Epigenetic Regulation of Adipogenic Differentiation by Histone Lysine Demethylation

 , ,
, ,  , ,
, ,
Abstract

1. Introduction

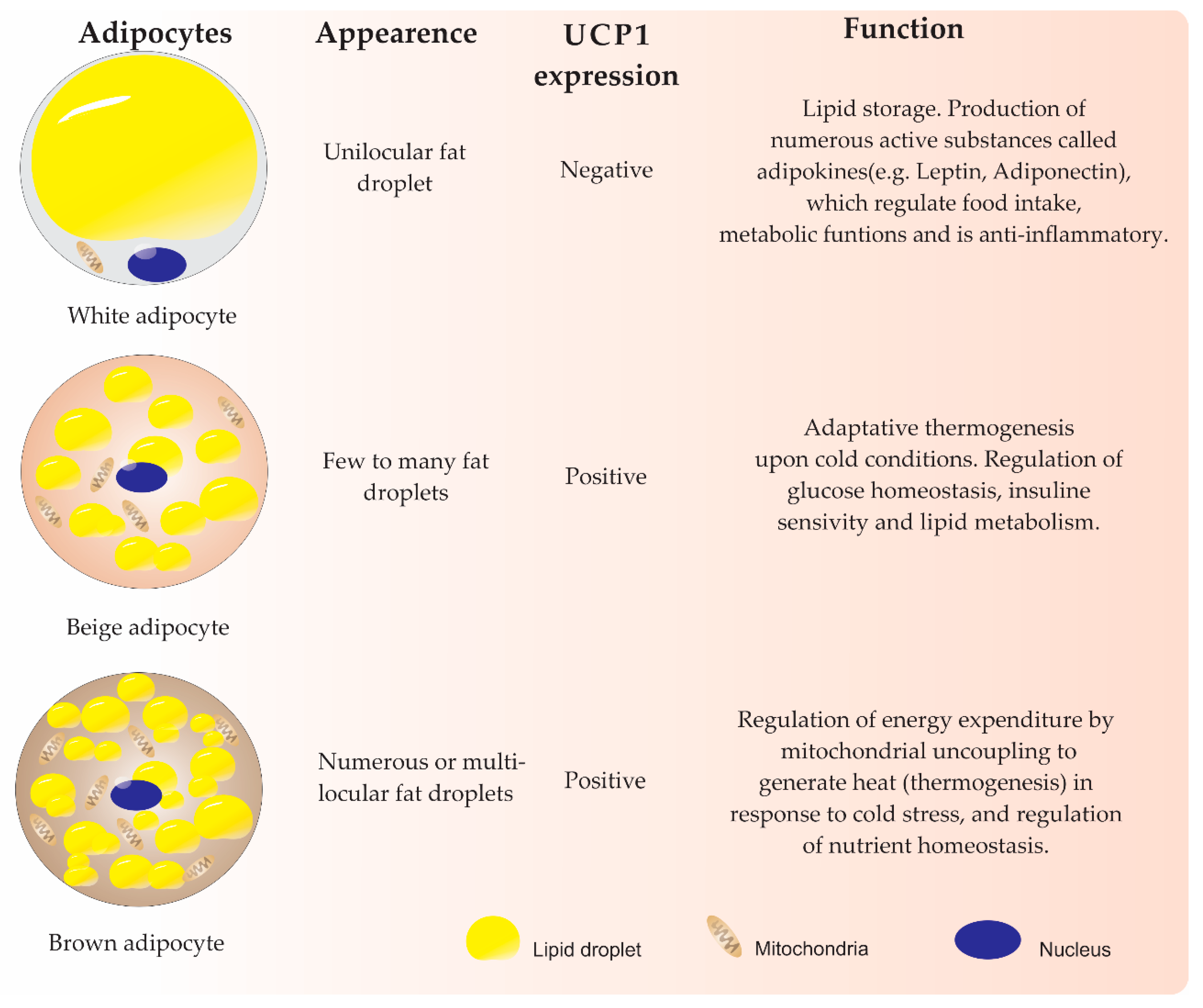
2. Adipogenesis Differentiation from MSCs
3. Epigenetic Regulation of Gene Expression
4. The Role of KMTs in the Adipogenic Differentiation of MSCs
5. Histone Lysine Demethylases (KDMs)
The Role of KDMs during Adipogenesis
6. Conclusions and Perspectives
Authors Contribution
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Obesity and Overweight; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Gonzalez-Muniesa, P.; Martinez-Gonzalez, M.A.; Hu, F.B.; Despres, J.P.; Matsuzawa, Y.; Loos, R.J.F.; Moreno, L.A.; Bray, G.A.; Martinez, J.A. Obesity. Nat. Rev. Dis. Primers 2017, 3, 17034. [Google Scholar] [CrossRef] [PubMed]
- De Soysa, A.K.H.; Klevjer, M.; Grill, V.; Mostad, I.L. Impact of a FTO gene risk variant on variables of energy metabolism in adults with obesity class 2 and 3. Metab. Open 2019, 1, 3–6. [Google Scholar] [CrossRef]
- Matsushita, K.; Dzau, V.J. Mesenchymal stem cells in obesity: Insights for translational applications. Lab. Investig. 2017, 97, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.; Rivera Gonzalez, G.; Ebmeier, S.; Grisotti, G.; Zwick, R.; Horsley, V. The Role of Adipocytes in Tissue Regeneration and Stem Cell Niches. Annu. Rev. Cell Dev. Biol. 2016, 32, 609–631. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wallace, M.; Sanchez-Gurmaches, J.; Hsiao, W.Y.; Li, H.; Lee, P.L.; Vernia, S.; Metallo, C.M.; Guertin, D.A. Adipose tissue mTORC2 regulates ChREBP-driven de novo lipogenesis and hepatic glucose metabolism. Nat. Commun. 2016, 7, 11365. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, J.M.; Stern, J.H.; Scherer, P.E. The cell biology of fat expansion. J. Cell Biol. 2015, 208, 501–512. [Google Scholar] [CrossRef]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef]
- Ma, X.; Lee, P.; Chisholm, D.J.; James, D.E. Control of adipocyte differentiation in different fat depots; implications for pathophysiology or therapy. Front. Endocrinol. 2015, 6, 1. [Google Scholar] [CrossRef]
- Carobbio, S.; Rosen, B.; Vidal-Puig, A. Adipogenesis: New insights into brown adipose tissue differentiation. J. Mol. Endocrinol. 2013, 51, T75–T85. [Google Scholar] [CrossRef]
- Ikeda, K.; Maretich, P.; Kajimura, S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol. Metab. 2018, 29, 191–200. [Google Scholar] [CrossRef]
- Giralt, M.; Villarroya, F. White, brown, beige/brite: Different adipose cells for different functions? Endocrinology 2013, 154, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chen, P.; Sun, L. Regulatory networks of non-coding RNAs in brown/beige adipogenesis. Biosci. Rep. 2015, 35, e00262. [Google Scholar] [CrossRef] [PubMed]
- Kwok, K.H.; Lam, K.S.; Xu, A. Heterogeneity of white adipose tissue: Molecular basis and clinical implications. Exp. Mol. Med. 2016, 48, e215. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gurmaches, J.; Hung, C.M.; Guertin, D.A. Emerging Complexities in Adipocyte Origins and Identity. Trends Cell Biol. 2016, 26, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Sakai, J.; Kajimura, S. Transcriptional and epigenetic control of brown and beige adipose cell fate and function. Nat. Rev. Mol. Cell Biol. 2016, 17, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Sarjeant, K.; Stephens, J.M. Adipogenesis. Cold Spring Harb Perspect. Biol. 2012, 4, a008417. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K. Adipocytes. Curr. Biol. 2014, 24, R988–R993. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kotzbeck, P.; Giordano, A.; Mondini, E.; Murano, I.; Severi, I.; Venema, W.; Cecchini, M.P.; Kershaw, E.E.; Barbatelli, G.; Haemmerle, G.; et al. Brown adipose tissue whitening leads to brown adipocyte death and adipose tissue inflammation. J. Lipid Res. 2018, 59, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Hammarstedt, A.; Hedjazifar, S.; Smith, U. Restricted adipogenesis in hypertrophic obesity: The role of WISP2, WNT, and BMP4. Diabetes 2013, 62, 2997–3004. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hanzu, F.A.; Musri, M.M.; Sanchez-Herrero, A.; Claret, M.; Esteban, Y.; Kaliman, P.; Gomis, R.; Parrizas, M. Histone demethylase KDM1A represses inflammatory gene expression in preadipocytes. Obesity 2013, 21, E616–E625. [Google Scholar] [CrossRef] [PubMed]
- Sambeat, A.; Gulyaeva, O.; Dempersmier, J.; Tharp, K.M.; Stahl, A.; Paul, S.M.; Sul, H.S. LSD1 Interacts with Zfp516 to Promote UCP1 Transcription and Brown Fat Program. Cell Rep. 2016, 15, 2536–2549. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, O.; Dempersmier, J.; Sul, H.S. Genetic and epigenetic control of adipose development. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. Transcriptional networks and chromatin remodeling controlling adipogenesis. Trends Endocrinol. Metab. 2012, 23, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jin, Q.; Lee, J.E.; Su, I.H.; Ge, K. Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7317–7322. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of adipogenesis by Wnt signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Haakonsson, A.K.; Madsen, J.G.S.; Larsen, M.; Forss, I.; Madsen, M.R.; Van Hauwaert, E.L.; Wiwie, C.; Jespersen, N.Z.; Tencerova, M.; et al. Osteogenesis depends on commissioning of a network of stem cell transcription factors that act as repressors of adipogenesis. Nat. Genet. 2019, 51, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Stricker, S.H.; Koferle, A.; Beck, S. From profiles to function in epigenomics. Nat. Rev. Genet. 2017, 18, 51–66. [Google Scholar] [CrossRef]
- Avgustinova, A.; Benitah, S.A. Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 2016, 17, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Rodas-Junco, B.A.; Canul-Chan, M.; Rojas-Herrera, R.A.; De-la-Pena, C.; Nic-Can, G.I. Stem Cells from Dental Pulp: What Epigenetics Can Do with Your Tooth. Front. Physiol. 2017, 8, 999. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, H.; Chen, M.; Ren, S.; Cheng, P.; Zhang, H. miR-301b~miR-130b-PPARgamma axis underlies the adipogenic capacity of mesenchymal stem cells with different tissue origins. Sci. Rep. 2017, 7, 1160. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Liao, L.; An, Y.; Su, X.; Liu, S.; Shuai, Y.; Zhang, X.; Jin, Y. Suppression of EZH2 Prevents the Shift of Osteoporotic MSC Fate to Adipocyte and Enhances Bone Formation During Osteoporosis. Mol. Ther. 2016, 24, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schurmann, A.; et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784 e776. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From stem cell to adipocyte. Annu. Rev. Biochem. 2012, 81, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Ahfeldt, T.; Schinzel, R.T.; Lee, Y.K.; Hendrickson, D.; Kaplan, A.; Lum, D.H.; Camahort, R.; Xia, F.; Shay, J.; Rhee, E.P.; et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat. Cell Biol. 2012, 14, 209–219. [Google Scholar] [CrossRef]
- Ruiz-Ojeda, F.J.; Rupérez, A.I.; Gomez-Llorente, C.; Gil, A.; Aguilera, C.M. Cell Models and Their Application for Studying Adipogenic Differentiation in Relation to Obesity: A Review. Int. J. Mol. Sci. 2016, 17, 1040. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef]
- Chen, Q.; Shou, P.; Zheng, C.; Jiang, M.; Cao, G.; Yang, Q.; Cao, J.; Xie, N.; Velletri, T.; Zhang, X.; et al. Fate decision of mesenchymal stem cells: Adipocytes or osteoblasts? Cell Death Differ. 2016, 23, 1128–1139. [Google Scholar] [CrossRef]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARgamma. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef]
- Pradhan, R.N.; Zachara, M.; Deplancke, B. A systems perspective on brown adipogenesis and metabolic activation. Obes. Rev. 2017, 18 (Suppl. 1), 65–81. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Gronborg, M.; Huang, H.; Kim, J.W.; Otto, T.C.; Pandey, A.; Lane, M.D. Sequential phosphorylation of CCAAT enhancer-binding protein beta by MAPK and glycogen synthase kinase 3beta is required for adipogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 9766–9771. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J., Jr.; Liu, X.S.; et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Basu-Roy, U.; Gunaratne, P.H.; Coarfa, C.; Lim, D.S.; Basilico, C.; Mansukhani, A. SOX2 regulates YAP1 to maintain stemness and determine cell fate in the osteo-adipo lineage. Cell Rep. 2013, 3, 2075–2087. [Google Scholar] [CrossRef] [PubMed]
- Casado-Diaz, A.; Anter, J.; Muller, S.; Winter, P.; Quesada-Gomez, J.M.; Dorado, G. Transcriptomic Analyses of Adipocyte Differentiation from Human Mesenchymal Stromal-Cells (MSC). J. Cell. Physiol. 2017, 232, 771–784. [Google Scholar] [CrossRef] [PubMed]
- van de Peppel, J.; Strini, T.; Tilburg, J.; Westerhoff, H.; van Wijnen, A.J.; van Leeuwen, J.P. Identification of Three Early Phases of Cell-Fate Determination during Osteogenic and Adipogenic Differentiation by Transcription Factor Dynamics. Stem Cell Rep. 2017, 8, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.D. Methylation and demethylation of DNA and histones in chromatin: The most complicated epigenetic marker. Exp. Mol. Med. 2017, 49, e321. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef]
- Zhang, T.Y.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Xu, W.; Lan, F. Histone lysine demethylases in mammalian embryonic development. Exp. Mol. Med. 2017, 49, e325. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Liao, B.; Hu, J.; Li, S.; Zhao, H.; Sun, M.; Gu, J.; Jin, Y. JMJD1C Ensures Mouse Embryonic Stem Cell Self-Renewal and Somatic Cell Reprogramming through Controlling MicroRNA Expression. Stem Cell Rep. 2017, 9, 927–942. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, K.; Suzuki, T.; Vargas, D.; Shibata, H.; Inagaki, T. Epigenetic regulation of beige adipocyte fate by histone methylation. Endocr. J. 2019, 66, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Xu, Z.; Zhang, X.; Wang, L.; Gimble, J.M.; Lander, E.S.; Rosen, E.D. Comparative epigenomic analysis of murine and human adipogenesis. Cell 2010, 143, 156–169. [Google Scholar] [CrossRef]
- Siersbaek, R.; Nielsen, R.; John, S.; Sung, M.H.; Baek, S.; Loft, A.; Hager, G.L.; Mandrup, S. Extensive chromatin remodelling and establishment of transcription factor ‘hotspots’ during early adipogenesis. EMBO J. 2011, 30, 1459–1472. [Google Scholar] [CrossRef]
- Siersbaek, R.; Rabiee, A.; Nielsen, R.; Sidoli, S.; Traynor, S.; Loft, A.; Poulsen, L.C.; Rogowska-Wrzesinska, A.; Jensen, O.N.; Mandrup, S. Transcription factor cooperativity in early adipogenic hotspots and super-enhancers. Cell Rep. 2014, 7, 1443–1455. [Google Scholar] [CrossRef]
- Xiao, H.; Leblanc, S.E.; Wu, Q.; Konda, S.; Salma, N.; Marfella, C.G.; Ohkawa, Y.; Imbalzano, A.N. Chromatin accessibility and transcription factor binding at the PPARgamma2 promoter during adipogenesis is protein kinase A-dependent. J. Cell. Physiol. 2011, 226, 86–93. [Google Scholar] [CrossRef]
- Waki, H.; Nakamura, M.; Yamauchi, T.; Wakabayashi, K.; Yu, J.; Hirose-Yotsuya, L.; Take, K.; Sun, W.; Iwabu, M.; Okada-Iwabu, M.; et al. Global mapping of cell type-specific open chromatin by FAIRE-seq reveals the regulatory role of the NFI family in adipocyte differentiation. PLoS Genet. 2011, 7, e1002311. [Google Scholar] [CrossRef]
- Zhuang, L.; Jang, Y.; Park, Y.K.; Lee, J.E.; Jain, S.; Froimchuk, E.; Broun, A.; Liu, C.; Gavrilova, O.; Ge, K. Depletion of Nsd2-mediated histone H3K36 methylation impairs adipose tissue development and function. Nat. Commun. 2018, 9, 1796. [Google Scholar] [CrossRef]
- Lee, J.E.; Wang, C.; Xu, S.; Cho, Y.W.; Wang, L.; Feng, X.; Baldridge, A.; Sartorelli, V.; Zhuang, L.; Peng, W.; et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2013, 2, e01503. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Park, Y.K.; Park, S.; Jang, Y.; Waring, N.; Dey, A.; Ozato, K.; Lai, B.; Peng, W.; Ge, K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017, 8, 2217. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Broun, A.; Wang, C.; Park, Y.K.; Zhuang, L.; Lee, J.E.; Froimchuk, E.; Liu, C.; Ge, K. H3.3K4M destabilizes enhancer H3K4 methyltransferases MLL3/MLL4 and impairs adipose tissue development. Nucleic Acids Res. 2018, 47, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Takada, I.; Mihara, M.; Suzawa, M.; Ohtake, F.; Kobayashi, S.; Igarashi, M.; Youn, M.Y.; Takeyama, K.; Nakamura, T.; Mezaki, Y.; et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-gamma transactivation. Nat. Cell Biol. 2007, 9, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, S.; Lee, J.E.; Baldridge, A.; Grullon, S.; Peng, W.; Ge, K. Histone H3K9 methyltransferase G9a represses PPARgamma expression and adipogenesis. EMBO J. 2013, 32, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Musri, M.M.; Carmona, M.C.; Hanzu, F.A.; Kaliman, P.; Gomis, R.; Parrizas, M. Histone demethylase LSD1 regulates adipogenesis. J. Biol. Chem. 2010, 285, 30034–30041. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; Tee, W.W.; Reinberg, D. A double take on bivalent promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef]
- Matsumura, Y.; Nakaki, R.; Inagaki, T.; Yoshida, A.; Kano, Y.; Kimura, H.; Tanaka, T.; Tsutsumi, S.; Nakao, M.; Doi, T.; et al. H3K4/H3K9me3 Bivalent Chromatin Domains Targeted by Lineage-Specific DNA Methylation Pauses Adipocyte Differentiation. Mol. Cell 2015, 60, 584–596. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Abdouh, M.; Hanna, R.; El Hajjar, J.; Flamier, A.; Bernier, G. The Polycomb Repressive Complex 1 Protein BMI1 Is Required for Constitutive Heterochromatin Formation and Silencing in Mammalian Somatic Cells. J. Biol. Chem. 2016, 291, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, E.; Turberfield, A.H.; Klose, R.J. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015, 16, 1620–1639. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Chen, Q.M.; Hong, C.; Wang, C.Y. Histone methyltransferases and demethylases: Regulators in balancing osteogenic and adipogenic differentiation of mesenchymal stem cells. Int. J. Oral Sci. 2015, 7, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Park, J.W.; Chun, Y.S. Jumonji histone demethylases as emerging therapeutic targets. Pharmacol. Res. 2016, 105, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Okada, Y.; Kallin, E.M.; Zhang, Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 2009, 458, 757–761. [Google Scholar] [CrossRef]
- Kang, C.; Saso, K.; Ota, K.; Kawazu, M.; Ueda, T.; Okada, H. JMJD2B/KDM4B inactivation in adipose tissues accelerates obesity and systemic metabolic abnormalities. Genes Cells 2018, 23, 767–777. [Google Scholar] [CrossRef]
- Chen, Y.; Ikeda, K.; Yoneshiro, T.; Scaramozza, A.; Tajima, K.; Wang, Q.; Kim, K.; Shinoda, K.; Sponton, C.H.; Brown, Z.; et al. Thermal stress induces glycolytic beige fat formation via a myogenic state. Nature 2019, 565, 180–185. [Google Scholar] [CrossRef]
- Abe, Y.; Fujiwara, Y.; Takahashi, H.; Matsumura, Y.; Sawada, T.; Jiang, S.; Nakaki, R.; Uchida, A.; Nagao, N.; Naito, M.; et al. Histone demethylase JMJD1A coordinates acute and chronic adaptation to cold stress via thermogenic phospho-switch. Nat. Commun. 2018, 9, 1566. [Google Scholar] [CrossRef]
- Verrier, L.; Vandromme, M.; Trouche, D. Histone demethylases in chromatin cross-talks. Biol. Cell 2011, 103, 381–401. [Google Scholar] [CrossRef] [PubMed]
- Duteil, D.; Metzger, E.; Willmann, D.; Karagianni, P.; Friedrichs, N.; Greschik, H.; Gunther, T.; Buettner, R.; Talianidis, I.; Metzger, D.; et al. LSD1 promotes oxidative metabolism of white adipose tissue. Nat. Commun. 2014, 5, 4093. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Jedrychowski, M.P.; Chen, Y.; Serag, S.; Lavery, G.G.; Gygi, S.P.; Spiegelman, B.M. Lysine-specific demethylase 1 promotes brown adipose tissue thermogenesis via repressing glucocorticoid activation. Genes Dev. 2016, 30, 1822–1836. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kim, J.; Zhang, R.; Yang, X.; Zhang, Y.; Fang, J.; Chen, Z.; Teng, L.; Chen, X.; Ge, H.; et al. Histone Demethylase LSD1 Promotes Adipocyte Differentiation through Repressing Wnt Signaling. Cell Chem. Biol. 2016, 23, 1228–1240. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhang, Q.; Liao, C.; Han, W.; Xu, F. Epigenomic Control of Thermogenic Adipocyte Differentiation and Function. Int. J. Mol. Sci. 2018, 19, 1793. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wang, E.; Huang, Y.; Guo, X.; Yu, Y.; Du, Q.; Ding, X.; Sun, Y. Inhibition of Lysine-Specific Demethylase-1 (LSD1/KDM1A) Promotes the Adipogenic Differentiation of hESCs Through H3K4 Methylation. Stem Cell Rev. 2016, 12, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Sese, B.; Boue, S.; Castano, J.; Paramonov, I.; Barrero, M.J.; Izpisua Belmonte, J.C. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat. Cell Biol. 2011, 13, 652–659. [Google Scholar] [CrossRef]
- Baba, A.; Ohtake, F.; Okuno, Y.; Yokota, K.; Okada, M.; Imai, Y.; Ni, M.; Meyer, C.A.; Igarashi, K.; Kanno, J.; et al. PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat. Cell Biol. 2011, 13, 668–675. [Google Scholar] [CrossRef]
- Okuno, Y.; Ohtake, F.; Igarashi, K.; Kanno, J.; Matsumoto, T.; Takada, I.; Kato, S.; Imai, Y. Epigenetic regulation of adipogenesis by PHF2 histone demethylase. Diabetes 2013, 62, 1426–1434. [Google Scholar] [CrossRef]
- Kawakami, E.; Tokunaga, A.; Ozawa, M.; Sakamoto, R.; Yoshida, N. The histone demethylase Fbxl11/Kdm2a plays an essential role in embryonic development by repressing cell-cycle regulators. Mech. Dev. 2015, 135, 31–42. [Google Scholar] [CrossRef]
- Dong, R.; Yao, R.; Du, J.; Wang, S.; Fan, Z. Depletion of histone demethylase KDM2A enhanced the adipogenic and chondrogenic differentiation potentials of stem cells from apical papilla. Exp. Cell Res. 2013, 319, 2874–2882. [Google Scholar] [CrossRef] [PubMed]
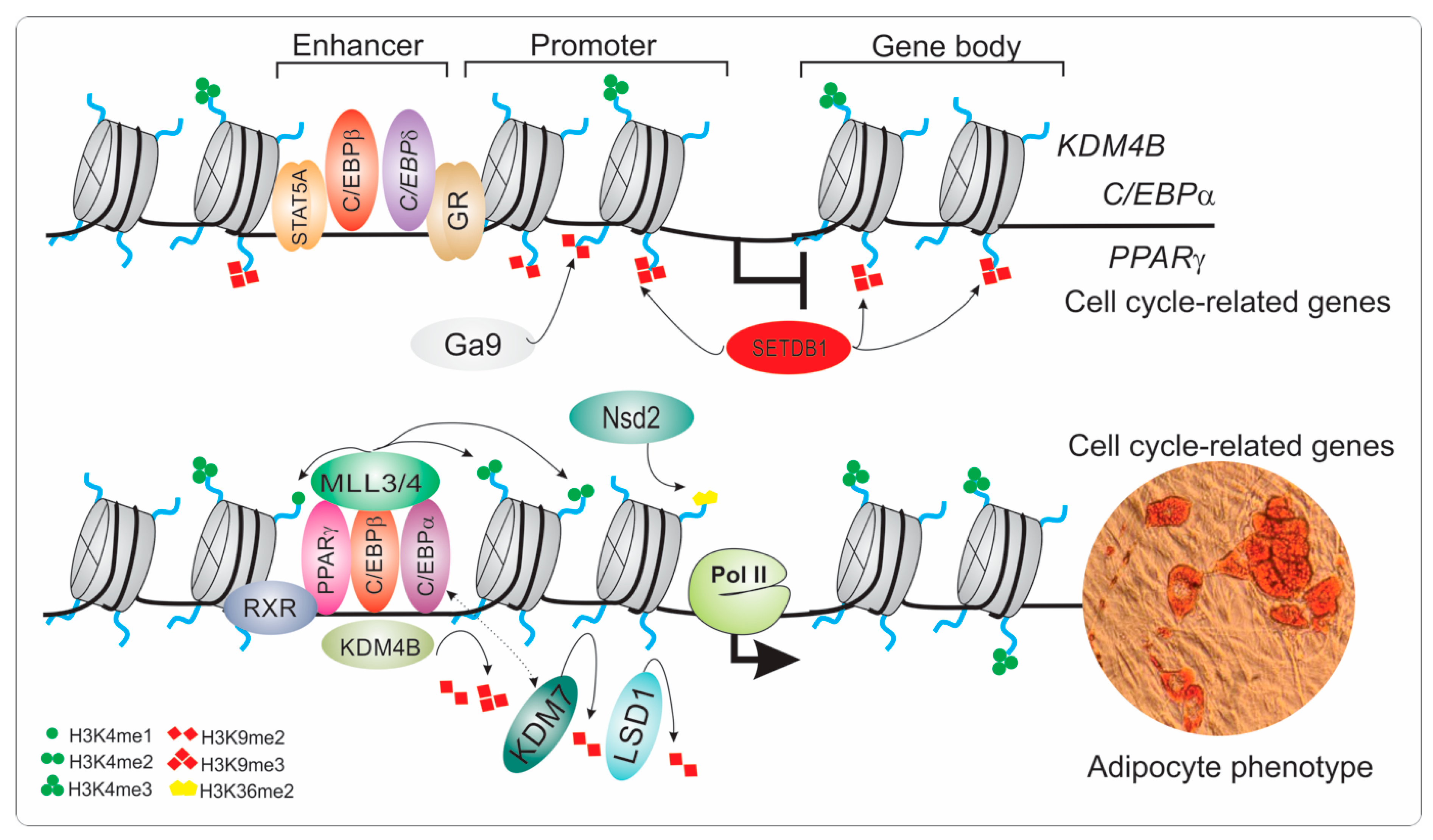
- Guo, L.; Li, X.; Huang, J.X.; Huang, H.Y.; Zhang, Y.Y.; Qian, S.W.; Zhu, H.; Zhang, Y.D.; Liu, Y.; Liu, Y.; et al. Histone demethylase Kdm4b functions as a co-factor of C/EBPbeta to promote mitotic clonal expansion during differentiation of 3T3-L1 preadipocytes. Cell Death Differ. 2012, 19, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Hao, W.; Xiao, C.; Wang, R.; Xu, X.; Lu, H.; Chen, W.; Deng, C.X. SIRT6 Is Essential for Adipocyte Differentiation by Regulating Mitotic Clonal Expansion. Cell Rep. 2017, 18, 3155–3166. [Google Scholar] [CrossRef] [PubMed]
- Brier, A.B.; Loft, A.; Madsen, J.G.S.; Rosengren, T.; Nielsen, R.; Schmidt, S.F.; Liu, Z.; Yan, Q.; Gronemeyer, H.; Mandrup, S. The KDM5 family is required for activation of pro-proliferative cell cycle genes during adipocyte differentiation. Nucleic Acids Res. 2017, 45, 1743–1759. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Iwasaki, S.; Matsumura, Y.; Kawamura, T.; Tanaka, T.; Abe, Y.; Yamasaki, A.; Tsurutani, Y.; Yoshida, A.; Chikaoka, Y.; et al. The FBXL10/KDM2B scaffolding protein associates with novel polycomb repressive complex-1 to regulate adipogenesis. J. Biol. Chem. 2015, 290, 4163–4177. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Yamaza, T.; Lee, J.S.; Yu, J.; Wang, S.; Fan, G.; Shi, S.; Wang, C.Y. BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat. Cell Biol. 2009, 11, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Kim, J.H.; Jung, M.H. Histone H3K9 Demethylase JMJD2B Activates Adipogenesis by Regulating H3K9 Methylation on PPARgamma and C/EBPalpha during Adipogenesis. PLoS ONE 2017, 12, e0168185. [Google Scholar] [CrossRef]
- Ye, L.; Fan, Z.; Yu, B.; Chang, J.; Al Hezaimi, K.; Zhou, X.; Park, N.H.; Wang, C.Y. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell 2012, 11, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Yuan, Q.; Vergnes, L.; Rong, X.; Youn, J.Y.; Li, J.; Yu, Y.; Liu, W.; Cai, H.; Lin, J.D.; et al. KDM4B protects against obesity and metabolic dysfunction. Proc. Natl. Acad. Sci. USA 2018, 115, E5566–E5575. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, F.; Romero, C.; Vargas, D. Regulation of adipogenesis by nuclear receptor PPARgamma is modulated by the histone demethylase JMJD2C. Genet. Mol. Biol. 2011, 34, 19–24. [Google Scholar] [CrossRef]
- Pack, L.R.; Yamamoto, K.R.; Fujimori, D.G. Opposing Chromatin Signals Direct and Regulate the Activity of Lysine Demethylase 4C (KDM4C). J. Biol. Chem. 2016, 291, 6060–6070. [Google Scholar] [CrossRef] [PubMed]
- Hemming, S.; Cakouros, D.; Isenmann, S.; Cooper, L.; Menicanin, D.; Zannettino, A.; Gronthos, S. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells 2014, 32, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Tong, K.I.; Goto, K.; Tomida, S.; Komuro, A.; Wang, Z.; Nishio, K.; Okada, H. The H3K27 demethylase, Utx, regulates adipogenesis in a differentiation stage-dependent manner. PLoS ONE 2017, 12, e0173713. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pan, D.; Huang, L.; Zhu, L.J.; Zou, T.; Ou, J.; Zhou, W.; Wang, Y.X. Jmjd3-Mediated H3K27me3 Dynamics Orchestrate Brown Fat Development and Regulate White Fat Plasticity. Dev. Cell 2015, 35, 568–583. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Tachibana, M.; Magoori, K.; Kudo, H.; Tanaka, T.; Okamura, M.; Naito, M.; Kodama, T.; Shinkai, Y.; Sakai, J. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 2009, 14, 991–1001. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromatin Modifier | Histone Targets | Type of Adipocytes | Roles in Adipose Tissue | Reference |
|---|---|---|---|---|
| LSD1 | H3K4me2, H3K9me2 | Brown, Beige, White | The adipose-specific deletion of LSD1 induces an increase in WAT-selective gene expression, impaired oxidative phosphorylation, reduced energy expenditure and increased fat mass. | [23,84] |
| KDM4B | H3K9me3 | Brown, White | Loss of KDM4B impairs energy expenditure, adaptive thermogenesis, and adipose tissue lipolysis, resulting in obesity and associated metabolic dysfunction. | [79,100] |
| KDM3A | H3K9me1, H3K9me2 | Brown, Beige | KDM3A regulates beige and brown adipogenesis by modulating the expression of metabolic genes. The loss of KDM3A function results in obesity and hyperlipidemia in mice. | [78,106] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nic-Can, G.I.; Rodas-Junco, B.A.; Carrillo-Cocom, L.M.; Zepeda-Pedreguera, A.; Peñaloza-Cuevas, R.; Aguilar-Ayala, F.J.; Rojas-Herrera, R.A. Epigenetic Regulation of Adipogenic Differentiation by Histone Lysine Demethylation. Int. J. Mol. Sci. 2019, 20, 3918. https://doi.org/10.3390/ijms20163918
Nic-Can GI, Rodas-Junco BA, Carrillo-Cocom LM, Zepeda-Pedreguera A, Peñaloza-Cuevas R, Aguilar-Ayala FJ, Rojas-Herrera RA. Epigenetic Regulation of Adipogenic Differentiation by Histone Lysine Demethylation. International Journal of Molecular Sciences. 2019; 20(16):3918. https://doi.org/10.3390/ijms20163918
Chicago/Turabian StyleNic-Can, Geovanny I., Beatriz A. Rodas-Junco, Leydi M. Carrillo-Cocom, Alejandro Zepeda-Pedreguera, Ricardo Peñaloza-Cuevas, Fernando J. Aguilar-Ayala, and Rafael A. Rojas-Herrera. 2019. "Epigenetic Regulation of Adipogenic Differentiation by Histone Lysine Demethylation" International Journal of Molecular Sciences 20, no. 16: 3918. https://doi.org/10.3390/ijms20163918
APA StyleNic-Can, G. I., Rodas-Junco, B. A., Carrillo-Cocom, L. M., Zepeda-Pedreguera, A., Peñaloza-Cuevas, R., Aguilar-Ayala, F. J., & Rojas-Herrera, R. A. (2019). Epigenetic Regulation of Adipogenic Differentiation by Histone Lysine Demethylation. International Journal of Molecular Sciences, 20(16), 3918. https://doi.org/10.3390/ijms20163918