Blockade of TRPV2 is a Novel Therapy for Cardiomyopathy in Muscular Dystrophy


Abstract
1. Introduction
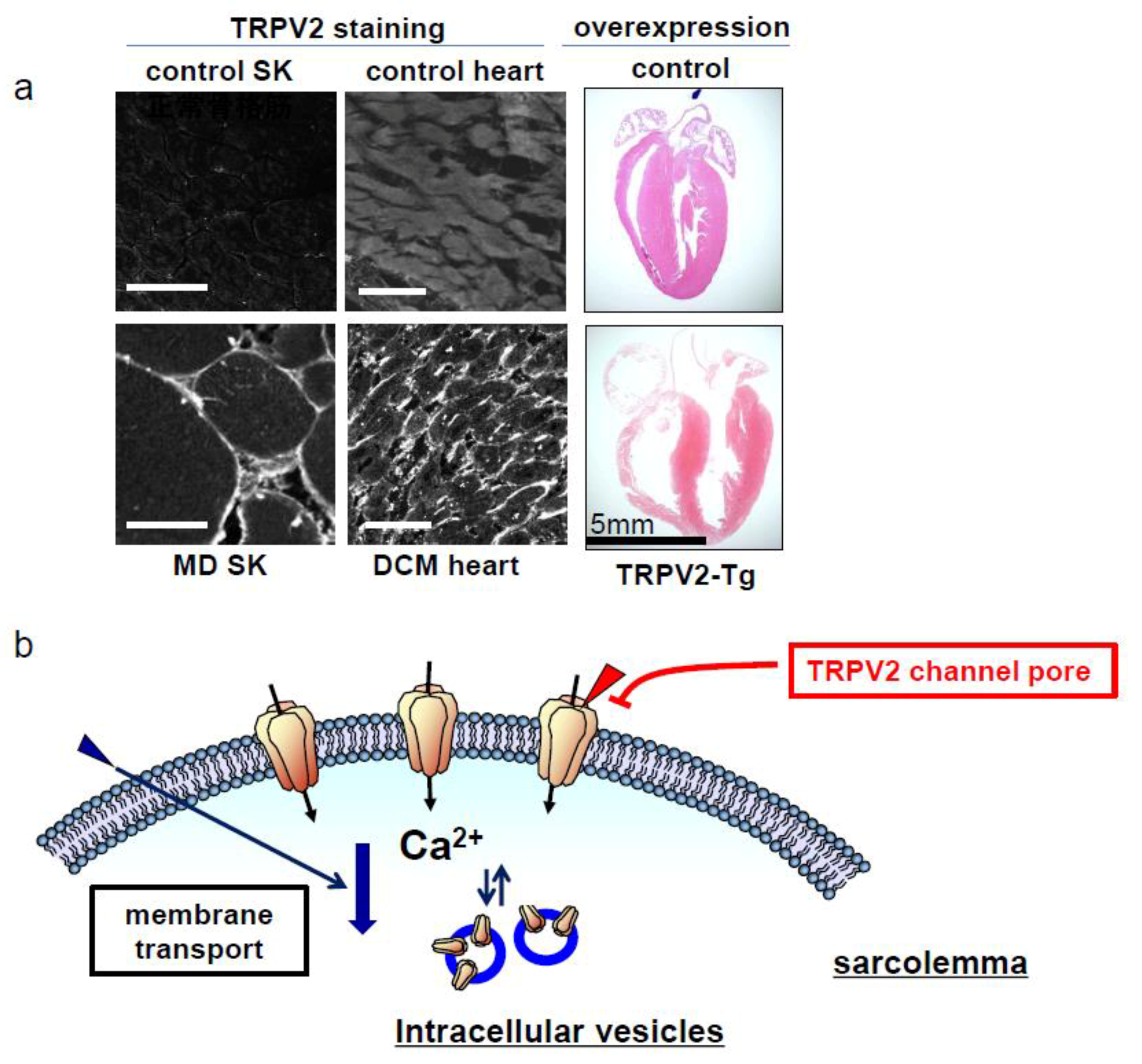
2. Muscular Dysgenesis and TRPV2
3. TRPV2 as a Drug Discovery Target
4. Effects of Various TRPV2 Blockers on Muscle Dysgenesis and Cardiac Dysfunction
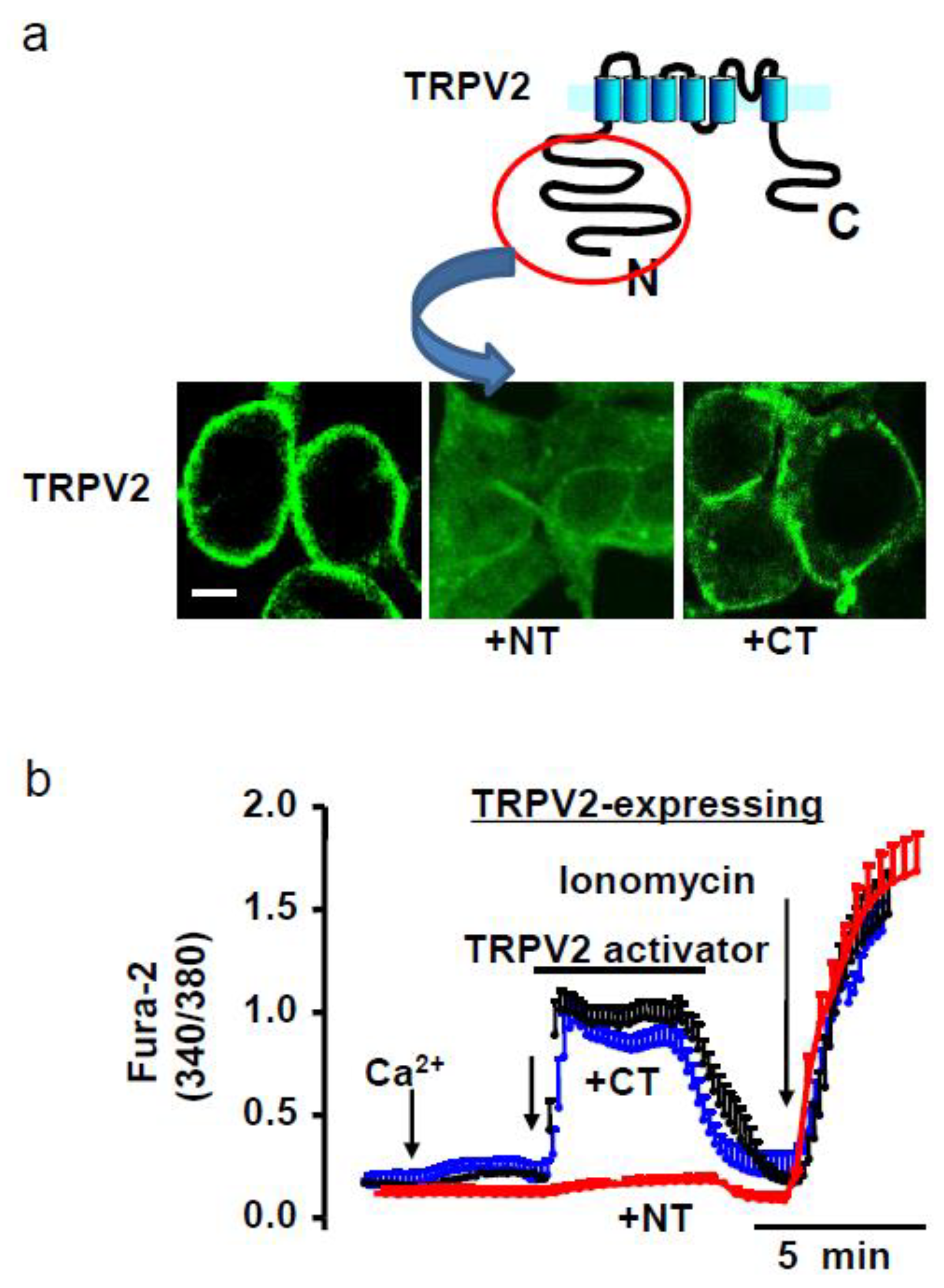
4.1. Amino Terminal (NT) Domain Expression
4.2. Dominant-Negative Strategy
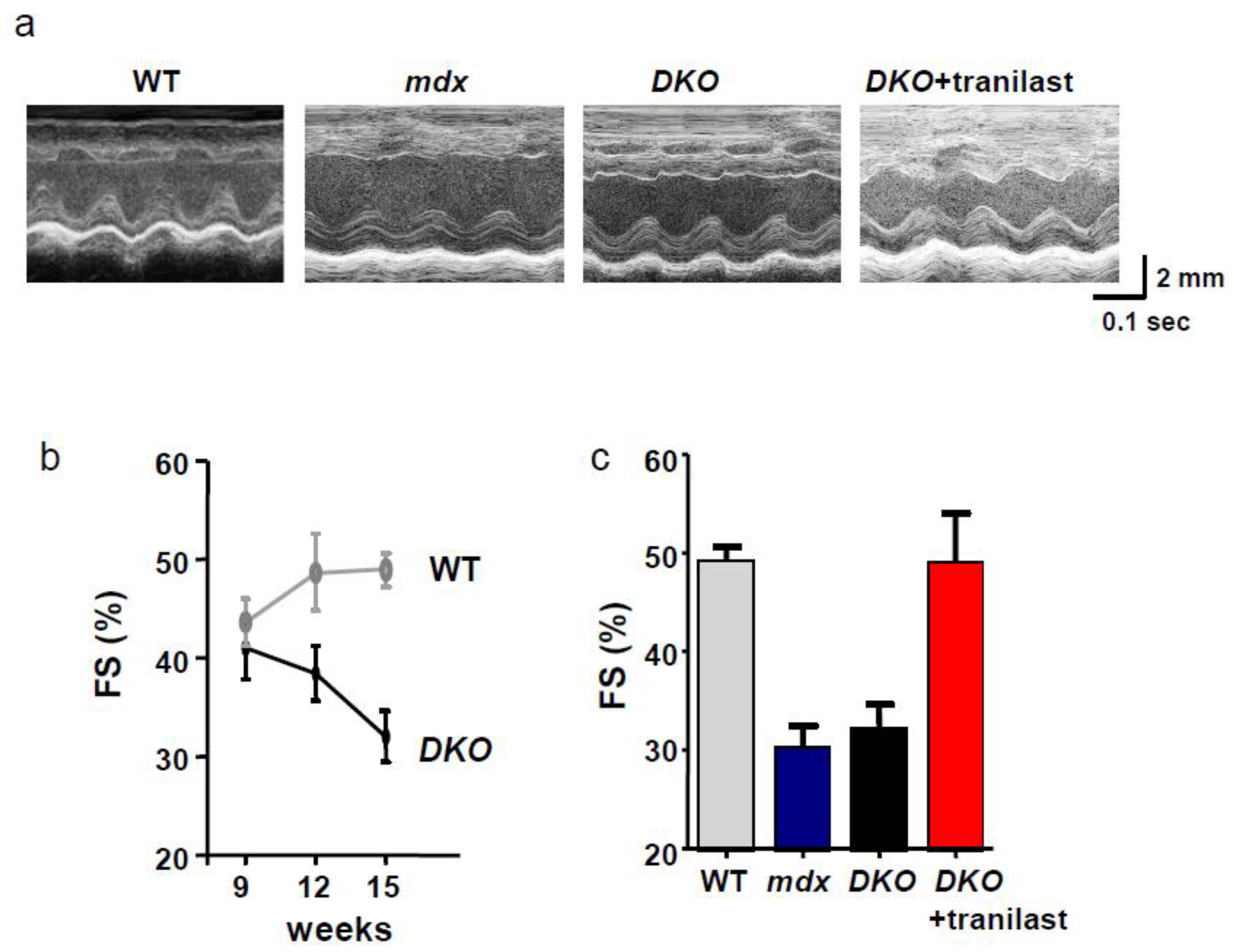
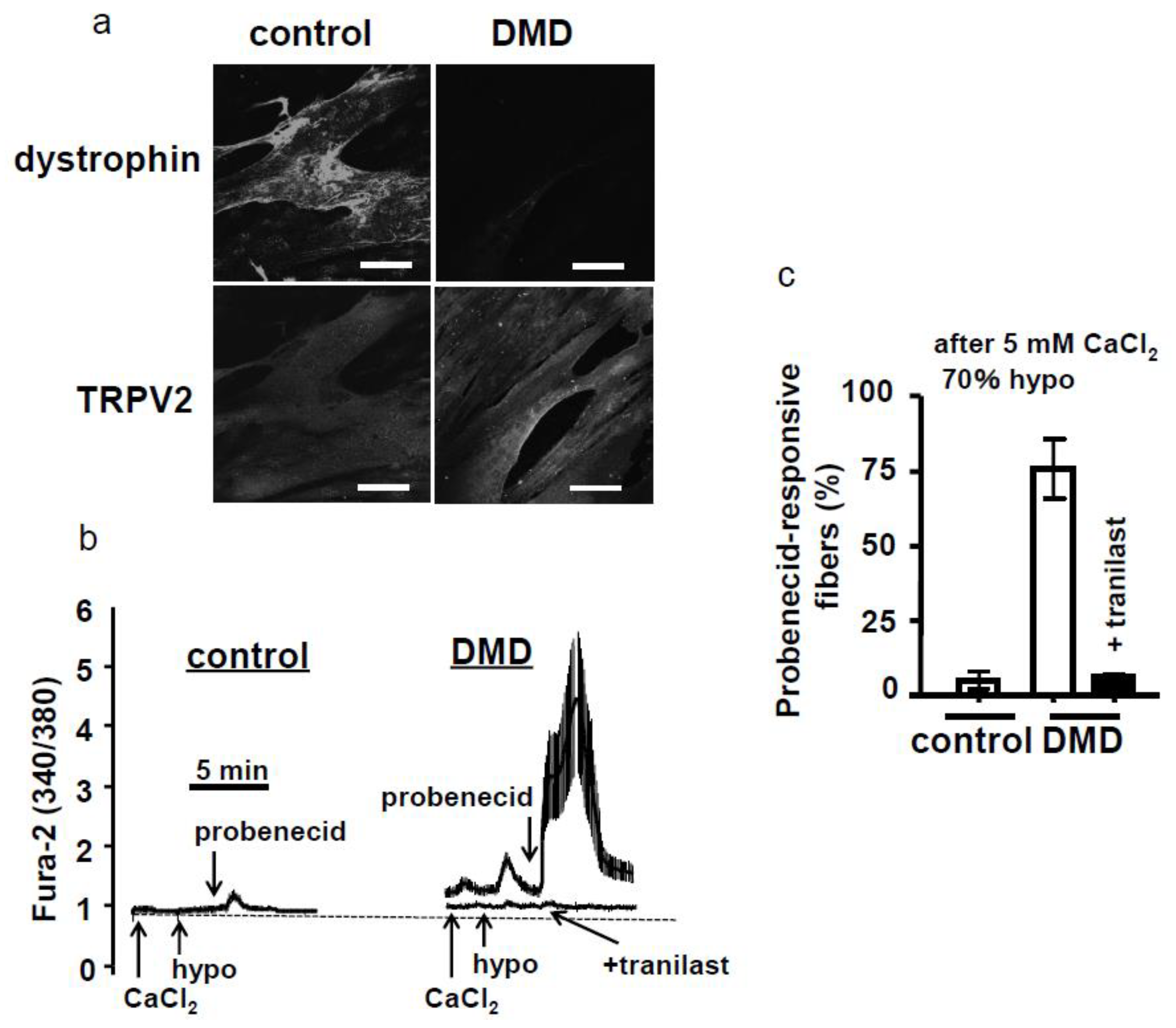
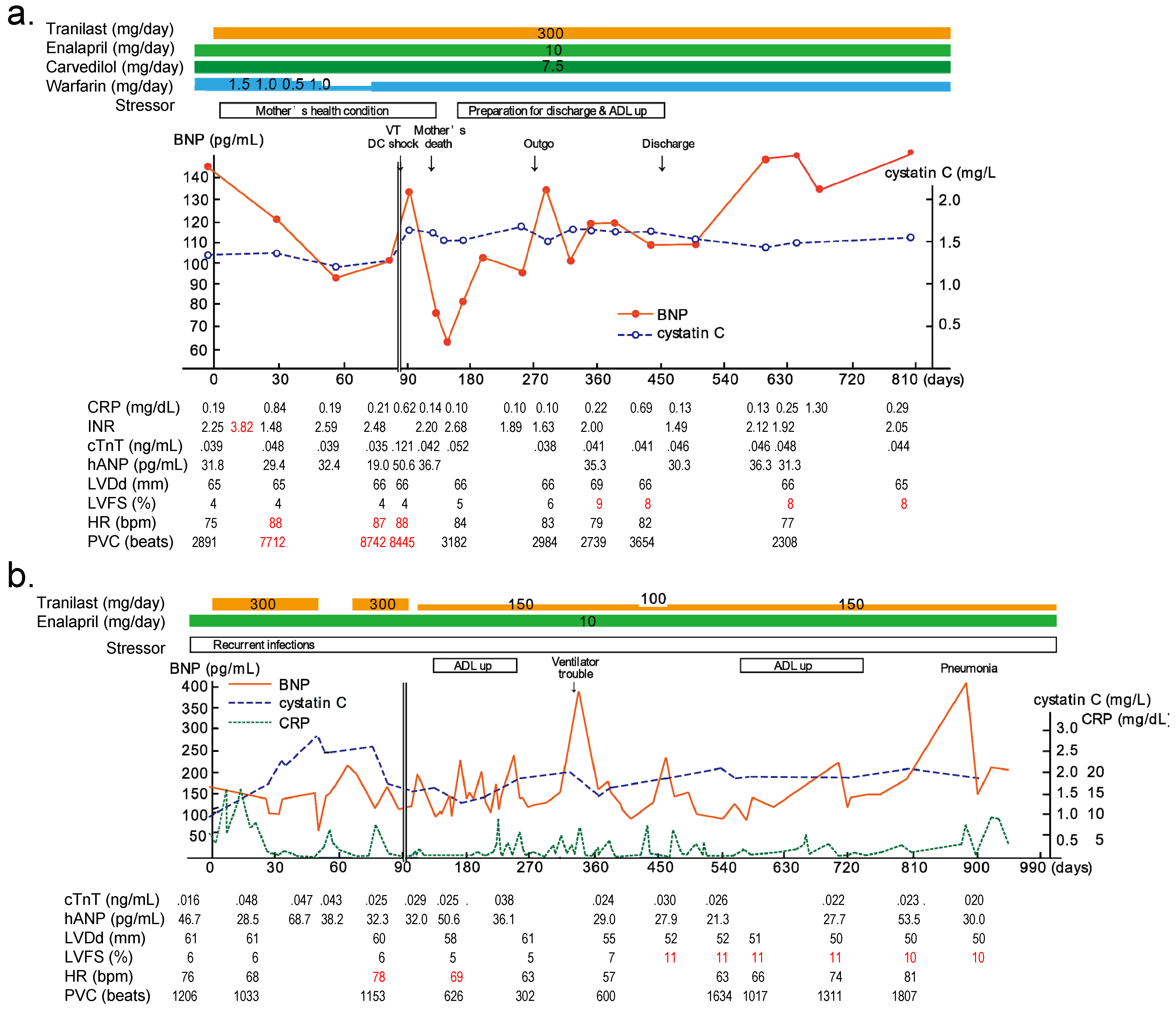
4.3. Functional Antibody and Tranilast
4.4. Gene Deletion
5. Development of TRPV2 Inhibitors
6. Clinical Trials of TRPV2 Inhibitors
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ervasti, J.M. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Boil. 1993, 122, 809–823. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Mestroni, L. Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Nakamura, H.; Mizuno, Y.; Yoshida, M.; Ozawa, E.; Shigekawa, M. Defective association of dystrophin with sarcolemmal glycoproteins in the cardiomyopathic hamster heart. FEBS Lett. 1993, 329, 227–231. [Google Scholar] [CrossRef]
- Campbell, K.P. Three muscular dystrophies: Loss of cytoskeleton-extracellular matrix linkage. Cell 1995, 80, 675–679. [Google Scholar] [CrossRef]
- Michalak, M.; Opas, M. Functions of dystrophin and dystrophin associated proteins. Curr. Opin. Neurol. 1997, 10, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Robert, V.; Massimino, M.L.; Tosello, V.; Marsault, R.; Cantini, M.; Sorrentino, V.; Pozzan, T. Alteration in calcium handling at the subcellular level in mdx myotubes. J. Biol. Chem. 2001, 276, 4647–4651. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.Y.; Iwata, Y.; Sampaolesi, M.; Hanada, H.; Saito, N.; Artman, M.; Coetzee, W.A.; Shigekawa, M. Stretch-activated cation channels in skeletal muscle myotubes from sarcoglycan-deficient hamsters. Am. J. Physiol. 2001, 281. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Katanosaka, Y.; Arai, Y.; Komamura, K.; Miyatake, K.; Shigekawa, M. A novel mechanism of myocyte degeneration involving the Ca2+-permeable growth factor-regulated channel. J. Cell Biol. 2003, 161, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Ohtake, H.; Suzuki, O.; Matsuda, J.; Komamura, K.; Wakabayashi, S. Blockade of sarcolemmal TRPV2 accumulation inhibits progression of dilated cardiomyopathy. Cardiovasc. Res. 2013, 99, 760–768. [Google Scholar] [CrossRef]
- Suzuki, O.; Kanai, T.; Nishikawa, T.; Yamamoto, Y.; Noguchi, A.; Takimoto, K.; Koura, M.; NoguchiHI, Y.; Uchio-Yamada, K.; Tsuji, S.; et al. Adult onset cardiac dilatation in a transgenic mouse line with Galbeta1, 3GalNAc alpha2,3-sialyltransferase II (ST3Gal-II) transgenes: A new model for dilated cardiomyopathy. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 2011, 87, 550–562. [Google Scholar] [CrossRef][Green Version]
- Du, C.-K.; Morimoto, S.; Nishii, K.; Minakami, R.; Ohta, M.; Tadano, N.; Lu, Q.-W.; Wang, Y.-Y.; Zhan, D.-Y.; Mochizuki, M.; et al. Knock-In Mouse Model of Dilated Cardiomyopathy Caused by Troponin Mutation. Circ. Res. 2007, 101, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.E.; Mann, A.; Jones, S.; Robbins, N.; AlKhattabi, A.; Worley, M.C.; Gao, X.; Lasko-Roiniotis, V.M.; Karani, R.; Fulford, L.; et al. Transient receptor potential vanilloid 2 function regulates cardiac hypertrophy via stretch-induced activation. J. Hypertens. 2017, 35, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Entin-Meer, M.; Levy, R.; Goryainov, P.; Landa, N.; Barshack, I.; Avivi, C.; Semo, J.; Keren, G. The Transient Receptor Potential Vanilloid 2 Cation Channel Is Abundant in Macrophages Accumulating at the Peri-Infarct Zone and May Enhance Their Migration Capacity towards Injured Cardiomyocytes following Myocardial Infarction. PLoS ONE 2014, 9, e105055. [Google Scholar] [CrossRef] [PubMed]
- Entin-Meer, M.; Cohen, L.; Hertzberg-Bigelman, E.; Levy, R.; Ben-Shoshan, J.; Keren, G. TRPV2 knockout mice demonstrate an improved cardiac performance following myocardial infarction due to attenuated activity of peri-infarct macrophages. PLoS ONE 2017, 12, e0177132. [Google Scholar] [CrossRef] [PubMed]
- Aguettaz, E.; Bois, P.; Cognard, C.; Sebille, S. Stretch-activated TRPV2 channels: Role in mediating cardiopathies. Prog. Biophys. Mol. Biol. 2017, 130, 273–280. [Google Scholar] [CrossRef]
- Jones, S.; Mann, A.; Worley, M.C.; Fulford, L.; Hall, D.; Karani, R.; Jiang, M.; Rubinstein, J.; Robbins, N.; Koch, S.E. The role of transient receptor potential vanilloid 2 channel in cardiac aging. Aging Clin. Exp. Res. 2017, 29, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Kamauchi, S.; Iwata, Y.; Du, C.-K.; Zhan, D.-Y.; Morimoto, S.; Shirai, M. Overexpression of the amino-terminal domain of TRPV2 has the beneficial effect for dilated cardiomyopathy mice with mutation of troponin T. J. Pharm. Sci. 2014, 124, 194. [Google Scholar]
- Iwata, Y.; Katanosaka, Y.; Arai, Y.; Shigekawa, M.; Wakabayashi, S. Dominant-negative inhibition of Ca2+ influx via TRPV2 ameliorates muscular dystrophy in animal models. Hum. Mol. Genet. 2009, 18, 824–834. [Google Scholar] [CrossRef]
- Zanou, N.; Iwata, Y.; Schakman, O.; Lebacq, J.; Wakabayashi, S.; Gailly, P. Essential role of TRPV2 ion channel in the sensitivity of dystrophic muscle to eccentric contractions. FEBS Lett. 2009, 583, 3600–3604. [Google Scholar] [CrossRef]
- Iwata, Y.; Wakabayashi, S.; Ito, S.; Kitakaze, M. Functional antibody against TRPV2 ameliorates dilated cardiomyopathy in animal models. Unpublished work (manuscript in preparation).
- Iwata, Y.; Hirayama, M.; Ito, S.; Kitakaze, M. Treatment with TRPV2 antibody ameliorates the severity of heart failure in dilated cardiomyopathic hamsters. WCP2018Kyoto. In Proceedings of the 18th World Congress of Basic and Clinical Pharmacology, Japan, Kyoto, 1–6 July 2018. [Google Scholar]
- Iwata, Y.; Hirayama, M.; Ito, S.; Kitakaze, M. Inhibition of TRPV2 prevents the progression of murine heart failure. Presented at the 92nd Annual Meeting of the Japanese Pharmacological Society, Osaka, Japan, 14–16 March 2019. [Google Scholar]
- Vögeli, I.; Lorin, C.; Niggli, E. Dystrophic cardiomyopathy: Role of TRPV2 channels in stretch-induced cell damage. Cardiovasc. Res. 2015, 106, 153–162. [Google Scholar]
- Aguettaz, E.; Lopez, J.; Krzesiak, A.; Constantin, B.; Cognard, C.; Sebille, S. Data on calcium increases depending on stretch in dystrophic cardiomyocytes. Data Brief. 2016, 8, 1443–1447. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aguettaz, E.; Lopez, J.J.; Krzesiak, A.; Lipskaia, L.; Adnot, S.; Hajjar, R.; Cognard, C.; Constantin, B.; Sebille, S. Axial stretch-dependent cation entry in dystrophic cardiomyopathy: Involvement of several TRPs channels. Cell Calcium 2016, 59, 145–155. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Komamura, K.; Iwata, Y. Tranilast, Orally Active TRPV2 Antagonist, Ameliorates End-stage Heart Failure in Mice with Dilated Cardiomyopathy. J. Card. Fail. 2014, 20, S198–S199. [Google Scholar] [CrossRef]
- Iwata, Y.; Komamura, K. Tranilast, Transient Receptor Potential Vanilloid 2 Antagonist, Ameliorates End-Stage Heart Failure of Mice with Dilated Cardiomyopathy. Circulation 2014, 130, A11977. [Google Scholar]
- Iwata, Y.; Katayama, Y.; Okuno, Y.; Wakabayashi, S. Novel inhibitor candidates of TRPV2 prevent damage of dystrophic myocytes and ameliorate against dilated cardiomyopathy in a hamster model. Oncotarget 2018, 9, 14042–14057. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Katanosaka, Y.; Shijun, Z.; Kobayashi, Y.; Hanada, H.; Shigekawa, M.; Wakabayashi, S. Protective effects of Ca2+ handling drugs against abnormal Ca2+ homeostasis and cell damage in myopathic skeletal muscle cells. Biochem. Pharm. 2005, 70, 740–751. [Google Scholar] [CrossRef]
- Mihara, H.; Boudaka, A.; Shibasaki, K.; Yamanaka, A.; Sugiyama, T.; Tominaga, M. Involvement of TRPV2 Activation in Intestinal Movement through Nitric Oxide Production in Mice. J. Neurosci. 2010, 30, 16536–16544. [Google Scholar] [CrossRef]
- Delfín, D.A.; Zang, K.E.; Schill, K.E.; Patel, N.T.; Janssen, P.M.; Raman, S.V.; Rafael-Fortney, J.A. Cardiomyopathy in the dystrophin/utrophin-deficient mouse model of severe muscular dystrophy is characterized by dysregulation of matrix metalloproteinases. Neuromuscul Disord. 2012, 22, 1006–1014. [Google Scholar] [CrossRef]
- Chai, H.; Cheng, X.; Zhou, B.; Zhao, L.; Lin, X.; Huang, D.; Lu, W.; Lv, H.; Tang, F.; Zhang, Q.; et al. Structure-based discovery of a subtype-selective inhibitor targeting a transient receptor potential vanilloid channel. J. Med. Chem. 2019, 62, 1373–1384. [Google Scholar] [CrossRef]
- Schiano, M.A.; Lopez, C.S.; Novo, F.O.; Eras, J.; Amodeo, P.; Canela-Garayoa, R.; Vitale, R.M.; Marzo, V.D.; Petrocellis, L.D. Elongation of the Hydrophobic Chain as a Molecular Switch: Discovery of Capsaicin Derivatives and Endogenous Lipids as Potent Transient Receptor Potential Vanilloid Channel 2 Antagonists. J. Med. Chem. 2018, 61, 8255–8281. [Google Scholar] [CrossRef]
- Iwata, Y.; Suzuki, N.; Ohtake, H.; Kamauchi, S.; Hashimoto, N.; Kiyono, T.; Wakabayashi, S. Cancer cachexia causes skeletal muscle damage via transient receptor potential vanilloid 2-independent mechanisms, unlike muscular dystrophy. J. Cachexia Sarcopenia Muscle 2016, 7, 366–376. [Google Scholar] [CrossRef]
- Uvin, V.; Penna, A.; Chemin, J.; Lin, Y.-L.; Rassendren, F.-A. Pharmacological Characterization and Molecular Determinants of the Activation of Transient Receptor Potential V2 Channel Orthologs by 2-Aminoethoxydiphenyl Borate. Mol. Pharm. 2007, 72, 1258–1268. [Google Scholar]
- Darakhshan, S.; Pour, A.B. Tranilast: A review of its therapeutic applications. Pharmacol. Res. 2015, 91, 15–28. [Google Scholar] [CrossRef]
- Hara, M.; Ono, K.; Hwang, M.-W.; Iwasaki, A.; Okada, M.; Nakatani, K.; Sasayama, S.; Matsumori, A. Evidence for a Role of Mast Cells in the Evolution to Congestive Heart Failure. J. Exp. Med. 2002, 195, 375–381. [Google Scholar] [CrossRef]
- Nakatani, Y.; Nishida, K.; Sakabe, M.; Kataoka, N.; Sakamoto, T.; Yamaguchi, Y.; Iwamoto, J.; Mizumaki, K.; Fujiki, A.; Inoue, H. Tranilast Prevents Atrial Remodeling and Development of Atrial Fibrillation in a Canine Model of Atrial Tachycardia and Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 2013, 61, 582–588. [Google Scholar] [CrossRef]
- Zhang, D.; Spielmann, A.; Wang, L.; Ding, G.; Huang, F.; Gu, Q.; Schwarz1, W. Mast-cell degranulation induced by physical stimuli involves the activation of transient-receptor-potential channel TRPV2. Physiol. Res. 2012, 61, 113–124. [Google Scholar]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- Matsumura, T.; Matsui, M.; Iwata, Y.; Asakura, M.; Saito, T.; Fujimura, H.; Sakoda, S. A Pilot Study of Tranilast for Cardiomyopathy of Muscular Dystrophy. Intern. Med. 2018, 57, 311–318. [Google Scholar] [CrossRef]
- Matsumura, T.; Matsui, M.; Iwata, Y.; Asakura, M.; Saito, T.; Fujimura, H.; Sakoda, S. Long-term effects of TRPV2 inhibition therapy for cardiomyopathy of muscular dystrophy. Neuromuscul Disord. 2018, 27, S114. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TRPV2 Blocker Gene, Agents | Animal Models/Cells for Human Disease | Strain | Background of Animals | Evaluation Index (Efficacy) | Reports |
|---|---|---|---|---|---|
| TRPV2-NT Transgenic oradenovirus | * DCM mice | 4C30 | sialyltransferase transgenic | cardiac function↑, heart weight↓, | [9] |
| fibrosis↓, survival↑ * CK↓,* ANP↓, * cTn-I↓ in serum | |||||
| DCM mice | * TNNT2 *ΔK210 | cTn-T mutant knock-in | cardiac function↑ | [17] | |
| DCM hamsters | J2N-k | δ-sarcoglycan defect | cardiac function↑ | [9] | |
| * DOX induced CM | BL6 | cardiotoxicity | cardiac function↑, *ROS production↓ | [9] | |
| * DN-TRPV2 Transgenicor adenovirus | * MD mice | mdx | dystrophin defect | fibrosis↓, serum CK↓, Ca2+ influx↓, recovery of muscle strength↑ | [18,19] |
| MD hamsters | BIO14.6 | δ-sarcoglycan defect | fibrosis↓, serum CK↓, Ca2+ influx↓ | [18] | |
| Functional antibody | DCM mice | 4C30 | sialyltransferase transgenic | cardiac function↑ | [20,21,22] |
| DCM hamsters | J2N-k | δ-sarcoglycan defect | cardiac function↑, serum CK↓ | ||
| MD cardiomyocytes | mdx | dystrophin defect | stretch induced Ca2+ influx↓ in isolated cardiomyocytes | [23,24,25] | |
| TAC mice | BL6 | hemodynamic stress | cardiac function↑ | [22] | |
| Inhibitorstranilast, * lumin | DCM mice | 4C30 | sialyltransferase transgenic | cardiac function↑, fibrosis↓ | [26,27] |
| DCM hamsters | J2N-k | δ-sarcoglycan defect | cardiac function↑, fibrosis↓ | [9,28] | |
| MD cardiomyocytes | mdx | dystrophin defect | stretch induced Ca2+ influx↓ | [23,24,25] | |
| MD mice | * DKO | utrophin/dystrophin defect | cardiac function↑ | Figure 3 | |
| Gene silencing | MD cardiomyocytes | mdx | dystrophin defect | Ca2+ influx↓ | [23] |
| antisense DNA,*siRNA | MD myotubes | BIO14.6 | δ-sarcoglycan defect | Ca2+ influx↓ | [8] |
| Gene deletion | * TAC mice | BL6/129S | hemodynamic stress | hypertrophy↓, cardiac function→ | [12] |
| (Knockout) | * MI mice | BL6/129S | ischemia stress | cardiac function↑ | [13,14] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwata, Y.; Matsumura, T. Blockade of TRPV2 is a Novel Therapy for Cardiomyopathy in Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 3844. https://doi.org/10.3390/ijms20163844
Iwata Y, Matsumura T. Blockade of TRPV2 is a Novel Therapy for Cardiomyopathy in Muscular Dystrophy. International Journal of Molecular Sciences. 2019; 20(16):3844. https://doi.org/10.3390/ijms20163844
Chicago/Turabian StyleIwata, Yuko, and Tsuyoshi Matsumura. 2019. "Blockade of TRPV2 is a Novel Therapy for Cardiomyopathy in Muscular Dystrophy" International Journal of Molecular Sciences 20, no. 16: 3844. https://doi.org/10.3390/ijms20163844
APA StyleIwata, Y., & Matsumura, T. (2019). Blockade of TRPV2 is a Novel Therapy for Cardiomyopathy in Muscular Dystrophy. International Journal of Molecular Sciences, 20(16), 3844. https://doi.org/10.3390/ijms20163844