, R.J.; King, V.R.; Bennett, G.S.; Patel, P.N.; Fawcett, J.W.; McMahon, S.B. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002, 416, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Kitagawa, H. Chondroitin sulfate and neuronal disorders. Front. Biosci. 2016, 21, 1330–1340. [Google Scholar]
- Brückner, G.; Grosche, J.; Schmidt, S.; Härtig, W.; Margolis, R.U.; Delpech, B.; Seidenbecher, C.I.; Czaniera, R.; Schachner, M. Postnatal development of perineuronal nets in wild-type mice and in a mutant deficient in tenascin-R. J. Comp. Neurol. 2000, 428, 616–629. [Google Scholar] [CrossRef]
- Carulli, D.; Pizzorusso, T.; Kwok, J.C.F.; Putignano, E.; Poli, A.; Forostyak, S.; Andrews, M.R.; Deepa, S.S.; Glant, T.T.; Fawcett, J.W. Animals lacking link protein have attenuated perineuronal nets and persistent Plasticity. Brain 2010, 133, 2331–2347. [Google Scholar] [CrossRef]
- Neal, A.P.; Guilarte, T.R. Molecular neurobiology of lead (Pb(2+)): Effects on synaptic function. Mol. Neurobiol. 2010, 42, 151–160. [Google Scholar] [CrossRef]
- Neal, A.P.; Stansfield, K.H.; Worley, P.F.; Thompson, R.E.; Guilarte, T.R. Lead exposure during synaptogenesis alters vesicular proteins and impairs vesicular release: Potential role of NMDA receptor-dependent BDNF signaling. Toxicol. Sci. 2010, 116, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Nelson, W.J. Synapses: Sites of cell recognition, adhesion, and functional specification. Annu. Rev. Biochem. 2007, 76, 267–294. [Google Scholar] [CrossRef] [PubMed]
- Sudhof, T.C. The synaptic vesicle cycle: A cascade of protein-protein interaction. Nature 1995, 375, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, F.; Pennuto, M.; Bonanomi, D.; Benfenati, F. Synaptophysin: Leading actor or walk-on role in synaptic vesicle exocytosis? Bioessays 2004, 26, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Booij, L.H.D.J.; Drobnik, L. Anatomy and physiology of neuromuscular transmission - part I. Anestezjol. Ratow. 2010, 4, 49–80. [Google Scholar]
- Chakrabarti, R.; Wichmann, C. Nanomachinery organizing release at neuronal and ribbon synapses. Int J. Mol. Sci. 2019, 20, E2147. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C.; Rizo, J. Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a005637. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C.; Rothman, J.E. Membrane fusion: Grappling with SNARE and SM proteins. Science 2009, 323, 474–477. [Google Scholar] [CrossRef]
- Südhof, T.C. The synaptic vesicle cycle. Annu. Rev. Neurosci. 2004, 27, 509–547. [Google Scholar] [CrossRef]
- Südhof, T.C. A molecular machine for neurotransmitter release: Synaptotagmin and beyond. Nat. Med. 2013, 19, 1227–1231. [Google Scholar] [CrossRef]
- Li, N.; Yu, Z.L.; Wang, L.; Zheng, Y.T.; Jia, J.X.; Wang, Q.; Zhu, M.J.; Liu, X.H.; Xia, X.; Li, W.J. Early-Life lead exposure affects the activity of TNF-a and expression of SNARE complex in hippocampus of mouse pups. Biol. Trace Elem. Res. 2009, 132, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Cupertino, R.B.; Kappel, D.B.; Bandeira, C.E.; Schuch, J.B.; da Silva, B.S.; Müller, D.; Bau, C.H.; Mota, N.R. SNARE complex in developmental psychiatry: Neurotransmitter exocytosis and beyond. J. Neural Transm. 2016, 123, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Woodman, P.G. The roles of NSF, SNAPs and SNAREs during membrane fusion. Biochim. Biophys. Acta. 1997, 1357, 155–172. [Google Scholar] [CrossRef]
- Gordon, S.L.; Cousin, M.A. The Sybtraps: Control of synaptobrevin traffic by synaptophysin, α-synuclein and AP-180. Traffic 2014, 15, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.I.; Vallejo, D.; Inestrosa, N.C. Emerging synaptic molecules as candidates in the etiology of neurological disorders. Neural Plast. 2017, 2017, 8081758. [Google Scholar] [CrossRef] [PubMed]
- Weimer, R.M.; Richmond, J.E. Synaptic vesicle docking: A putative role for the Munc18/Sec1 protein family. Cur. Top. Dev. Biol. 2005, 65, 83–113. [Google Scholar]
- Pevsner, J.; Scheller, R.H. Mechanisms of vesicle docking and fusion: Insights from the nervous system. Curr. Opin. Cell Biol. 1994, 6, 555–560. [Google Scholar] [CrossRef]
- Littleton, J.T.; Bellen, H.J. Synaptotagmin controls and modulates synaptic-vesicle fusion in a Ca2+ -dependent manner. Trends Neurosci. 1995, 18, 177–183. [Google Scholar] [CrossRef]
- Südhof, T.C. Towards an understanding of synapse formation. Neuron 2018, 100, 276–293. [Google Scholar] [CrossRef]
- Nimchinsky, E.A.; Sabatini, B.L.; Svoboda, K. Structure and function of dendritic spines. Annu. Rev. Physiol. 2002, 64, 313–353. [Google Scholar] [CrossRef]
- Boeckers, T.M. The postsynaptic density. Cell Tissue Res. 2006, 326, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Kim, M.J. Postsynaptic signaling and plasticity mechanisms. Science 2002, 298, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.B. Signal-processing machines at the postsynaptic density. Science 2000, 290, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Valtschanoff, J.G.; Weinberg, R.J. Laminar organization of the NMDA receptor complex within the postsynaptic density. J. Neurosci. 2001, 21, 1211–1217. [Google Scholar] [CrossRef]
- Fagni, L.; Worley, P.F.; Ango, F. Homer as both a scaffold and transduction molecule. Sci. STKE 2002, 137, RE8. [Google Scholar] [CrossRef]
- Gomperts, S.N. Clustering membrane proteins: It’s all coming together with the PSD-95/SAP90 protein family. Cell 1996, 84, 659–662. [Google Scholar] [CrossRef]
- Béïque, J.C.; Andrade, R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J. Physiol. 2003, 546, 859–867. [Google Scholar] [CrossRef]
- Peng, J.; Kim, M.J.; Cheng, D.; Duong, D.M.; Gygi, S.P.; Sheng, M. Semiquantitative Proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J. Biol. Chem. 2004, 279, 21003–21011. [Google Scholar] [CrossRef]
- Steiner, P.; Higley, M.J.; Xu, W.; Czervionke, B.L.; Malenka, R.C.; Sabatini, B.L. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron 2008, 60, 788–802. [Google Scholar] [CrossRef]
- Torres, L.H.; Garcia, R.C.T.; Blois, A.M.M.; Dati, L.M.M.; Durão, A.C.; Alves, A.S.; Pacheco-Neto, M.; Mauad, T.; Britto, L.R.B.; Xavier, G.F.; et al. Exposure of neonatal mice to tobacco smoke disturbs synaptic proteins and spatial learning and memory from late infancy to early adulthood. PLoS ONE 2015, 10, 1–21. [Google Scholar] [CrossRef]
- Hering, H.; Sheng, M. Dentritic spines: Structure, dynamics and regulation. Nat. Rev. Neuroscience 2001, 2, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Kim, E. The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol. 2011, 3, a005678. [Google Scholar] [CrossRef] [PubMed]
- Fellin, T. Communication between neurons and astrocytes: Relevance to the modulation of synaptic and network activity. J. Neurochem. 2009, 108, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bosiacka, I.; Gutowska, I.; Rybicka, M.; Nowacki, P.; Chlubek, D. Neurotoxicity of lead: Hypothetical molecular mechanisms of synaptic function disorders. Neurol. Neurochir. Pol. 2012, 46, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.; Bramham, C.R.; Duarte, C.B. BDNF and hippocampal synaptic plasticity. Vitam. Horm. 2017, 104, 153–195. [Google Scholar] [PubMed]
- Chao, D.L.; Ma, L.; Shen, K. Transient cell-cell interactions in neural circuit formation. Nat. Rev. Neurosci. 2009, 10, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Christian, K.; Lu, B. BDNF: A key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol. Learn. Mem. 2008, 89, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Walz, C.; Jüngling, K.; Lessmann, V.; Gottmann, K. Presynaptic plasticity in an immature neocortical network requires NMDA receptor activation and BDNF release. J. Neurophysiol. 2006, 96, 3512–3516. [Google Scholar] [CrossRef]
- Tartaglia, N.; Du, J.; Tyler, W.J.; Neale, E.; Pozzo-Miller, L.; Lu, B. Protein synthesis-dependent and independent regulation of hippocampal synapses by brain-derived neurotrophic factor. J. Biol. Chem. 2001, 276, 37585–37593. [Google Scholar] [CrossRef]
- Moretto, G.; Xu, R.Y.; Walker, D.G.; Kim, S.U. Co-expression of mRNA for neurotrophic factors in human neurons and glial cells in culture. J. Neuropath. Exp. Neur. 1994, 53, 78–85. [Google Scholar] [CrossRef]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Tampellini, D. Synaptic activity and Alzheimer’s disease: A critical update. Front. Neurosci. 2015, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Van Spronsen, M.; Hoogenraad, C.C. Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep. 2010, 10, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Pham, E.; Crews, L.; Ubhi, K.; Hansen, L.; Adame, A.; Cartier, A.; Salmon, D.; Galasko, D.; Michael, S.; Savas, J.N.; et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010, 277, 3051–3067. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Kohno, R.; Sawada, H.; Kawamoto, Y.; Uemura, K.; Shibasaki, H.; Shimohama, S. BDNF is induced by wild-type a-synuclein but not by the two mutants, A30P or A53T, in glioma cell line. Biochem. Biophys. Res. Commun. 2004, 318, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Fukumoto, H.; Orne, J.; Klucken, J.; Raju, S.; Vanderburg, C.R.; Irizarry, M.C.; Hyman, B.T.; Ingelsson, M. Decreased levels of BDNF protein in Alzheimer temporal cortex are independent of BDNF polymorphisms. Exp. Neurol. 2005, 194, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Murer, M.G.; Boissiere, F.; Yan, Q. An immunohistochemical study of the distribution of brain-derived neurotrophic factor in the adult human brain, with particular reference to Alzheimer’s disease. Neuroscience 1999, 88, 1015–1032. [Google Scholar] [CrossRef]
- Markiewicz, R.; Kozioł, M.; Olajossy, M.; Masiak, J. Can brain-derived neurotrophic factor (BDNF) be an indicator of effective rehabilitation interventions in schizophrenia? Psychiatr. Pol. 2018, 52, 819–834. [Google Scholar] [CrossRef]
- Rijal Upadhaya, A.; Capetillo-Zarate, E.; Kosterin, I.; Abramowski, D.; Kumar, S.; Yamaguchi, H.; Walter, J.; Fändrich, M.; Staufenbiel, M.; Thal, D.R. Dispersible amyloid β-protein oligomers, protofibrils, and fibrils represent diffusible but not soluble aggregates: Their role in neurodegeneration in amyloid precursor protein (APP) transgenic mice. Neurobiol. Aging 2012, 33, 2641–2660. [Google Scholar] [CrossRef]
- Lisik, M.Z. Molecular aspects of autism spectrum disorders. Psychiatr. Pol. 2014, 48, 689–700. [Google Scholar]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology involved in Autism Spectrum Disorder. Front. Cell Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef]
- Sala, C.; Vicidomini, C.; Bigi, I.; Mossa, A.; Verpelli, C. Shank synaptic scaffold proteins: Keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 2015, 135, 849–858. [Google Scholar] [CrossRef]
- Arons, M.H.; Thynne, C.J.; Grabrucker, A.M.; Li, D.; Schoen, M.; Cheyne, J.E.; Boeckers, T.M.; Montgomery, J.M.; Garner, C.C. Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J. Neurosci. 2012, 32, 14966–14978. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.M.; Madison, D.V. Discrete synaptic states define a major mechanism of synapse plasticity. Trends Neurosci. Engl. 2004, 27, 744–750. [Google Scholar] [CrossRef]
- Dzwonek, J.; Rylski, M.; Kaczmarek, L. Matrix metalloproteinases and their endogenous inhibitors in neuronal physiology of the adult brain. FEBS Lett. 2004, 567, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Corvetti, L.; Rossi, F. Degradation of chondroitin sulfate proteoglycans induces sprouting of intact purkinje axons in the cerebellum of the adult rat. J. Neurosci. 2005, 25, 7150–7158. [Google Scholar] [CrossRef]
- Saroja, S.R.; Sase, A.; Kircher, S.G.; Wan, J.; Berger, J.; Höger, H.; Pollak, A.; Lubec, G. Hippocampal proteoglycans brevican and versican are linked to spatial memory of Sprague-Dawley rats in the morris water maze. J. Neurochem. 2014, 130, 797–804. [Google Scholar] [CrossRef]
- Milev, P.; Maurel, P.; Chiba, A.; Mevissen, M.; Popp, S.; Yamaguchi, Y.; Margolis, R.K.; Margolis, R.U. Differential regulation of expression of hyaluronan-binding proteoglycans in developing brain: Aggrecan, versican, neurocan, and brevican. Biochem. Biophys. Res. Commun. 1998, 247, 207–212. [Google Scholar] [CrossRef]
- Dino, M.R.; Harroch, S.; Hockfield, S.; Matthews, R.T. Monoclonal antibody Cat-315 detects a glycoform of receptor protein tyrosine phosphatase beta/phosphacan early in CNS development that localizes to extrasynaptic sites prior to synapse formation. Neuroscience 2006, 142, 1055–1069. [Google Scholar] [CrossRef]
- Gogolla, N.; Caroni, P.; Luthi, A.; Herry, C. Perineuronal nets protect fear memories from erasure. Science 2009, 325, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- Sethi, M.K.; Zaia, J. Extracellular matrix proteomics in schizophrenia and Alzheimer’s disease. Anal. Bioanal. Chem. 2012, 409, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J.; Cummings, B.J.; Cotman, C.W. Early association of reactive astrocytes with senile plaques in Alzheimer’s disease. Exp. Neurol. 1995, 132, 172–179. [Google Scholar] [CrossRef]
- Wiese, S.; Karus, M.; Faissner, A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front. Pharmacol. 2012, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, I.; Irintchev, A.; Schachner, M. Tenascin-R restricts post traumatic remodeling of motoneuron innervation and functional recoveryafter spinal cord injury in adult mice. J. Neurosci. 2006, 26, 7849–7859. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Martínez-Sáez, E.; Gutiérrez-Franco, A.; Eixarch, H.; Castro, Z.; Ortega-Aznar, A.; Ramón, Y.; Cajal, S.; Montalban, X.; Espejo, C. Expression of semaphorin 3A, semaphorin 7A and theirreceptors in multiple sclerosis lesions. Mult. Scler. 2015, 21, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- McKeon, R.J.; Jurynec, M.J.; Buck, C.R. The chondroitin sulfate proteoglycansneurocan and phosphacan are expressed by reactive astrocytes in the chronicCNS glial scar. J. Neurosci. 1999, 19, 10778–10788. [Google Scholar] [CrossRef]
- Bovolenta, P.; Fernaud-Espinosa, I.; Méndez-Otero, R.; Nieto-Sampedro, M. Neurite outgrowth inhibitor of gliotic brain tissue. Model of action and cellular localization, studied with specific monoclonal antibodies. Eur, J. Neurosci. 1997, 9, 977–989. [Google Scholar] [CrossRef]
- Fawcett, J.W. The extracellular matrix in plasticity and regeneration after CNS injury and neurodegenerative disease. Prog. Brain Res. 2015, 218, 213–226. [Google Scholar]
- Howell, M.D.; Bailey, L.A.; Cozart, M.A.; Gannon, B.M.; Gottschall, P.E. Hippocampal administration of chondroitinase ABCincreases plaque-adjacent synaptic marker and diminishes amyloid burden inaged APPswe/PS1dE9 mice. Acta. Neuropathol. Commun. 2015, 3, 54. [Google Scholar] [CrossRef]
- Végh, M.J.; Heldring, C.M.; Kamphuis, W.; Hijazi, S.; Timmerman, A.J.; Li, K.W.; van Nierop, P.; Mansvelder, H.D.; Hol, E.M.; Smit, A.B.; et al. Reducing hippocampal extracellular matrix reverses earlymemory deficits in a mouse model of Alzheimer’s disease. Acta. Neuropathol. Commun. 2014, 2, 76. [Google Scholar] [PubMed]
- Bonneh-Barkay, D.; Willey, C.A. Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009, 19, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Hemphill, M.A.; Dauth, S.; Yu, C.J.; Dabiri, B.E.; Parker, K.K. Traumatic brain injury and the neuronal microenvironment: A potential role for neuropathological mechanotransduction. Neuron 2015, 85, 1177–1192. [Google Scholar] [CrossRef] [PubMed]
- Baig, S.; Wilcock, G.K.; Love, S. Loss of perineuronal net N-acetylgalactosamine in Alzheimer’s disease. Acta. Neuropathol. 2005, 110, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.G.; Mironova, Y.A.; Hovda, D.A.; Sutton, R.L. Pericontusion axon sprouting is spatially and temporally consistent with a growth-permissive environment after traumatic brain injury. J. Neuropathol. Exp. Neurol. 2010, 69, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.H.; Binder, D.K.; Ethell, I.M.; Razak, K.A. The Perineuronal ‘Safety’ Net? Perineuronal net abnormalities in neurological disorders. Front. Mol. Neurosci. 2018, 11, 270. [Google Scholar] [CrossRef] [PubMed]
- Fitch, M.T.; Silver, J. CNS injury, glial scars, and inflammation: Inhibitory extracellular matrices and regeneration failure. Exp. Neurol. 2008, 209, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Dyck, S.M.; Karimi-Abdolrezaee, S. Chondroitin sulfate proteoglycans: Key modulators in the developing and pathologic central nervous system. Exp. Neurol. 2015, 269, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Trendelenburg, G.; Dirnagl, U. Neuroprotective role of astrocytes in cerebral ischemia: Focus on ischemic preconditioning. Glia 2005, 50, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Quirico-Santos, T.; Fonseca, C.O.; Lagrota-Candido, J. Brain sweet brain Importance of sugars for the cerebral microenvironment and tumor development. Arq. Neuropsiquiatr. 2010, 68, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Lendvai, D.; Morawski, M.; Négyessy, L.; Gáti, G.; Jäger, C.; Baksa, G.; Glasz, T.; Attems, J.; Tanila, H.; Arendt, T.; et al. Neurochemical mapping of the human hippocampus reveals perisynaptic matrix around functional synapses in Alzheimer’s disease. Acta. Neuropathol. 2013, 125, 215–229. [Google Scholar] [CrossRef] [PubMed]
- McRae, P.A.; Porter, B.E. The perineuronal net component of the extracellular matrix in plasticity and epilepsy. Neurochem. Int. 2012, 61, 963–972. [Google Scholar] [CrossRef] [PubMed]
- McRae, P.A.; Baranov, E.; Rogers, S.L.; Porter, B.E. Persistent decrease in multiple components of the perineuronal net following status epilepticus. Eur. J. Neurosci. 2012, 36, 3471–3482. [Google Scholar] [CrossRef] [PubMed]
- McRae, P.A.; Baranov, E.; Sarode, S.; Brooks-Kayal, A.R.; Porter, B.E. Aggrecan expression, a component of the inhibitory interneuron perineuronal net, is altered following an early-life seizure. Neurobiol. Dis. 2010, 39, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Brückner, G.; Jäger, C.; Seeger, G.; Matthews, R.T.; Arendt, T. Involvement of perineuronal and perisynaptic extracellular matrix in Alzheimer’s Disease neuropathology. Brain Pathol. 2012, 22, 547–561. [Google Scholar]
- Burket, J.A.; Urbano, M.R.; Deutsch, S.I. Sugarcoated perineuronal nets regulate “GABAergic” transmission: Bittersweet hypothesis in Autism Spectrum Disorder. Clin. Neuropharmacol. 2017, 40, 120–130. [Google Scholar] [CrossRef]
- Fox, K.; Caterson, B. Freeing the Brain from the Perineuronal Net. Science 2002, 298, 1187–1189. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Neuron | Astrocyte | Oligodendrocyte | Reactive Astrocytes |
|---|---|---|---|
| aggrecan | aggrecan | phosphacan | hyaluronan |
| brevican | brevican | versican | brevican |
| neurocan | neurocan | Tn-R | neurocan |
| phosphacan | phosphacan | MMPs | phosphacan |
| hyaluronan | hyaluronan | versican | |
| Hapln1 | versican | Tn-C | |
| Hapln4 | Tn-R | ||
| Tn-R | Tn-C | ||
| Tn-C | MMPs | ||
| MMPs | |||
| semaphorin 3A |
| Base Unit | ||||
|---|---|---|---|---|
| Galactose (Gal) or | Uronic Acid | + Amino sugar | ||
| Glucuronic Acid (GlcA) | Induronic Acid (IdoA) | N-Acetylgalactoseamine (GalNAc) | N-Acetylglucosamine (GalNAc) | |
| Composition of Glycosaminoglycans (GAGs) | ||||
| Glycosaminoglycan | Repeated Units | |||
| Hyaluronan (HA) | GlcA | + GlcNAc | ||
| Chondroitin Sulphate | GlcA | + GalNAc | ||
| Heparan Sulphate | GlcA | + GlcNAc | ||
| Keratan Sulphate | Gal | + GlcNAc | ||
| Dermatan Sulphate | IdoA | + GalNAc | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosiacki, M.; Gąssowska-Dobrowolska, M.; Kojder, K.; Fabiańska, M.; Jeżewski, D.; Gutowska, I.; Lubkowska, A. Perineuronal Nets and Their Role in Synaptic Homeostasis. Int. J. Mol. Sci. 2019, 20, 4108. https://doi.org/10.3390/ijms20174108
Bosiacki M, Gąssowska-Dobrowolska M, Kojder K, Fabiańska M, Jeżewski D, Gutowska I, Lubkowska A. Perineuronal Nets and Their Role in Synaptic Homeostasis. International Journal of Molecular Sciences. 2019; 20(17):4108. https://doi.org/10.3390/ijms20174108
Chicago/Turabian StyleBosiacki, Mateusz, Magdalena Gąssowska-Dobrowolska, Klaudyna Kojder, Marta Fabiańska, Dariusz Jeżewski, Izabela Gutowska, and Anna Lubkowska. 2019. "Perineuronal Nets and Their Role in Synaptic Homeostasis" International Journal of Molecular Sciences 20, no. 17: 4108. https://doi.org/10.3390/ijms20174108
APA StyleBosiacki, M., Gąssowska-Dobrowolska, M., Kojder, K., Fabiańska, M., Jeżewski, D., Gutowska, I., & Lubkowska, A. (2019). Perineuronal Nets and Their Role in Synaptic Homeostasis. International Journal of Molecular Sciences, 20(17), 4108. https://doi.org/10.3390/ijms20174108