Analysis of IFITM-IFITM Interactions by a Flow Cytometry-Based FRET Assay

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
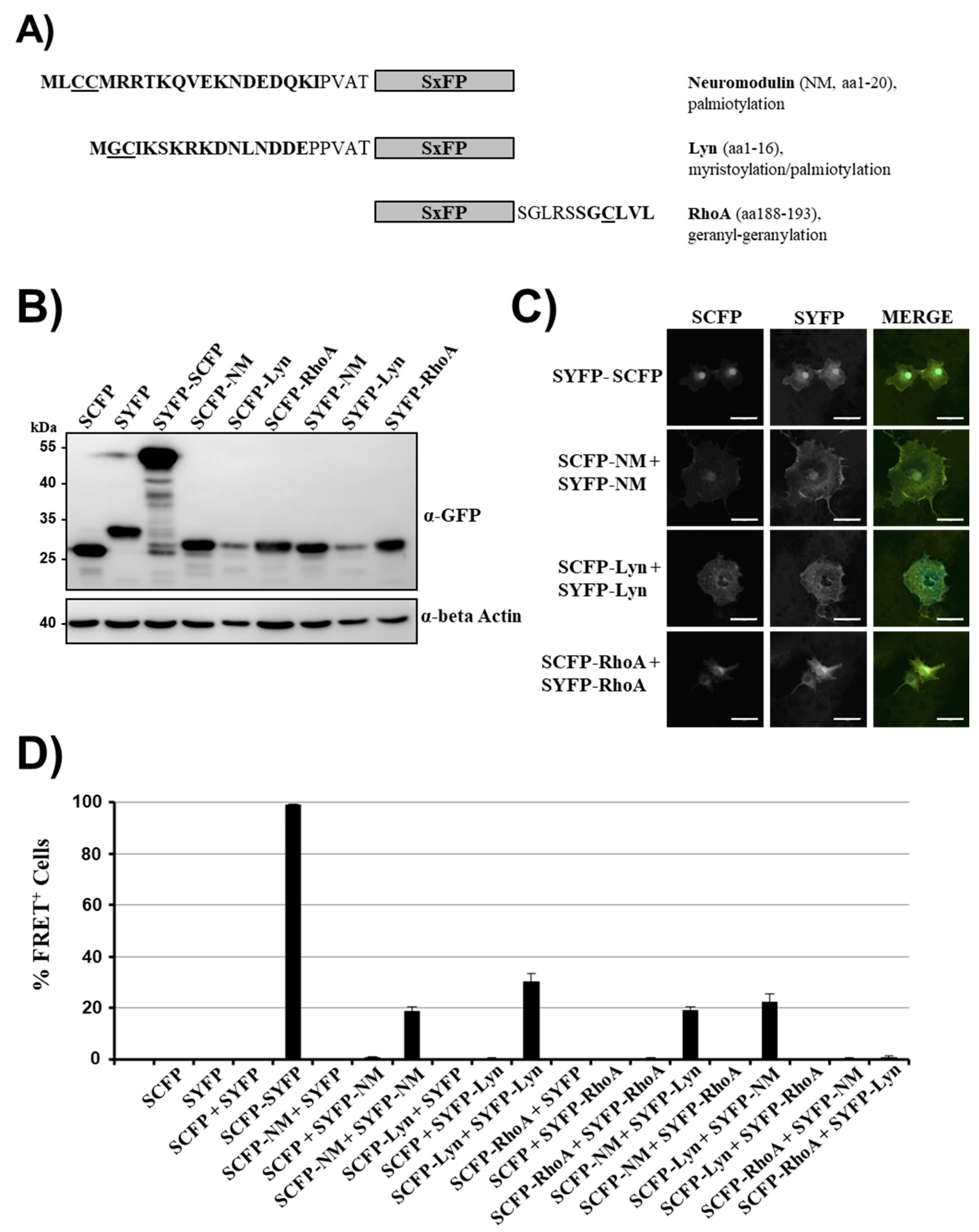
2.1. Establishment of FRET Measurements by Targeting of Non-Interacting, Cytosolic Proteins to Membranes
2.2. Homotypic and Heterotypic Interactions between IFITM Proteins
2.3. F75 and F78 Are Not Required for Normal Membrane Targeting of IFITM3 and for IFITM3/IFITM3 Interactions
2.4. F75 and F78 are Dispensable for IFITM3-IFITM3 Interactions as Determined by Co-Immunoprecipitation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmid Construction
4.3. Oligonucleotides
4.4. Antibodies
4.5. Immunofluorescence
4.6. Immunoblot
4.7. Production of Retroviral Vectors and Transduction Experiments
4.8. Coimmunoprecipitation
4.9. Flow-Cytometry and FRET
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EBOV | Ebola virus |
| eCFP | enhanced cyan fluorescent protein |
| eYFP | enhanced yellow fluorescent protein |
| FLUAV | Influenza A virus |
| FRET | Fluorescence resonance energy transfer |
| IFITM | Interferon-induced transmembrane protein |
| MLV | Murine leukemia virus |
| SCFP | super cyan fluorescent protein |
| SYFP | super yellow fluorescent protein |
| wt | wildtype |
References
- Shi, G.; Schwartz, O.; Compton, A.A. More than meets the I: The diverse antiviral and cellular functions of interferon-induced transmembrane proteins. Retrovirology 2017, 14, 53. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef] [PubMed]
- Feeley, E.M.; Sims, J.S.; John, S.P.; Chin, C.R.; Pertel, T.; Chen, L.M.; Gaiha, G.D.; Ryan, B.J.; Donis, R.O.; Elledge, S.J.; et al. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 2011, 7, e1002337. [Google Scholar] [CrossRef] [PubMed]
- Everitt, A.R.; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; Adams, D.J.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012, 484, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Jeong, B.H. No Correlation of the Disease Severity of Influenza A Virus Infection with the rs12252 Polymorphism of the Interferon-Induced Transmembrane Protein 3 Gene. Intervirology 2017, 60, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Mills, T.C.; Rautanen, A.; Elliott, K.S.; Parks, T.; Naranbhai, V.; Ieven, M.M.; Butler, C.C.; Little, P.; Verheij, T.; Garrard, C.S.; et al. IFITM3 and susceptibility to respiratory viral infections in the community. J. Infect. Dis. 2014, 209, 1028–1031. [Google Scholar] [CrossRef]
- Bailey, C.C.; Huang, I.C.; Kam, C.; Farzan, M. Ifitm3 limits the severity of acute influenza in mice. PLoS Pathog. 2012, 8, e1002909. [Google Scholar] [CrossRef]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. IFITM proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of Transmitted Founder HIV-1 to IFITM-Mediated Restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef]
- Roesch, F.; OhAinle, M.; Emerman, M. A CRISPR screen for factors regulating SAMHD1 degradation identifies IFITMs as potent inhibitors of lentiviral particle delivery. Retrovirology 2018, 15, 26. [Google Scholar] [CrossRef]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology 2014, 11, 103. [Google Scholar] [CrossRef]
- Poddar, S.; Hyde, J.L.; Gorman, M.J.; Farzan, M.; Diamond, M.S. The Interferon-Stimulated Gene IFITM3 Restricts Infection and Pathogenesis of Arthritogenic and Encephalitic Alphaviruses. J. Virol. 2016, 90, 8780–8794. [Google Scholar] [CrossRef]
- Weston, S.; Czieso, S.; White, I.J.; Smith, S.E.; Wash, R.S.; Diaz-Soria, C.; Kellam, P.; Marsh, M. Alphavirus Restriction by IFITM Proteins. Traffic 2016, 17, 997–1013. [Google Scholar] [CrossRef]
- Wrensch, F.; Karsten, C.B.; Gnirss, K.; Hoffmann, M.; Lu, K.; Takada, A.; Winkler, M.; Simmons, G.; Pöhlmann, S. Interferon-Induced Transmembrane Protein-Mediated Inhibition of Host Cell Entry of Ebolaviruses. J. Infect. Dis. 2015, 212 (Suppl. 2), S210–S218. [Google Scholar] [CrossRef]
- Chan, Y.K.; Huang, I.C.; Farzan, M. IFITM proteins restrict antibody-dependent enhancement of dengue virus infection. PLoS ONE 2012, 7, e34508. [Google Scholar] [CrossRef]
- Savidis, G.; Perreira, J.M.; Portmann, J.M.; Meraner, P.; Guo, Z.; Green, S.; Brass, A.L. The IFITMs Inhibit Zika Virus Replication. Cell Rep. 2016, 15, 2323–2330. [Google Scholar] [CrossRef]
- Gorman, M.J.; Poddar, S.; Farzan, M.; Diamond, M.S. The Interferon-Stimulated Gene Ifitm3 Restricts West Nile Virus Infection and Pathogenesis. J. Virol. 2016, 90, 8212–8225. [Google Scholar] [CrossRef]
- Xie, M.; Xuan, B.; Shan, J.; Pan, D.; Sun, Y.; Shan, Z.; Zhang, J.; Yu, D.; Li, B.; Qian, Z. Human cytomegalovirus exploits interferon-induced transmembrane proteins to facilitate morphogenesis of the virion assembly compartment. J. Virol. 2015, 89, 3049–3061. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, F.; Liu, F.; Cuconati, A.; Chang, J.; Block, T.M.; Guo, J.T. Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc. Natl. Acad. Sci. USA 2014, 111, 6756–6761. [Google Scholar] [CrossRef]
- Zhao, X.; Sehgal, M.; Hou, Z.; Cheng, J.; Shu, S.; Wu, S.; Guo, F.; Le Marchand, S.J.; Lin, H.; Chang, J.; et al. Identification of Residues Controlling Restriction versus Enhancing Activities of IFITM Proteins on Entry of Human Coronaviruses. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef]
- Li, K.; Markosyan, R.M.; Zheng, Y.M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C.; et al. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.C.; Farzan, M.; Jung, J.U. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 2013, 13, 452–464. [Google Scholar] [CrossRef]
- Lin, T.Y.; Chin, C.R.; Everitt, A.R.; Clare, S.; Perreira, J.M.; Savidis, G.; Aker, A.M.; John, S.P.; Sarlah, D.; Carreira, E.M.; et al. Amphotericin B increases influenza A virus infection by preventing IFITM3-mediated restriction. Cell Rep. 2013, 5, 895–908. [Google Scholar] [CrossRef]
- John, S.P.; Chin, C.R.; Perreira, J.M.; Feeley, E.M.; Aker, A.M.; Savidis, G.; Smith, S.E.; Elia, A.E.; Everitt, A.R.; Vora, M.; et al. The CD225 domain of IFITM3 is required for both IFITM protein association and inhibition of influenza A virus and dengue virus replication. J. Virol. 2013, 87, 7837–7852. [Google Scholar] [CrossRef]
- Yount, J.S.; Karssemeijer, R.A.; Hang, H.C. S-palmitoylation and ubiquitination differentially regulate interferon-induced transmembrane protein 3 (IFITM3)-mediated resistance to influenza virus. J. Biol. Chem. 2012, 287, 19631–19641. [Google Scholar] [CrossRef]
- Yount, J.S.; Moltedo, B.; Yang, Y.Y.; Charron, G.; Moran, T.M.; Lopez, C.B.; Hang, H.C. Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat. Chem. Biol. 2010, 6, 610–614. [Google Scholar] [CrossRef]
- McMichael, T.M.; Zhang, L.; Chemudupati, M.; Hach, J.C.; Kenney, A.D.; Hang, H.C.; Yount, J.S. The palmitoyltransferase ZDHHC20 enhances interferon-induced transmembrane protein 3 (IFITM3) palmitoylation and antiviral activity. J. Biol. Chem. 2017, 292, 21517–21526. [Google Scholar] [CrossRef]
- Banning, C.; Votteler, J.; Hoffmann, D.; Koppensteiner, H.; Warmer, M.; Reimer, R.; Kirchhoff, F.; Schubert, U.; Hauber, J.; Schindler, M. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS ONE 2010, 5, e9344. [Google Scholar] [CrossRef]
- Hagen, N.; Bayer, K.; Rosch, K.; Schindler, M. The intraviral protein interaction network of hepatitis C virus. Mol. Cell. Proteom. 2014, 13, 1676–1689. [Google Scholar] [CrossRef] [PubMed]
- Gondim, M.V.; Wiltzer-Bach, L.; Maurer, B.; Banning, C.; Arganaraz, E.; Schindler, M. AP-2 Is the Crucial Clathrin Adaptor Protein for CD4 Downmodulation by HIV-1 Nef in Infected Primary CD4+ T Cells. J. Virol. 2015, 89, 12518–12524. [Google Scholar] [CrossRef] [PubMed]
- Suffner, S.; Gerstenberg, N.; Patra, M.; Ruibal, P.; Orabi, A.; Schindler, M.; Bruss, V. Domains of the Hepatitis B Virus Small Surface Protein S Mediating Oligomerization. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Kremers, G.J.; Goedhart, J.; van Munster, E.B.; Gadella, T.W., Jr. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Forster radius. Biochemistry 2006, 45, 6570–6580. [Google Scholar] [CrossRef] [PubMed]
- Skene, J.H.; Virag, I. Posttranslational membrane attachment and dynamic fatty acylation of a neuronal growth cone protein, GAP-43. J. Cell Biol. 1989, 108, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Sudo, Y.; Valenzuela, D.; Beck-Sickinger, A.G.; Fishman, M.C.; Strittmatter, S.M. Palmitoylation alters protein activity: Blockade of G(o) stimulation by GAP-43. EMBO J. 1992, 11, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- Kovarova, M.; Tolar, P.; Arudchandran, R.; Draberova, L.; Rivera, J.; Draber, P. Structure-function analysis of Lyn kinase association with lipid rafts and initiation of early signaling events after Fcepsilon receptor I aggregation. Mol. Cell. Biol. 2001, 21, 8318–8328. [Google Scholar] [CrossRef]
- Adamson, P.; Paterson, H.F.; Hall, A. Intracellular localization of the P21rho proteins. J. Cell Biol. 1992, 119, 617–627. [Google Scholar] [CrossRef]
- Hori, Y.; Kikuchi, A.; Isomura, M.; Katayama, M.; Miura, Y.; Fujioka, H.; Kaibuchi, K.; Takai, Y. Post-translational modifications of the C-terminal region of the rho protein are important for its interaction with membranes and the stimulatory and inhibitory GDP/GTP exchange proteins. Oncogene 1991, 6, 515–522. [Google Scholar]
- Seibel, N.M.; Eljouni, J.; Nalaskowski, M.M.; Hampe, W. Nuclear localization of enhanced green fluorescent protein homomultimers. Anal. Biochem. 2007, 368, 95–99. [Google Scholar] [CrossRef]
- De Almeida, R.F.; Loura, L.M.; Fedorov, A.; Prieto, M. Lipid rafts have different sizes depending on membrane composition: A time-resolved fluorescence resonance energy transfer study. J. Mol. Biol. 2005, 346, 1109–1120. [Google Scholar] [CrossRef]
- Ben-Johny, M.; Yue, D.N.; Yue, D.T. Detecting stoichiometry of macromolecular complexes in live cells using FRET. Nat. Commun. 2016, 7, 13709. [Google Scholar] [CrossRef]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.L.; Liang, C. The IFITM proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef]
- Narayana, S.K.; Helbig, K.J.; McCartney, E.M.; Eyre, N.S.; Bull, R.A.; Eltahla, A.; Lloyd, A.R.; Beard, M.R. The Interferon-induced Transmembrane Proteins, IFITM1, IFITM2, and IFITM3 Inhibit Hepatitis C Virus Entry. J. Biol. Chem. 2015, 290, 25946–25959. [Google Scholar] [CrossRef]
- Anafu, A.A.; Bowen, C.H.; Chin, C.R.; Brass, A.L.; Holm, G.H. Interferon-inducible transmembrane protein 3 (IFITM3) restricts reovirus cell entry. J. Biol. Chem. 2013, 288, 17261–17271. [Google Scholar] [CrossRef]
- Bailey, C.C.; Zhong, G.; Huang, I.C.; Farzan, M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef]
- Dirks, W.G.; Drexler, H.G. STR DNA typing of human cell lines: Detection of intra- and interspecies cross-contamination. Methods Mol. Biol. 2013, 946, 27–38. [Google Scholar] [CrossRef]
- Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef]
- Wrensch, F.; Hoffmann, M.; Gartner, S.; Nehlmeier, I.; Winkler, M.; Pöhlmann, S. Virion Background and Efficiency of Virion Incorporation Determine Susceptibility of Simian Immunodeficiency Virus Env-Driven Viral Entry to Inhibition by IFITM Proteins. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Bertram, S.; Dijkman, R.; Habjan, M.; Heurich, A.; Gierer, S.; Glowacka, I.; Welsch, K.; Winkler, M.; Schneider, H.; Hofmann-Winkler, H.; et al. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J. Virol. 2013, 87, 6150–6160. [Google Scholar] [CrossRef]
- Teasdale, R.D.; Loci, D.; Houghton, F.; Karlsson, L.; Gleeson, P.A. A large family of endosome-localized proteins related to sorting nexin 1. Biochem. J. 2001, 358, 7–16. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelieres, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc 2006, 224, 213–232. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winkler, M.; Wrensch, F.; Bosch, P.; Knoth, M.; Schindler, M.; Gärtner, S.; Pöhlmann, S. Analysis of IFITM-IFITM Interactions by a Flow Cytometry-Based FRET Assay. Int. J. Mol. Sci. 2019, 20, 3859. https://doi.org/10.3390/ijms20163859
Winkler M, Wrensch F, Bosch P, Knoth M, Schindler M, Gärtner S, Pöhlmann S. Analysis of IFITM-IFITM Interactions by a Flow Cytometry-Based FRET Assay. International Journal of Molecular Sciences. 2019; 20(16):3859. https://doi.org/10.3390/ijms20163859
Chicago/Turabian StyleWinkler, Michael, Florian Wrensch, Pascale Bosch, Maike Knoth, Michael Schindler, Sabine Gärtner, and Stefan Pöhlmann. 2019. "Analysis of IFITM-IFITM Interactions by a Flow Cytometry-Based FRET Assay" International Journal of Molecular Sciences 20, no. 16: 3859. https://doi.org/10.3390/ijms20163859
APA StyleWinkler, M., Wrensch, F., Bosch, P., Knoth, M., Schindler, M., Gärtner, S., & Pöhlmann, S. (2019). Analysis of IFITM-IFITM Interactions by a Flow Cytometry-Based FRET Assay. International Journal of Molecular Sciences, 20(16), 3859. https://doi.org/10.3390/ijms20163859