Extracellular Signal-Regulated Kinase: A Regulator of Cell Growth, Inflammation, Chondrocyte and Bone Cell Receptor-Mediated Gene Expression

{kind=link}
Abstract
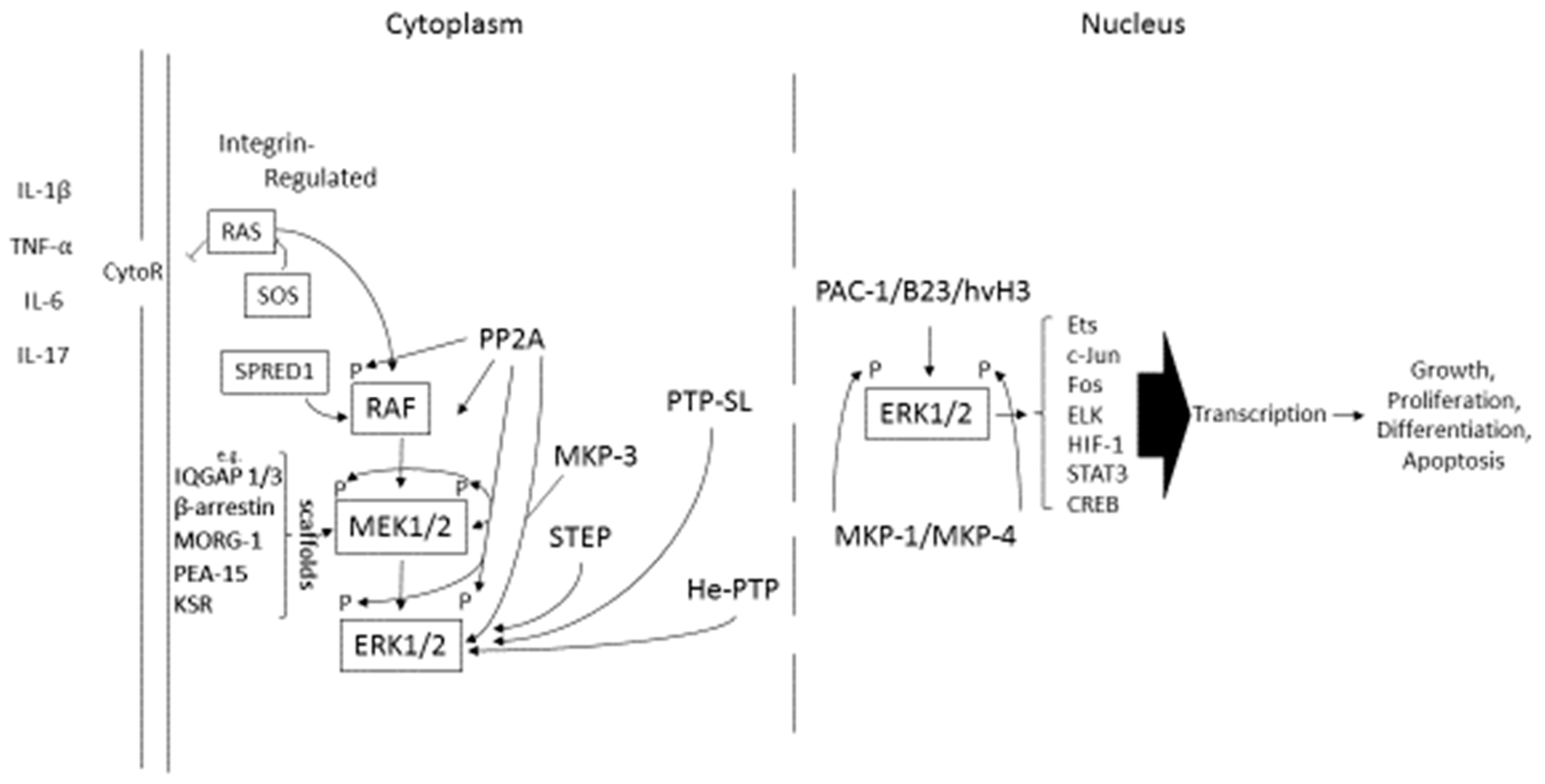
1. An Overview of ERK Signaling Components
1.1. Regulation of the RAS-RAF-ERK Pathway
1.2. Inhibition of the ERK1/2 Pathway
1.3. Role of the ERK1/2 Pathway in Inflammation
TPL2/Pro-Inflammatory Cytokines/Inflammation
2. Novel Insights into the Regulation of ERK Activation
3. Upstream Inhibitors of ERK
4. Role of ERK Signaling as a Regulator of Chondrocyte Receptor-Mediated Gene Expression
4.1. Regulation of MMPs and A Disintegrin and Metalloproteinase with Thrombospondin Motif (ADAMTS)
4.2. Epigenetic Regulation of ERK1/2
4.3. ERK1/2 and Neoangiogenesis
4.4. ERK1/2 and OA Chondrocytes
4.5. ERK1/2 Signaling Pathway in Osteoblasts and Osteoclasts
4.6. Interaction between Bone Cells and Articular Chondrocytes
4.7. Other Aspects of ERK1/2 in Relation to Chondrocyte Homeostasis
4.8. Regulation of ERK1/2 by MicroRNA
5. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Perander, M.; Al-Mahdl, R.; Jensen, T.C.; Kildasen, H.; Johansen, B.; Gabrielsen, M.; Seternes, O.M. Regulation of atypical MAP kinases ERK3 and ERK4 by the phosphatase DUSP2. Sci. Rep. 2017, 7, 43471. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.K.; Kahle, K.T.; Saelzler, M.P.; Orth, K.; Dixon, J.E.; Rosner, M.R. ERK7 is an autoactivated member of the MAPK family. J. Biol. Chem. 2001, 276, 21272–21279. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, P.; Meloche, S. Atypical mitogen-activated protein kinase: Structure, regulation and functions. Biochim. Biophys. Acta 2007, 1773, 1376–1387. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.A.; Burow, M.E.; Beckman, B.S. MEK5/ERK5 pathway: The first fifteen years. Biochim. Biophys. Acta 2012, 1825, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef]
- McKay, M.M.; Morrison, D.K. Proteomic analysis of scaffold proteins in the ERK cascade. Methods Mol. Biol. 2010, 661, 323–334. [Google Scholar] [PubMed]
- White, C.D.; Brown, M.D.; Sacks, D.B. IQGAPs in cancer: A family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009, 583, 1817–1824. [Google Scholar] [CrossRef]
- Matsunaga-Udagawa, R.; Fujita, Y.; Yoshiki, S.; Terai, K.; Kamioka, Y.; Kiyokawa, E.; Yugi, K.; Aoki, K.; Matsuda, M. The scaffold protein Shoc/SUR-8 accelerates the interaction between Ras and Raf. J. Biol. Chem. 2010, 285, 7818–7826. [Google Scholar] [CrossRef]
- Magee, J.; Cygler, M. Interactions between kinase scaffold MP1/p14 and its endosomal anchoring protein p18. Biochemistry 2011, 50, 3696–3705. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vetterkind, S.; Poythress, R.H.; Lin, Q.Q.; Morgan, K.G. Hierarchical scaffolding of an ERK1/2 activation pathway. Cell. Commun. Signal. 2013, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Teichert, I.; Steffens, E.K.; Schnaß, N.; Fränzel, B.; Krisp, C.; Wolters, D.A.; Kück, U. PRO40 is a scaffold protein of the cell wall integrity pathway, linking the MAP kinase module to the upstream activator protein kinase C. PLoS Genet. 2014, 10, e1004582. [Google Scholar] [CrossRef] [PubMed]
- Luan, Z.; He, Y.; Alattar, M.; Chen, Z.; He, F. Targeting the prohibitin scaffold-CRAF kinase interaction in the RAS-ERK-driven pancreatic ductal adenocarcinoma. Mol. Cancer 2014, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Posada, I.M.; Serulla, M.; Zhou, Y.; Oetken-Lindholm, C.; Abankwa, D.; Lectez, B. ASPP2 is a novel pan-Ras nanocluster scaffold. PLoS ONE 2016, 11, e0159677. [Google Scholar] [CrossRef] [PubMed]
- Von Kriegsheim, A.; Pitt, A.; Grindlay, G.J.; Kolch, W.; Dhillon, A.S. Regulation of the Raf-MEK-ERK pathway by protein phosphatase 5. Nat. Cell Biol. 2006, 8, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Forrmstecher, E.; Ramos, J.W.; Fauquet, M.; Calderwood, D.A.; Hsieh, J.C.; Canton, B.; Nguyen, X.T.; Barnier, J.V.; Ginsburg, M.H.; Chneiweiss, H. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell 2001, 1, 239–250. [Google Scholar] [CrossRef]
- Sondermann, H.; Soisson, S.M.; Boykevisch, S.; Yang, S.S.; Bar-Sagi, D.; Kuriyan, J. Structural analysis of autoinhibition in the Ras activator Son of sevenless. Cell 2004, 119, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Geissler, K.J.; Jung, M.J.; Riecken, L.B.; Sperka, T.; Cui, Y.; Schacke, S.; Breithaupt, C. Regulation of Son of sevenless by the membrane-actin linker protein ezrin. Proc. Natl. Acad. Sci. USA 2013, 110, 20587–20592. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Chiou, C.H.; Liu, S.C.; Hu, S.L.; Su, C.M.; Tsai, C.H.; Tang, C.H. Melatonin attenuates TNF-αand IL-1β expression in synovial fibroblasts and diminishes cartilage degradation: Implications for the treatment of rheumatoid arthritis. J. Pineal Res. 2019, 66, e12560. [Google Scholar] [CrossRef]
- Rodriguez-Berriguete, G.; Prieto, A.; Fraile, B.; Bouraoui, Y.; de Bethencourt, F.R.; Martinez-Onsurbe, P.; Royuela, M. Relationship between IL-6/ERK and NF-κB: A study in normal and pathological human prostate gland. Eur. Cytokine Netw. 2010, 21, 241–250. [Google Scholar] [PubMed]
- Huang, H.; Kim, H.J.; Chang, E.J.; Lee, Z.H.; Hwang, S.J.; Kim, H.M.; Kim, H.H. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells; implications for bone remodeling. Cell Death Differ. 2009, 16, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Berti, D.A.; Seger, R. The nuclear translocation of ERK. Methods. Mol. Biol. 2017, 1487, 175–194. [Google Scholar] [PubMed]
- Maik-Rachline, G.; Hacohen-Lev-Ran, A.; Seger, R. Nuclear ERK: Mechanism of translocation, substrates, and role in cancer. Int. J. Mol. Sci. 2019, 20, 1194. [Google Scholar] [CrossRef] [PubMed]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cellular proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Wainstein, E.; Seger, R. The dynamic subcellular localization of ERK: Mechanisms of translocation and role in various organelles. Curr. Opin. Cell Biol. 2016, 39, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Zuckerbraun, B.S.; Shapiro, R.A.; Billiar, T.R.; Tzeng, E. Rho influences the nuclear localization of extracellular signal-regulated kinases to modulate p21Waf/Cip1 expression. Circulation 2003, 108, 876–881. [Google Scholar] [CrossRef]
- Nishimoto, S.; Nishida, E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 2006, 7, 782–786. [Google Scholar] [CrossRef]
- Flores, K.; Yadav, S.S.; Katz, A.A.; Seger, R. The nuclear translocation of mitogen-activated protein kinases: Molecular mechanism and use as novel therapeutic target. Neuroendocrinology 2019, 108, 121–131. [Google Scholar] [CrossRef]
- Poltorak, M.; Meinart, I.; Stone, J.C.; Schraven, B.; Simeon, L. Sos1 regulates sustained TCR-mediated ERK activation. Eur. J. Immunol. 2014, 44, 1535–1540. [Google Scholar] [CrossRef]
- Denaver, E.; Ahmed, T.; Brems, H.; Van Woerden, G.; Borgesius, N.Z.; Callaerts-Vegh, Z.; Legius, E. Spred1 is required for synaptic plasticity and hippocampus-dependent learning. J. Neurosci. 2008, 28, 14443–14449. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Morita, R.; Hirata, Y.; Shichita, T.; Yoshimura, A. Spred 1, a suppressor of the Ras-ERK pathway, negatively regulates expansion and function of Group 2 innate lymphoid cells. J. Immunol. 2015, 195, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenov, S.K. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [PubMed]
- Vomastek, T.; Schaeffer, H.J.; Tarcsafalvi, A.; Smolkin, M.E.; Bissonette, E.A.; Weber, M.J. Modular construction of a signaling scaffold: MORG1 interacts with components of the ERK cascade and links ERK signaling to specific agonists. Proc. Natl. Acad. Sci. USA 2004, 101, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Ohmachi, M.; Rocheleau, C.E.; Church, D.; Lambie, E.; Schedl, T.; Sundaram, M.V.C. elegans ksr-1 and ksr-2 have both unique and redundant functions and are required for MPK-1 ERK phosphorylation. Curr. Biol. 2002, 12, 427–433. [Google Scholar] [CrossRef]
- Frodyma, D.; Neilsen, B.; Costanzo-Garvey, O.; Fisher, K.; Lewis, R. Coordinating ERK signaling via the molecular scaffold kinase suppressor of Ras. F1000Research 2017, 6, 1621. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, A.J.; Lagunes, L.; Zebarjedi, B.; Bardwell, L. The WW domain of the scaffolding protein IQGAP1 is neither necessary nor sufficient for binding to the MAPKs ERK1 and ERK2. J. Biol. Chem. 2017, 292, 8750–8761. [Google Scholar] [CrossRef] [PubMed]
- Nojima, H.; Adachi, M.; Matsui, T.; Okawa, K.; Tsukita, S.; Tsukita, S. IQGAP3 regulates cell proliferation through the Ras/ERK signalling cascade. Nat. Cell Biol. 2008, 10, 971–978. [Google Scholar] [CrossRef]
- Greig, F.H.; Nixon, G.F. Phosphoprotein enriched in astrocytes (PEA)-15: A potential therapeutic target in multiple disease states. Pharmacol. Ther. 2014, 143, 265–274. [Google Scholar] [CrossRef]
- Johnson, M.A.; Lombroso, P.J. A common STEP in the synaptic pathology of diverse neuropsychiatric disorders. Yale J. Biol. Med. 2012, 85, 481–490. [Google Scholar]
- Torii, S.; Kusakabe, M.; Yamamoto, T.; Maekawa, M.; Nishida, E. Sef is a spatial regulator for Ras/MAP kinase signaling. Dev. Cell 2004, 7, 33–44. [Google Scholar] [CrossRef]
- Malemud, C.J.; Pearlman, E. Targeting JAK/STAT signaling pathway in inflammatory diseases. Curr. Signal Transduct. Ther. 2009, 4, 201–221. [Google Scholar] [CrossRef]
- Patterson, K.; Brummer, T.; O’Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef]
- Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008, 27, 253–261. [Google Scholar] [CrossRef]
- Perander, M.; Keyse, S.M.; Seternes, T. Does MK5 reconcile classical and atypical MAP kinases? Front. Biosci. 2008, 13, 4617–4623. [Google Scholar] [CrossRef] [PubMed]
- Piserchio, A.; Warthaka, M.; Kaoud, T.S.; Callaway, K.; Dalby, K.N.; Ghose, R. Local destabilization, rigid body, and fuzzy docking facilitates the phosphorylation of the transcription factor Ets-1 by the mitogen-activated protein kinase ERK2. Proc. Natl. Acad. Sci. USA 2017, 114, E6287–E6296. [Google Scholar] [CrossRef]
- Shin, Y.; Kim, C.G.; Lim, Y.; Lee, K.H. The ETS transcription factor ELK-1 regulates induction of the cell. cycle-regulatory gene p21 (Waf1/Cip1) and the BAX gene in sodium-arsenite-exposed human keratinocyte HaCaT cells. J. Biol. Chem. 2011, 286, 26860–26872. [Google Scholar] [CrossRef]
- Durchdewald, M.; Angel, P.; Hess, J. The transcription factor Fos: A Janus-type regulator in health and disease. Histol. Histopathol. 2009, 24, 1451–1461. [Google Scholar]
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.J.; Xu, J.; Zheng, W. FOXO signaling pathways as therapeutic targets in cancer. Int. J. Biol. Sci. 2017, 13, 815–827. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Stivala, F. Roles of Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Ünal, E.B.; Uhlitz, F.; Blüthgen, N. A compendium of ERK targets. FEBS Lett. 2017, 591, 2607–2615. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signaling. Nat. Rev. Mol. Cell Biol. 2015, 16, 2810–2898. [Google Scholar] [CrossRef]
- Dorard, C.; Vucak, G.; Baccarini, M. Deciphering the RAS/ERK pathway in vivo. Biochem. Soc. Trans. 2017, 45, 27–36. [Google Scholar] [CrossRef]
- An, S.; Yang, Y.; Ward, R.; Liu, Y.; Guo, X.X.; Xu, T.R. Raf-interactome in tuning the complexity and diversity of Raf function. FEBS Lett. 2015, 282, 32–53. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Yang, Y.; Ward, R.; Liu, Y.; Guo, X.X.; Xu, T.R. A-Raf: A new star of the family of raf kinases. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 520–531. [Google Scholar] [CrossRef]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Sanz-Garcia, E.; Argiles, G.; Elez, E.; Tabenero, J. BRAF mutant colorectal cancer: Prognosis, treatment, and new perspectives. Ann. Oncol. 2017, 28, 2648–2657. [Google Scholar] [CrossRef]
- Baik, C.S.; Myall, N.J.; Wakelee, H.A. Targeting BRAF-mutant non-small cell lung cancer: From molecular profiling to rationally designed therapy. Oncologist 2017, 22, 786–796. [Google Scholar] [CrossRef]
- Fujimura, T.; Hidake, T.; Kambayashi, Y.; Aiba, S. BRAF kinase inhibitors for treatment of melanoma: Developments from early-stage animal studies to Phase II clinical trials. Expert. Opin. Investig. Drugs. 2019, 28, 143–148. [Google Scholar] [CrossRef]
- Broman, K.K.; Dossett, L.A.; Sun, J.; Eroglu, Z.; Zager, J.S. Update on BRAF and MEK inhibition for treatment of melanoma in metastatic, unresectable, and adjuvant settings. Expert Opin. Drug Saf. 2019, 18, 381–392. [Google Scholar] [CrossRef]
- Yao, Z.; Seger, R. The ERK signaling cascade—Views from different subcellular compartments. Biofactors 2009, 35, 407–416. [Google Scholar] [CrossRef]
- Buscà, R.; Pouysségur, J.; Lenormand, P. ERK1 and ERK2 Map kinases: Specific roles or functionary redundancy? Front. Cell Dev. Biol. 2016, 4, 53. [Google Scholar] [CrossRef]
- Grant, S. Cotargeting survival signaling pathways in cancer. J. Clin. Investig. 2008, 118, 3003–3006. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTor pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Chetram, M.A.; Hinton, C.V. PTEN regulation of ERK1/2 signaling in cancer. J. Recept. Signal Transduct. Res. 2012, 32, 190–195. [Google Scholar]
- Malemud, C.J. PI3K/Akt/PTEN/mTOR signaling: A fruitful target for inducing cell death in rheumatoid arthritis? Future Med. Chem. 2015, 7, 1137–1347. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Singh, R. ATGs: Scaffolds for MAPK/ERK signaling. Autophagy 2014, 10, 535–537. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef]
- Kohno, M.; Tanimura, S.; Osaki, K. Targeting the extracellular signal-regulated kinase pathway in cancer therapy. Biol. Pharm. Bull. 2011, 34, 1781–1784. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signaling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Asati, V.; Bharti, S.K.; Mahapatra, D.K. Mutant B-RAF kinase inhibitors as anticancer agents. Antican. Agents Med. Chem. 2016, 16, 1558–1575. [Google Scholar] [CrossRef]
- Kidger, A.M.; Keyse, S.M. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases. Semin. Cell Dev. Biol. 2016, 50, 125–132. [Google Scholar] [CrossRef]
- Khotskaya, Y.B.; Holla, V.R.; Farago, A.F.; Mills Shaw, K.B.; Meric-Bernstam, F.; Hong, D.S. Targeting TRK family proteins in cancer. Pharmacol. Ther. 2017, 173, 58–66. [Google Scholar] [CrossRef]
- Malemud, C.J. The biological basis of osteoarthritis: State of the evidence. Curr. Opin. Rheumatol. 2015, 27, 289–294. [Google Scholar] [CrossRef]
- Lang, R.; Hammer, M.; Mages, J. DUSP meets immunology: Dual specificity MAPK phosphatases in control of the inflammatory response. J. Immunol. 2006, 177, 497–504. [Google Scholar] [CrossRef]
- Mandrekar, P.; Szabo, G. Signaling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009, 50, 1258–1266. [Google Scholar] [CrossRef]
- Xu, D.; Matsumoto, M.L.; McKenzie, B.S.; Zarrin, A.A. TPL2 kinase action and control of inflammation. Pharmacol. Res. 2018, 129, 188–193. [Google Scholar] [CrossRef]
- Gantke, T.; Sriskantharajah, S.; Sadowski, M.; Ley, S.C. IκB kinase regulation of the TPL-2/ERK MAPK pathway. Immunol. Rev. 2012, 246, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signaling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Finotto, S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011, 22, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159. [Google Scholar] [CrossRef]
- Principi, M.; Giorgio, F.; Losurdo, G.; Neve, V.; Contaldo, A.; Di Leo, A.; Ierardi, E. Fibrogenesis and fibrosis in inflammatory bowel diseases: Good and bad side of same coin? World J. Gastrointest. Pathophysiol. 2013, 4, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, S.; Takeda, K. ERK signaling as a regulator of cell motility. J. Biochem. 2017, 162, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Gaestel, M. MAPK-activated protein kinases (MKs): Novel insights and challenges. Front. Cell Dev. Biol. 2016, 3, 88. [Google Scholar] [CrossRef]
- Masliah-Planchon, J.; Garinet, S.; Pasmant, E. RAS-MAPK pathway regulates epigenetic activation in cancer: miRNAs in action. Oncotarget 2016, 7, 38892–38907. [Google Scholar] [CrossRef]
- Granovsky, A.E.; Rosner, M.R. Raf kinase inhibitory protein: A signal transduction modulator and metastasis suppressor. Cell Res. 2008, 18, 452–457. [Google Scholar] [CrossRef]
- Vandamme, D.; Herrero, A.; Al-Mulla, F.; Kolch, W. Regulation of the MAPK pathway by raf kinase inhibitory protein. Crit. Rev. Oncog. 2014, 19, 405–415. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, S.; Abrams, S.L.; Chappell, W.H.; Russo, S.; Ove, R.; Milella, M.; Tafuri, A.; Lunghi, P.; Stivala, F.; et al. Emerging MEK inhibitors. Expert Opin. Emerg. Drugs 2010, 15, 203–223. [Google Scholar] [CrossRef]
- Zhao, Y.; Adjei, A.A. The clinical development of MEK inhibitors. Nat. Rev. Clin. Oncol. 2014, 11, 385–400. [Google Scholar] [CrossRef]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef]
- Uehling, D.E.; Harris, P.A. Recent progress on MAP kinase pathway inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 4047–4056. [Google Scholar] [CrossRef]
- Wu, P.K.; Park, J.I. MEK1/2 inhibitors: Molecular activity and resistance mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef]
- Luke, J.J.; Ott, P.A.; Shapiro, G.I. The biology and clinical development of MEK inhibitors for cancer. Drugs 2014, 74, 2111–2118. [Google Scholar] [CrossRef]
- Hong, S.K.; Wu, P.K.; Karkhanis, M.; Park, G.I. ERK1/2 can feedback-regulate cellular MEK1/2 levels. Cell Signal. 2015, 27, 1939–1948. [Google Scholar] [CrossRef]
- Thalhamer, T.; McGrath, M.A.; Harnett, M.M. MAPKS and their relevance to arthritis and inflammation. Rheumatology 2008, 47, 409–414. [Google Scholar] [CrossRef]
- Huang, G.; Chubinskaya, S.; Liao, W.; Loesser, R.F. Wnt5a induces catabolic signaling and matrix metalloproteinase production in human articular chondrocytes. Osteoarthr. Cartil. 2017, 25, 1505–1515. [Google Scholar] [CrossRef]
- Beier, F.; Loesser, R.F. Biology and pathology of Rho, GTPase, PI-3 kinase-Akt and MAP kinase signaling pathways in chondrocytes. J. Cell. Biochem. 2010, 110, 573–580. [Google Scholar] [CrossRef]
- Malemud, C.J. Regulation of chondrocyte matrix metalloproteinase gene expression. In Role of Proteases in Cellular Dysfunction; Dhalla, N.S., Chakraborti, S., Eds.; Springer Science: London, UK, 2013; pp. 63–77. [Google Scholar]
- Malemud, C.J. Matrix metalloproteases and synovial joint pathology. In Matrix Metalloproteinases and Tissue Remodeling in Health and Disease Part II. Target Tissues and Therapy; Khalil, R.A., Ed.; Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2017; pp. 306–325. [Google Scholar]
- Malemud, C.J. MAP kinases. OA, Inflammation and Degradation: A Continuum; Buckwalter, J., Lotz, M., Stoltz, J.F., Eds.; Biomedical and Health Research; IOS Press: Amsterdam, The Netherlands, 2007; Volume 70, pp. 99–117. [Google Scholar]
- Prasadam, I.; Crawford, R.; Xiao, Y. Aggravation of ADAMTS5 and matrix metalloproteinase production and role of ERK1/2 pathway in the interaction of osteoarthritic subchondral bone osteoblasts and articular cartilage chondrocytes – possible pathogenic role of osteoarthritis. J. Rheumatol. 2012, 39, 621–634. [Google Scholar] [CrossRef]
- Hashizume, M.; Mihara, M. High molecular weight hyaluronic acid inhibits IL-6-induced MMP production from human chondrocytes by up-regulating the ERK inhibitor, MKP-1. Biochem. Biophys. Res. Commun. 2010, 403, 184–189. [Google Scholar] [CrossRef]
- Sinha, K.M.; Zhou, X. Genetic and molecular control of osterix in skeletal formation. J. Cell. Biochem. 2013, 114, 975–984. [Google Scholar] [CrossRef]
- Carpio, L.R.; Bradley, E.W.; Westendorf, J.J. Histone deacetylase 3 suppresses Erk phosphorylation and matrix metalloproteinase (Mmp) -13 activity in chondrocytes. Connect. Tissue Res. 2017, 58, 27–36. [Google Scholar] [CrossRef]
- Wang, P.; Mao, Z.; Pan, Q.; Lu, R.; Huang, X.; Shang, X.; Zhang, R.; You, H. Histone deacetylase-4 and histone deactylase-8 regulate interleukin-1β-induced cartilage catabolic degradation through MAPK/JNK and ERK pathways. Int. J. Mol. Med. 2018, 41, 2117–2127. [Google Scholar]
- Saito, T.; Nishida, K.; Furumatsu, T.; Yoshida, A.; Ozawa, M.; Ozaki, T. Histone deacetylase inhibitors suppress mechanical stress-induced expression of RUNX-2 and ADAMTS-5 through the inhibition of the MAPK signaling pathway in cultured human chondrocytes. Osteoarthr. Cartil. 2013, 21, 165–174. [Google Scholar] [CrossRef]
- Ma, D.; Kou, X.; Jin, J.; Xu, T.; Wu, M.; Deng, L.; Fu, L.; Liu, Y.; Wu, G.; Lu, H. Hydrostatic compress force enhances the viability and decreases the apoptosis of condylar chondrocytes through integrin-FAK-ERK/PI3K pathway. Int. J. Mol. Sci. 2016, 17, 1847. [Google Scholar] [CrossRef]
- Islam, N.; Haqqi, T.M.; Jepsen, K.J.; Kraay, M.; Welter, J.F.; Goldberg, V.M.; Malemud, C.J. Hydrostatic pressure induces apoptosis in human chondrocytes from osteoarthritic cartilage through up-regulation of tumor necrosis factor-α, inducible nitric oxide synthase, p53, c-myc and bax-α and suppression of bcl-2. J. Cell. Biochem. 2002, 87, 266–278. [Google Scholar] [CrossRef]
- Ding, L.; Heying, E.; Nicholson, N.; Stroud, N.J.; Homandberg, G.A.; Buckwalter, J.A.; Guo, D.; Martin, J.A. Mechanical impact induces cartilage degradation via mitogen activated protein kinases. Osteoarthr. Cartil. 2010, 18, 1509–1517. [Google Scholar] [CrossRef]
- Zhong, H.M.; Ding, Q.H.; Chen, W.P.; Luo, R.B. Vorinostat, a HDAC inhibitor, showed anti-osteoarthritic activities through inhibition of iNOS and MMP expression, p38 and ERK phosphorylation and blocking NF-κB nuclear translocation. Int. Immunopharmacol. 2013, 17, 329–335. [Google Scholar] [CrossRef]
- Weng, L.H.; Ko, J.Y.; Wang, C.J.; Sun, Y.C.; Wang, F.S. Dkk-1 promotes angiogenic responses and cartilage matrix proteinase secretion in synovial fibroblasts from osteoarthritic joints. Arthritis Rheum. 2012, 64, 3267–3277. [Google Scholar] [CrossRef]
- Liu, Z.; Cai, H.; Zheng, X.; Zhang, B.; Xia, C. The involvement of mutual inhibition of ERK and mTOR in PLCγ1-mediated MMP-13 expression in human osteoarthritis chondrocytes. Int. J. Mol. Sci. 2015, 16, 17857–17869. [Google Scholar] [CrossRef]
- Raggat, L.J.; Jefcoat, S.C., Jr.; Choudhury, J.; Williams, S.; Tiku, M.; Partridge, N.C. Matrix metalloproteinase-13 influences ERK signalling in articular rabbit chondrocytes. Osteoarthr. Cartil. 2006, 14, 680–689. [Google Scholar] [CrossRef]
- Yin, J.; Hao, Z.; Ma, Y.; Liao, S.; Li, X.; Fu, J.; Wu, Y.; Shen, J.; Zhang, P.; Li, X.; et al. Concomitant activation of the PI3K/Akt and ERK1/2 signalling is involved in cyclic compressive force-induced IL-6 secretion in MLO-Y4 cells. Cell Biol. Int. 2014, 38, 591–598. [Google Scholar] [CrossRef]
- Prasadam, I.; Friis, T.; Shi, W.; Van Gennip, S.; Crawford, R.; Xiao, Y. Osteoarthritic cartilage chondrocytes alter subchondral bone osteoblast differentiation via MAPK signalling pathway involving ERK1/2. Bone 2010, 46, 226–235. [Google Scholar] [CrossRef]
- Choi, Y.H.; Gu, Y.M.; Oh, J.W.; Lee, K.Y. Osterix is regulated by Erk1/2 during osteoblast differentiation. Biochem. Biophys. Res. Commun. 2011, 415, 472–478. [Google Scholar] [CrossRef]
- Nakai, K.; Kawato, T.; Morita, T.; Linuma, T.; Kamio, N.; Zhao, N.; Maeno, M. Angiotensin II induces the production of MMP-3 and MMP-13 through the MAPK signaling pathways via the AT1 receptor in osteoblasts. Biochimie 2013, 95, 922–933. [Google Scholar] [CrossRef]
- Guo, R.W.; Yang, L.X.; Wang, H.; Liu, B.; Wang, L. Angiotensin II induces matrix metalloproteinase—9 expression via a nuclear factor-κB-dependent pathway in vascular smooth muscle cells. Regul. Pept. 2008, 147, 37–44. [Google Scholar] [CrossRef]
- Shimizu, H.; Nakagami, H.; Osako, M.K.; Hanayama, R.; Kunugiza, Y.; Kizawa, T.; Tomita, T.; Yoshikawa, H.; Ogihara, T.; Morishita, R. Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB J. 2008, 22, 2465–2475. [Google Scholar] [CrossRef]
- Tsai, K.S.; Kao, S.Y.; Wang, C.Y.; Wang, Y.J.; Wang, J.P.; Hung, S.C. Type I collagen promotes proliferation and osteogenesis of human mesenchymal stem cells via activation of ERK and Akt pathways. J. Biomed. Mater. Res. Part A 2010, 94, 673–682. [Google Scholar] [CrossRef]
- Asagiri, M.; Takayanagi, H. The molecular understanding of osteoclast differentiation. Bone 2007, 40, 251–264. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current understanding of RANK signaling in osteoclast differentiation and maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar]
- Yue, L.; Haroun, S.; Parent, J.L.; de Brum-Fernandes, A.J. Prostaglandin (D2) induces apoptosis of human osteoclasts through ERK1/2 and Akt signaling pathways. Bone 2014, 60, 112–121. [Google Scholar] [CrossRef]
- Oh, J.H.; Lee, N.K. Up-regulation of RANK expression via ERK1/2 by insulin contributes to the enhancement of osteoclast differentiation. Mol. Cells 2017, 40, 371–377. [Google Scholar]
- Kim, B.H.; Oh, J.H.; Lee, N.K. The inactivation of ERK1/2, p38 and NF-κB is involved in the down-regulation of osteoclastogenesis and function by A2B adenosine receptor stimulation. Mol. Cells 2017, 40, 752–760. [Google Scholar]
- Findlay, D.M.; Atkins, G.J. Osteoblast-chondrocyte interactions in osteoarthritis. Curr. Osteoporos. Res. 2014, 12, 127–134. [Google Scholar] [CrossRef]
- Prasadam, I.; Van Gennip, S.; Friis, T.; Shi, W.; Crawford, R.; Xiao, Y. ERK-1/2 and p38 in the regulation of hypertrophic changes of normal articular cartilage chondrocytes induced by osteoarthritic subchondral osteoblasts. Arthritis Rheum. 2010, 62, 1349–1360. [Google Scholar] [CrossRef]
- Yang, X.; Harkins, L.K.; Zubanova, O.; Harrington, A.; Kovalenko, D.; Nadeau, R.J.; Chen, P.Y.; Toher, J.L.; Lindner, V.; Liaw, R.; et al. Overexpression of Spry1 in chondrocytes causes attenuated FGFR ubquitination and sustained ERK activation resulting in chondrodysplasia. Dev. Biol. 2008, 321, 64–76. [Google Scholar] [CrossRef]
- Zhu, Y.; Gu, J.; Zhu, T.; Jin, C.; Hu, X.; Wang, X. Crosstalk between Smad2/3 and specific isoforms of ERK in TGF-β1-induced TIMP-3 expression in rat chondrocytes. J. Cell. Mol. Med. 2017, 21, 1781–1790. [Google Scholar] [CrossRef]
- Thiel, M.J.; Schaefer, C.J.; Lesch, M.E.; Mobley, J.L.; Dudley, D.T.; Tecle, H.; Barrett, S.D.; Schrier, D.J.; Flory, C.M. Central role of MEK/ERK MAP kinase pathway in a mouse model of rheumatoid arthritis: Potential proinflammatory mechanisms. Arthritis Rheum. 2007, 56, 3347–3357. [Google Scholar] [CrossRef]
- Malemud, C.J. MicroRNAs and osteoarthritis. Cells 2018, 7, 92. [Google Scholar] [CrossRef]
- Feng, Q.; Zheng, S.; Zheng, J. The emerging role of microRNAs in bone remodeling and its therapeutic implications for osteoporosis. Biosci. Rep. 2018, 36, 453. [Google Scholar] [CrossRef]
- Bluhm, B.; Ehlen, H.W.A.; Holzer, T.; Georgieva, V.S.; Heilig, J.; Pitzler, L.; Etich, J.; Bortecen, T.; Frie, C.; Probst, K.; et al. miR-322 stabilizes MEK1 expression to inhibit RAF/MEK/ERK pathway activation in cartilage. Development 2017, 144, 3562–3577. [Google Scholar] [CrossRef]
- Malemud, C.J.; Lewis, A.C.; Meszaros, E.C.; Wylie, M.A.; Skomorovska-Prokvolit, Y.; Mesiano, S. Inhibition of ERK1/2 phosphorylation by U0126 increases tumor necrosis factor-α-induced apoptosis, but not interleukin-6-induced apoptosis in C28/I2 human chondrocytes. J. Autoimmun. Disord. 2015, 1, 1. [Google Scholar] [CrossRef]
- Zhang, W.; Cheng, P.; Hu, W.; Yin, W.; Guo, F.; Chen, A.; Huang, H. Downregulated microRNA-340-5p promotes proliferation and inhibits apoptosis of chondrocytes in osteoarthritis mice through inhibiting the extracellular signal-regulated kinase signaling pathway by negatively targeting the FMOD gene. J. Cell. Physiol. 2018, 234, 927–939. [Google Scholar] [CrossRef]
- Kalamajski, S.; Bihan, D.; Bonna, A.; Rubin, K.; Farndale, R.W. Fibromodulin interacts with collagen cross-linking sites and activates lysyl oxidase. J. Biol. Chem. 2016, 291, 7951–7960. [Google Scholar] [CrossRef]
- Dobin-Sears, I.; Roberts, J.; O’Reilly, D.D.; Rahman, P. Ustekinumab in psoriatic arthritis and related phenotypes. Ther. Adv. Chronic Dis. 2018, 9, 191–198. [Google Scholar] [CrossRef]
- Al-Salama, Z.T.; Scott, L.J. Guselkumab: A review of moderate to severe plaque psoriasis. Am. J. Clin. Dermatol. 2018, 19, 907–918. [Google Scholar] [CrossRef]
- Garcia-Montoya, L.; Marzo-Ortega, H. The role of secukinumab in the treatment of psoriatic arthritis and ankylosing spondylitis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 169–180. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 signaling pathway and the treatment of psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef]
- Malemud, C.J. Inhibition of MMPs and ADAM/ADAMTS. Biochem. Pharmacol. 2019, 165, 33–40. [Google Scholar] [CrossRef]
- Cuenda, A.; Sanz-Ezquerro, J.J. p38γ and p38δ: From spectators to key physiological players. Trends Biochem. Sci. 2017, 42, 431–442. [Google Scholar] [CrossRef]
- Malemud, C.J. Dysfunctional immune-mediated inflammation in rheumatoid arthritis dictates that development of anti-rheumatic drugs target multiple intracellular signaling pathways. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2011, 10, 78–84. [Google Scholar] [CrossRef]
- Malemud, C.J.; Sun, Y.; Pearlman, E.; Ginley, N.M.; Awadallah, A.; Wisler, B.A.; Dennis, J.E. Monosodium urate and tumor necrosis factor-α increase apoptosis in human chondrocyte cultures. Rheumatolology 2012, 2, 113. [Google Scholar] [CrossRef]
- Malemud, C.J. Small molecular weight inhibitors of stress-activated and mitogen-activated protein kinases. Mini Rev. Med. Chem. 2006, 6, 689–698. [Google Scholar] [CrossRef]
- Montgaut, C.; Settleman, J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009, 283, 125–134. [Google Scholar] [CrossRef]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles Heel of the MAPK pathway, in cancer Therapy. Arch. Pharm. Sin. B. 2018, 8, 552–562. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Mungo, E.; Gazzano, E.; Kopecka, J.; Riganti, C. ERK is a pivotal player of chemo-immune-Resistance in cancer. Int. J. Mol. Sci. 2019, 20, 2505. [Google Scholar] [CrossRef]
- Allen, L.F.; Sebolt-Leopold, J.; Meyer, M.B. Cl-1040 (PD184353), a targeted signal transduction inhibitor of MEK (MAPKK). Semin. Oncol. 2003, 30, 105–116. [Google Scholar] [CrossRef]
- Miller, C.R.; Oliver, K.E.; Farley, J.H. MEK1/2 inhibitors in the treatment of gynecologic malignancies. Gynecol. Oncol. 2014, 133, 128–137. [Google Scholar] [CrossRef]
- Roskowski, R., Jr. Allosteric MEK1/2 inhibitors including cobimetanib and trametinib in the treatment of cutaneous melanoma. Pharmacol. Res. 2017, 117, 20–31. [Google Scholar] [CrossRef]
- Rice, K.D.; Aay, N.; Anand, N.K.; Blazey, C.M.; Bowles, O.J.; Bussenius, J.; Costanzo, S.; Curtis, J.K.; Defina, S.C.; Dubenko, L.; et al. Novel carboxamide-based allosteric MEK inhibitors: Discovery and optimization efforts toward XL518 (GDC-0973). ACS Med. Chem. Lett. 2012, 3, 416–421. [Google Scholar] [CrossRef]
- Deming, D.A.; Cavalcante, L.L.; Lubner, S.J.; Mulkerin, D.L.; LoConte, N.K.; Eickhoff, J.C.; Kolesar, J.M.; Fiorvanti, S.; Greten, T.F.; Compton, K.; et al. A phase 1 study of selumetinib (AZD6244/ARRY-142866). A MEK1/2 inhibitor, in combination with cetuximab in refractory solid tumors and KRAS mutant colorectal. cancer. Investig. New Drugs 2016, 34, 168–175. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2019, 142, 151–168. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Song, K.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, D.J.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; Morales, T.; et al. RAF inhibitors prime-wild type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 421–435. [Google Scholar] [CrossRef]
- Ammar, U.M.; Abdel-Maksoud, M.S.; Oh, C.H. Recent advances of RAF (rapidly accelerated fibrosarcoma) inhibitors as anti-cancer agents. Eur. J. Med. Chem. 2018, 158, 144–166. [Google Scholar] [CrossRef]
- Sanchez-Laorden, B.; Viros, A.; Marais, R. Mind the IQGAP. Cancer Cell 2013, 23, 715–717. [Google Scholar] [CrossRef]
- Yu, Z.; Chen, Z.; Su, Q.; Ye, S.; Yuan, H.; Kuai, M.; Lv, M.; Tu, Z.; Yang, X.; Liu, R.; et al. Dual inhibitors of RAF-MEK-ERK and PI3K-PDK1-AKT pathways: Design, synthesis and preliminary anticancer activity studies of 3-substituted-5-(phenylamino) indole derivatives. Bioorg. Med. Chem. 2019, 27, 944–954. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, N.; Malemud, C.J. Extracellular Signal-Regulated Kinase: A Regulator of Cell Growth, Inflammation, Chondrocyte and Bone Cell Receptor-Mediated Gene Expression. Int. J. Mol. Sci. 2019, 20, 3792. https://doi.org/10.3390/ijms20153792
Lu N, Malemud CJ. Extracellular Signal-Regulated Kinase: A Regulator of Cell Growth, Inflammation, Chondrocyte and Bone Cell Receptor-Mediated Gene Expression. International Journal of Molecular Sciences. 2019; 20(15):3792. https://doi.org/10.3390/ijms20153792
Chicago/Turabian StyleLu, Nathan, and Charles J. Malemud. 2019. "Extracellular Signal-Regulated Kinase: A Regulator of Cell Growth, Inflammation, Chondrocyte and Bone Cell Receptor-Mediated Gene Expression" International Journal of Molecular Sciences 20, no. 15: 3792. https://doi.org/10.3390/ijms20153792
APA StyleLu, N., & Malemud, C. J. (2019). Extracellular Signal-Regulated Kinase: A Regulator of Cell Growth, Inflammation, Chondrocyte and Bone Cell Receptor-Mediated Gene Expression. International Journal of Molecular Sciences, 20(15), 3792. https://doi.org/10.3390/ijms20153792