Arachidonic Acid Metabolism and Kidney Inflammation

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Regulation of the AA Metabolism in the Kidney
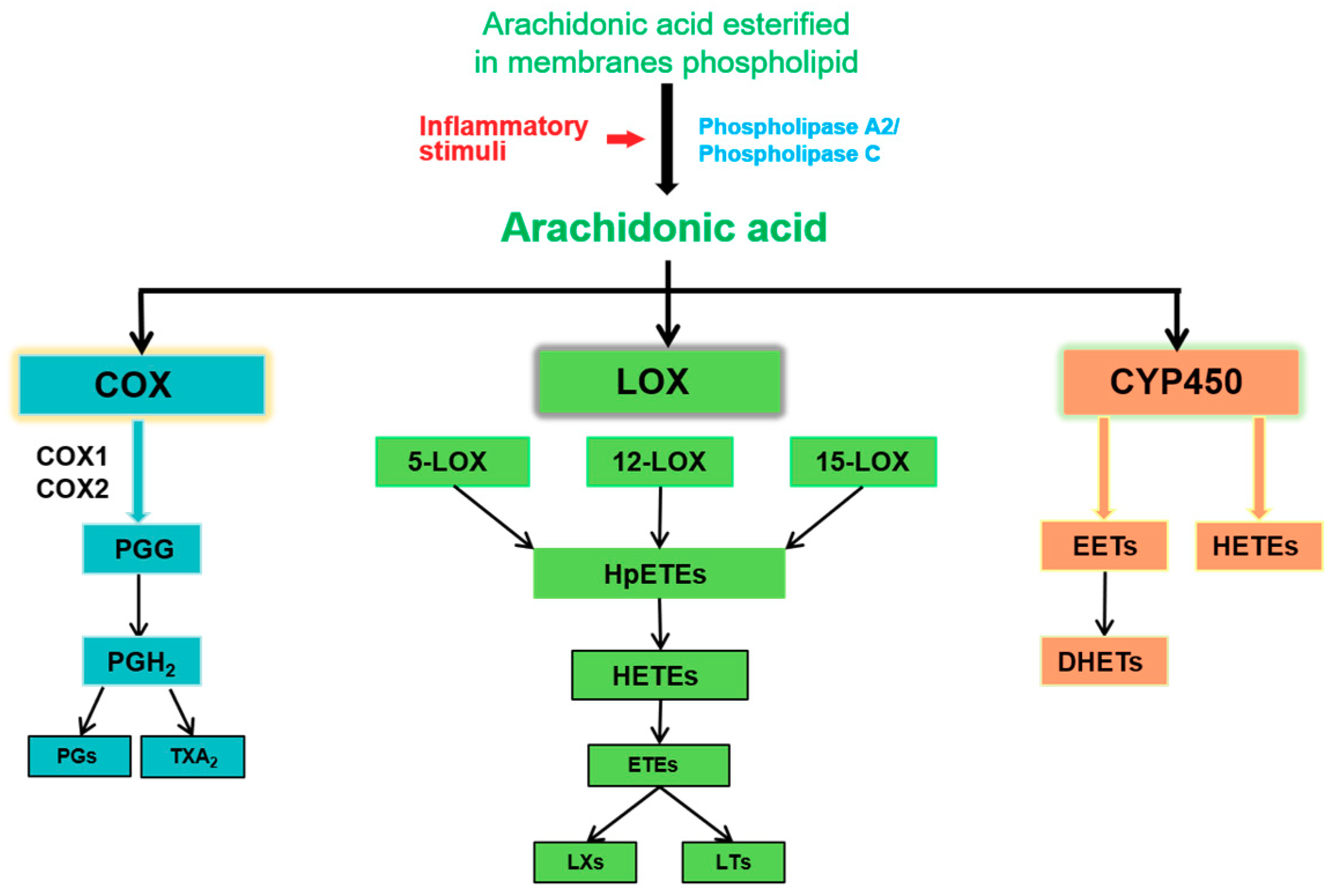
2.1. The Release of AA
2.2. COX Pathway
2.3. LOX Pathway
2.4. Cytochrome P450 (CYP450) Pathway
3. Mechanism of AA-induced Renal Inflammation
3.1. PGs and Renal Inflammation
3.2. HETEs and Renal Inflammation
3.3. LTs/LXs and Renal Inflammation
3.4. EETs and Renal Inflammation
4. Treatments
4.1. Phospholipase-Associated Therapy
4.2. COX-Associated Therapy
4.3. LOX-Associated Therapy
4.4. CYP450/sEH-Associated Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Van Dorp, D.A. Essential fatty acid metabolism. Proc. Nutr. Soc. 1975, 34, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.I.; Benincaso, A.I.; Knoell, C.T.; Larkin, J.K.; Austen, K.F.; Robinson, D.R. Dietary omega-3 polyunsaturated fatty acids inhibit phosphoinositide formation and chemotaxis in neutrophils. J. Clin. Investig. 1993, 91, 651–660. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, H.W.; Dekkers, D.H.; Lamers, J.M. Polyunsaturated fatty acids and signalling via phospholipase C-beta and A2 in myocardium. Mol. Cell. Biochem. 1996, 157, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim. Biophys. Acta 2015, 1851, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.M.; Calder, P.C.; Ed Rainger, G. Pharmacology and therapeutics of omega-3 polyunsaturated fatty acids in chronic inflammatory disease. Pharmacol. Ther. 2014, 141, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Rae, S.A.; Davidson, E.M.; Smith, M.J. Leukotriene B4, an inflammatory mediator in gout. Lancet 1982, 2, 1122–1124. [Google Scholar] [CrossRef]
- Arachidonic acid, analgesics, and asthma. Lancet 1981, 2, 1266–1267.
- Rand, A.A.; Barnych, B.; Morisseau, C.; Cajka, T.; Lee, K.S.S.; Panigrahy, D.; Hammock, B.D. Cyclooxygenase-derived proangiogenic metabolites of epoxyeicosatrienoic acids. Proc. Natl. Acad. Sci. USA 2017, 114, 4370–4375. [Google Scholar] [CrossRef]
- Kopp, B.T.; Thompson, R.; Kim, J.; Konstan, R.; Diaz, A.; Smith, B.; Shrestha, C.; Rogers, L.K.; Hayes, D., Jr.; Tumin, D.; et al. Secondhand smoke alters arachidonic acid metabolism and inflammation in infants and children with cystic fibrosis. Thorax 2019, 74, 237–246. [Google Scholar] [CrossRef]
- Chauhan, G.; Roy, K.; Kumar, G.; Kumari, P.; Alam, S.; Kishore, K.; Panjwani, U.; Ray, K. Distinct influence of COX-1 and COX-2 on neuroinflammatory response and associated cognitive deficits during high altitude hypoxia. Neuropharmacology 2019, 146, 138–148. [Google Scholar] [CrossRef]
- Goldstein, A.R.; White, R.H.; Akuse, R.; Chantler, C. Long-term follow-up of childhood Henoch-Schonlein nephritis. Lancet 1992, 339, 280–282. [Google Scholar] [CrossRef]
- Murakami, M.; Nakatani, Y.; Kuwata, H.; Kudo, I. Cellular components that functionally interact with signaling phospholipase A(2)s. Biochim. Biophys. Acta 2000, 1488, 159–166. [Google Scholar] [CrossRef]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Van De Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Lambert, I.H.; Hoffmann, E.K.; Christensen, P. Role of prostaglandins and leukotrienes in volume regulation by Ehrlich ascites tumor cells. J. Membr. Biol. 1987, 98, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Lipsky, P.E.; Brooks, P.; Crofford, L.J.; DuBois, R.; Graham, D.; Simon, L.S.; van de Putte, L.B.; Abramson, S.B. Unresolved issues in the role of cyclooxygenase-2 in normal physiologic processes and disease. Arch. Intern. Med. 2000, 160, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; Yu, Y.; Grosser, T.; Fitzgerald, G.A. COX-2, the dominant source of prostacyclin. Proc. Natl. Acad. Sci. USA 2013, 110, E183. [Google Scholar] [CrossRef] [PubMed]
- Thuresson, E.D.; Lakkides, K.M.; Rieke, C.J.; Sun, Y.; Wingerd, B.A.; Micielli, R.; Mulichak, A.M.; Malkowski, M.G.; Garavito, R.M.; Smith, W.L. Prostaglandin endoperoxide H synthase-1: The functions of cyclooxygenase active site residues in the binding, positioning, and oxygenation of arachidonic acid. J. Biol. Chem. 2001, 276, 10347–10357. [Google Scholar] [CrossRef]
- Hawkey, C.J. COX-2 inhibitors. Lancet 1999, 353, 307–314. [Google Scholar] [CrossRef]
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim. Biophys. Acta 2015, 1851, 414–421. [Google Scholar] [CrossRef]
- Smith, W.L.; Garavito, R.M.; DeWitt, D.L. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J. Biol. Chem. 1996, 271, 33157–33160. [Google Scholar] [CrossRef]
- Naraba, H.; Murakami, M.; Matsumoto, H.; Shimbara, S.; Ueno, A.; Kudo, I.; Oh-ishi, S. Segregated coupling of phospholipases A2, cyclooxygenases, and terminal prostanoid synthases in different phases of prostanoid biosynthesis in rat peritoneal macrophages. J. Immunol. 1998, 160, 2974–2982. [Google Scholar] [PubMed]
- Smith, W.L.; Song, I. The enzymology of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediat. 2002, 68–69, 115–128. [Google Scholar] [CrossRef]
- Bahia, M.S.; Katare, Y.K.; Silakari, O.; Vyas, B.; Silakari, P. Inhibitors of microsomal prostaglandin E2 synthase-1 enzyme as emerging anti-inflammatory candidates. Med. Res. Rev. 2014, 34, 825–855. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Haurand, M.; Ullrich, V.; Diczfalusy, U.; Hammarstrom, S. Products, kinetics, and substrate specificity of homogeneous thromboxane synthase from human platelets: Development of a novel enzyme assay. Arch. Biochem. Biophys. 1987, 254, 124–135. [Google Scholar] [CrossRef]
- Goetzl, E.J.; An, S.; Smith, W.L. Specificity of expression and effects of eicosanoid mediators in normal physiology and human diseases. FASEB J. 1995, 9, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Stenson, W.F. The universe of arachidonic acid metabolites in inflammatory bowel disease: Can we tell the good from the bad? Curr. Opin. Gastroenterol. 2014, 30, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Alhouayek, M.; Muccioli, G.G. COX-2-derived endocannabinoid metabolites as novel inflammatory mediators. Trends Pharmacol. Sci. 2014, 35, 284–292. [Google Scholar] [CrossRef]
- Li, H.S.; Hebda, P.A.; Kelly, L.A.; Ehrlich, G.D.; Whitcomb, D.C.; Dohar, J.E. Up-regulation of prostaglandin EP4 receptor messenger RNA in fetal rabbit skin wound. Arch. Otolaryngol. Head Neck Surg. 2000, 126, 1337–1343. [Google Scholar] [CrossRef][Green Version]
- Breyer, R.M.; Bagdassarian, C.K.; Myers, S.A.; Breyer, M.D. Prostanoid receptors: Subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 661–690. [Google Scholar] [CrossRef]
- Sandulache, V.C.; Chafin, J.B.; Li-Korotky, H.S.; Otteson, T.D.; Dohar, J.E.; Hebda, P.A. Elucidating the role of interleukin 1beta and prostaglandin E2 in upper airway mucosal wound healing. Arch. Otolaryngol. Head Neck Surg. 2007, 133, 365–374. [Google Scholar] [CrossRef][Green Version]
- Bauer, J.; Ripperger, A.; Frantz, S.; Ergun, S.; Schwedhelm, E.; Benndorf, R.A. Pathophysiology of isoprostanes in the cardiovascular system: Implications of isoprostane-mediated thromboxane A2 receptor activation. Br. J. Pharmacol. 2014, 171, 3115–3131. [Google Scholar] [CrossRef] [PubMed]
- Ekambaram, P.; Lambiv, W.; Cazzolli, R.; Ashton, A.W.; Honn, K.V. The thromboxane synthase and receptor signaling pathway in cancer: An emerging paradigm in cancer progression and metastasis. Cancer Metast. Rev. 2011, 30, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Sun, C.; Tilley, S.L.; Mustafa, S.J. Mechanisms underlying uridine adenosine tetraphosphate-induced vascular contraction in mouse aorta: Role of thromboxane and purinergic receptors. Vasc. Pharmacol. 2015, 73, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Good, R.L.; Roupe, K.A.; Yanez, J.A. Cyclooxygenase-3: Axiom, dogma, anomaly, enigma or splice error?—Not as easy as 1, 2, 3. J. Pharm. Pharm. Sci. 2004, 7, 217–226. [Google Scholar] [PubMed]
- Chandrasekharan, N.V.; Dai, H.; Roos, K.L.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression. Proc. Natl. Acad. Sci. USA 2002, 99, 13926–13931. [Google Scholar] [CrossRef] [PubMed]
- Schwab, J.M.; Beiter, T.; Linder, J.U.; Laufer, S.; Schulz, J.E.; Meyermann, R.; Schluesener, H.J. COX-3—A virtual pain target in humans? FASEB J. 2003, 17, 2174–2175. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, D.A.; Moore, A.R.; Colville-Nash, P.R. COX-1, COX-2, and COX-3 and the future treatment of chronic inflammatory disease. Lancet 2000, 355, 646–648. [Google Scholar] [CrossRef]
- Radmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a key enzyme for leukotriene biosynthesis in health and disease. Biochim. Biophys. Acta 2015, 1851, 331–339. [Google Scholar] [CrossRef]
- Mittal, M.; Kumar, R.B.; Balagunaseelan, N.; Hamberg, M.; Jegerschold, C.; Radmark, O.; Haeggstrom, J.Z.; Rinaldo-Matthis, A. Kinetic investigation of human 5-lipoxygenase with arachidonic acid. Bioorganic Med. Chem. Lett. 2016, 26, 3547–3551. [Google Scholar] [CrossRef]
- Powell, W.S.; Gravelle, F.; Gravel, S. Metabolism of 5(S)-hydroxy-6,8,11,14-eicosatetraenoic acid and other 5(S)-hydroxyeicosanoids by a specific dehydrogenase in human polymorphonuclear leukocytes. J. Biol. Chem. 1992, 267, 19233–19241. [Google Scholar] [PubMed]
- Mastyugin, V.; Aversa, E.; Bonazzi, A.; Vafaes, C.; Mieyal, P.; Schwartzman, M.L. Hypoxia-induced production of 12-hydroxyeicosanoids in the corneal epithelium: Involvement of a cytochrome P-4504B1 isoform. J. Pharmacol. Exp. Ther. 1999, 289, 1611–1619. [Google Scholar] [PubMed]
- Powell, W.S.; Rokach, J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim. Biophys. Acta 2015, 1851, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Radmark, O.; Samuelsson, B. Enzyme with dual lipoxygenase activities catalyzes leukotriene A4 synthesis from arachidonic acid. Proc. Natl. Acad. Sci. USA 1984, 81, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Green, A.R.; Freedman, C.; Tena, J.; Tourdot, B.E.; Liu, B.; Holinstat, M.; Holman, T.R. 5 S,15 S-Dihydroperoxyeicosatetraenoic Acid (5,15-diHpETE) as a Lipoxin Intermediate: Reactivity and Kinetics with Human Leukocyte 5-Lipoxygenase, Platelet 12-Lipoxygenase, and Reticulocyte 15-Lipoxygenase-1. Biochemistry 2018, 57, 6726–6734. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Watanabe, T.; Ohishi, N.; Toda, A.; Asano, K.; Taniguchi, S.; Nosaka, K.; Noiri, E.; Suzuki, T.; Sakai, T.; et al. Ubiquitous localization of leukotriene A4 hydrolase in the rat nephron. Kidney Int. 1999, 55, 100–108. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baba, T.; Black, K.L.; Ikezaki, K.; Chen, K.N.; Becker, D.P. Intracarotid infusion of leukotriene C4 selectively increases blood-brain barrier permeability after focal ischemia in rats. J. Cereb. Blood Flow Metab. 1991, 11, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Porro, B.; Songia, P.; Squellerio, I.; Tremoli, E.; Cavalca, V. Analysis, physiological and clinical significance of 12-HETE: A neglected platelet-derived 12-lipoxygenase product. J. Chromatogr. B 2014, 964, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Witola, W.H.; Liu, S.R.; Montpetit, A.; Welti, R.; Hypolite, M.; Roth, M.; Zhou, Y.; Mui, E.; Cesbron-Delauw, M.F.; Fournie, G.J.; et al. ALOX12 in human toxoplasmosis. Infect. Immun. 2014, 82, 2670–2679. [Google Scholar] [CrossRef]
- Serhan, C.N.; Sheppard, K.A. Lipoxin formation during human neutrophil-platelet interactions. Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J. Clin. Investig. 1990, 85, 772–780. [Google Scholar] [CrossRef]
- Fiore, S.; Ryeom, S.W.; Weller, P.F.; Serhan, C.N. Lipoxin recognition sites. Specific binding of labeled lipoxin A4 with human neutrophils. J. Biol. Chem. 1992, 267, 16168–16176. [Google Scholar] [PubMed]
- Edenius, C.; Haeggstrom, J.; Lindgren, J.A. Transcellular conversion of endogenous arachidonic acid to lipoxins in mixed human platelet-granulocyte suspensions. Biochem. Biophys. Res. Commun. 1988, 157, 801–807. [Google Scholar] [CrossRef]
- Sheppard, K.A.; Greenberg, S.M.; Funk, C.D.; Romano, M.; Serhan, C.N. Lipoxin generation by human megakaryocyte-induced 12-lipoxygenase. Biochim. Biophys. Acta 1992, 1133, 223–234. [Google Scholar] [CrossRef]
- Moreno, J.J. New aspects of the role of hydroxyeicosatetraenoic acids in cell growth and cancer development. Biochem. Pharmacol. 2009, 77, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Lasker, J.M.; Chen, W.B.; Wolf, I.; Bloswick, B.P.; Wilson, P.D.; Powell, P.K. Formation of 20-hydroxyeicosatetraenoic acid, a vasoactive and natriuretic eicosanoid, in human kidney. Role of Cyp4F2 and Cyp4A11. J. Biol. Chem. 2000, 275, 4118–4126. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.F.; Zhang, C.C.; Chou, K.C.; Wei, D.Q. Structure of cytochrome p450s and personalized drug. Curr. Med. Chem. 2009, 16, 232–244. [Google Scholar] [CrossRef]
- Nebert, D.W.; Russell, D.W. Clinical importance of the cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Tacconelli, S.; Patrignani, P. Inside epoxyeicosatrienoic acids and cardiovascular disease. Front. Pharmacol. 2014, 5, 239. [Google Scholar] [CrossRef]
- Aspromonte, N.; Monitillo, F.; Puzzovivo, A.; Valle, R.; Caldarola, P.; Iacoviello, M. Modulation of cardiac cytochrome P450 in patients with heart failure. Expert Opin. Drug Metab. Toxicol. 2014, 10, 327–339. [Google Scholar] [CrossRef]
- Shahabi, P.; Siest, G.; Visvikis-siest, S. Influence of inflammation on cardiovascular protective effects of cytochrome P450 epoxygenase-derived epoxyeicosatrienoic acids. Drug Metab. Rev. 2014, 46, 33–56. [Google Scholar] [CrossRef]
- Alsaad, A.M.; Zordoky, B.N.; Tse, M.M.; El-Kadi, A.O. Role of cytochrome P450-mediated arachidonic acid metabolites in the pathogenesis of cardiac hypertrophy. Drug Metab. Rev. 2013, 45, 173–195. [Google Scholar] [CrossRef]
- Bellien, J.; Joannides, R. Epoxyeicosatrienoic acid pathway in human health and diseases. J. Cardiovasc. Pharmacol. 2013, 61, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Makita, K.; Falck, J.R.; Capdevila, J.H. Cytochrome P450, the arachidonic acid cascade, and hypertension: New vistas for an old enzyme system. FASEB J. 1996, 10, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Zeldin, D.C.; DuBois, R.N.; Falck, J.R.; Capdevila, J.H. Molecular cloning, expression and characterization of an endogenous human cytochrome P450 arachidonic acid epoxygenase isoform. Arch. Biochem. Biophys. 1995, 322, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Zeldin, D.C.; Blaisdell, J.A.; Hodgson, E.; Goldstein, J.A. Cloning and expression of murine CYP2Cs and their ability to metabolize arachidonic acid. Arch. Biochem. Biophys. 1998, 357, 45–57. [Google Scholar] [CrossRef]
- Wu, S.; Moomaw, C.R.; Tomer, K.B.; Falck, J.R.; Zeldin, D.C. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J. Biol. Chem. 1996, 271, 3460–3468. [Google Scholar] [CrossRef]
- Ma, J.; Qu, W.; Scarborough, P.E.; Tomer, K.B.; Moomaw, C.R.; Maronpot, R.; Davis, L.S.; Breyer, M.D.; Zeldin, D.C. Molecular cloning, enzymatic characterization, developmental expression, and cellular localization of a mouse cytochrome P450 highly expressed in kidney. J. Biol. Chem. 1999, 274, 17777–17788. [Google Scholar] [CrossRef]
- Zhao, X.; Imig, J.D. Kidney CYP450 enzymes: Biological actions beyond drug metabolism. Curr. Drug Metab. 2003, 4, 73–84. [Google Scholar] [CrossRef]
- Yu, Z.; Xu, F.; Huse, L.M.; Morisseau, C.; Draper, A.J.; Newman, J.W.; Parker, C.; Graham, L.; Engler, M.M.; Hammock, B.D.; et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ. Res. 2000, 87, 992–998. [Google Scholar] [CrossRef]
- Quigley, R.; Baum, M.; Reddy, K.M.; Griener, J.C.; Falck, J.R. Effects of 20-HETE and 19(S)-HETE on rabbit proximal straight tubule volume transport. Am. J. Physiol. Ren. Physiol. 2000, 278, F949–F953. [Google Scholar] [CrossRef] [PubMed]
- Snider, N.T.; Kornilov, A.M.; Kent, U.M.; Hollenberg, P.F. Anandamide metabolism by human liver and kidney microsomal cytochrome p450 enzymes to form hydroxyeicosatetraenoic and epoxyeicosatrienoic acid ethanolamides. J. Pharmacol. Exp. Ther. 2007, 321, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Collins, X.H.; Harmon, S.D.; Kaduce, T.L.; Berst, K.B.; Fang, X.; Moore, S.A.; Raju, T.V.; Falck, J.R.; Weintraub, N.L.; Duester, G.; et al. Omega-oxidation of 20-hydroxyeicosatetraenoic acid (20-HETE) in cerebral microvascular smooth muscle and endothelium by alcohol dehydrogenase 4. J. Biol. Chem. 2005, 280, 33157–33164. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.; Wang, M.H.; Reddy, K.M.; Falck, J.R.; Schwartzman, M.L. Kinetic profile of the rat CYP4A isoforms: Arachidonic acid metabolism and isoform-specific inhibitors. Am. J. Physiol. 1999, 276, R1691–R1700. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Yadav, A.; Misra, R.; Aggarwal, A. Urinary prostaglandin D synthase as biomarker in lupus nephritis: A longitudinal study. Clin. Exp. Rheumatol. 2015, 33, 694–698. [Google Scholar] [PubMed]
- Somparn, P.; Hirankarn, N.; Leelahavanichkul, A.; Khovidhunkit, W.; Thongboonkerd, V.; Avihingsanon, Y. Urinary proteomics revealed prostaglandin H(2)D-isomerase, not Zn-alpha2-glycoprotein, as a biomarker for active lupus nephritis. J. Proteom. 2012, 75, 3240–3247. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.D.; Eleuteri, A.; Goilav, B.; Lewandowski, L.; Phuti, A.; Rubinstein, T.; Wahezi, D.; Jones, C.A.; Marks, S.D.; Corkhill, R.; et al. A Markov Multi-State model of lupus nephritis urine biomarker panel dynamics in children: Predicting changes in disease activity. Clin. Immunol. 2019, 198, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Pellefigues, C.; Dema, B.; Lamri, Y.; Saidoune, F.; Chavarot, N.; Loheac, C.; Pacreau, E.; Dussiot, M.; Bidault, C.; Marquet, F.; et al. Prostaglandin D2 amplifies lupus disease through basophil accumulation in lymphoid organs. Nat. Commun. 2018, 9, 725. [Google Scholar] [CrossRef]
- Pettipher, R.; Hansel, T.T.; Armer, R. Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nat. Rev. Drug Discov. 2007, 6, 313–325. [Google Scholar] [CrossRef]
- Rajakariar, R.; Hilliard, M.; Lawrence, T.; Trivedi, S.; Colville-Nash, P.; Bellingan, G.; Fitzgerald, D.; Yaqoob, M.M.; Gilroy, D.W. Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyDelta12 14 PGJ2. Proc. Natl. Acad. Sci. USA 2007, 104, 20979–20984. [Google Scholar] [CrossRef]
- Hirai, H.; Tanaka, K.; Yoshie, O.; Ogawa, K.; Kenmotsu, K.; Takamori, Y.; Ichimasa, M.; Sugamura, K.; Nakamura, M.; Takano, S.; et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J. Exp. Med. 2001, 193, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Togel, F.; Isaac, J.; Hu, Z.; Weiss, K.; Westenfelder, C. Renal SDF-1 signals mobilization and homing of CXCR4-positive cells to the kidney after ischemic injury. Kidney Int. 2005, 67, 1772–1784. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.; Wang, Y.; Chew, W.K.; Lima, R.; Gonzalez, N.A.; Mattar, C.N.; Chong, S.Z.; Schlitzer, A.; Bakocevic, N.; Chew, S.; et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J. Exp. Med. 2013, 210, 2321–2336. [Google Scholar] [CrossRef] [PubMed]
- Daza, L.; Kornhauser, C.; Zamora, L.; Flores, J. Captopril effect on prostaglandin E2, thromboxane B2 and proteinuria in lupus nephritis patients. Prostaglandins Other Lipid Mediat. 2005, 78, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Yan, X.; Nagata, N.; Aritake, K.; Katsumata, Y.; Matsuhashi, T.; Nakamura, M.; Hirai, H.; Urade, Y.; Asano, K.; et al. PGD2-CRTH2 pathway promotes tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2012, 23, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhou, Q.; Zhong, F.; Guo, S.; Hao, X.; Li, C.; Wang, W.; Chen, N. 15-Deoxy-Delta(12,14)-prostaglandin J(2) modulates lipopolysaccharide-induced chemokine expression by blocking nuclear factor-kappaB activation via peroxisome proliferator activated receptor-gamma-independent mechanism in renal tubular epithelial cells. Nephron Exp. Nephrol. 2013, 123, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hart, D.; Lifschitz, M.D. Renal physiology of the prostaglandins and the effects of nonsteroidal anti-inflammatory agents on the kidney. Am. J. Nephrol. 1987, 7, 408–418. [Google Scholar] [CrossRef]
- Dunn, M.J.; Hood, V.L. Prostaglandins and the kidney. Am. J. Physiol. 1977, 233, 169–184. [Google Scholar] [CrossRef]
- Nasrallah, R.; Hassouneh, R.; Hebert, R.L. Chronic kidney disease: Targeting prostaglandin E2 receptors. Am. J. Physiol. Ren. Physiol. 2014, 307, F243–F250. [Google Scholar] [CrossRef]
- Breyer, M.D.; Breyer, R.M. Prostaglandin E receptors and the kidney. Am. J. Physiol. Ren. Physiol. 2000, 279, F12–F23. [Google Scholar] [CrossRef]
- Kotnik, P.; Nielsen, J.; Kwon, T.H.; Krzisnik, C.; Frokiaer, J.; Nielsen, S. Altered expression of COX-1, COX-2, and mPGES in rats with nephrogenic and central diabetes insipidus. Am. J. Physiol. Ren. Physiol. 2005, 288, F1053–F1068. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, T.; Moreno, J.J. Role of EP(1) and EP(4) PGE(2) subtype receptors in serum-induced 3T6 fibroblast cycle progression and proliferation. Am. J. Physiol. Cell Physiol. 2002, 282, C280–C288. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Ballinger, M.N.; White, E.S.; Green, M.E.; Herrygers, A.B.; Wilke, C.A.; Toews, G.B.; Peters-Golden, M. Bleomycin-induced E prostanoid receptor changes alter fibroblast responses to prostaglandin E2. J. Immunol. 2005, 174, 5644–5649. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, U.; Iwatsubo, K.; Umemura, M.; Fujita, T.; Ishikawa, Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol. Rev. 2013, 65, 1010–1052. [Google Scholar] [CrossRef] [PubMed]
- Breyer, M.D.; Davis, L.; Jacobson, H.R.; Breyer, R.M. Differential localization of prostaglandin E receptor subtypes in human kidney. Am. J. Physiol. 1996, 270, F912–F918. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, J.F.; Nasrallah, R.; Carter, A.; He, Y.; Touyz, R.; Hebert, R.L.; Kennedy, C.R.J. PTGER1 deletion attenuates renal injury in diabetic mouse models. Am. J. Pathol. 2013, 183, 1789–1802. [Google Scholar] [CrossRef]
- Rahal, S.; McVeigh, L.I.; Zhang, Y.; Guan, Y.; Breyer, M.D.; Kennedy, C.R. Increased severity of renal impairment in nephritic mice lacking the EP1 receptor. Can. J. Physiol. Pharmacol. 2006, 84, 877–885. [Google Scholar] [CrossRef]
- Tang, L.; Loutzenhiser, K.; Loutzenhiser, R. Biphasic actions of prostaglandin E(2) on the renal afferent arteriole: Role of EP(3) and EP(4) receptors. Circ. Res. 2000, 86, 663–670. [Google Scholar] [CrossRef]
- Srivastava, T.; Alon, U.S.; Cudmore, P.A.; Tarakji, B.; Kats, A.; Garola, R.E.; Duncan, R.S.; McCarthy, E.T.; Sharma, R.; Johnson, M.L.; et al. Cyclooxygenase-2, prostaglandin E2, and prostanoid receptor EP2 in fluid flow shear stress-mediated injury in the solitary kidney. Am. J. Physiol. Ren. Physiol. 2014, 307, F1323–F1333. [Google Scholar] [CrossRef]
- Mohamed, R.; Jayakumar, C.; Ranganathan, P.V.; Ganapathy, V.; Ramesh, G. Kidney proximal tubular epithelial-specific overexpression of netrin-1 suppresses inflammation and albuminuria through suppression of COX-2-mediated PGE2 production in streptozotocin-induced diabetic mice. Am. J. Pathol. 2012, 181, 1991–2002. [Google Scholar] [CrossRef]
- Hassouneh, R.; Nasrallah, R.; Zimpelmann, J.; Gutsol, A.; Eckert, D.; Ghossein, J.; Burns, K.D.; Hebert, R.L. PGE2 receptor EP3 inhibits water reabsorption and contributes to polyuria and kidney injury in a streptozotocin-induced mouse model of diabetes. Diabetologia 2016, 59, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Frolich, S.; Olliges, A.; Kern, N.; Schreiber, Y.; Narumiya, S.; Nusing, R.M. Temporal expression of the PGE2 synthetic system in the kidney is associated with the time frame of renal developmental vulnerability to cyclooxygenase-2 inhibition. Am. J. Physiol. Ren. Physiol. 2012, 303, F209–F219. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nakagawa, N.; Yuhki, K.; Kawabe, J.; Fujino, T.; Takahata, O.; Kabara, M.; Abe, K.; Kojima, F.; Kashiwagi, H.; Hasebe, N.; et al. The intrinsic prostaglandin E2-EP4 system of the renal tubular epithelium limits the development of tubulointerstitial fibrosis in mice. Kidney Int. 2012, 82, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Fujino, H.; Xu, W.; Regan, J.W. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J. Biol. Chem. 2003, 278, 12151–12156. [Google Scholar] [CrossRef] [PubMed]
- Faour, W.H.; Gomi, K.; Kennedy, C.R. PGE(2) induces COX-2 expression in podocytes via the EP(4) receptor through a PKA-independent mechanism. Cell Signal. 2008, 20, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.L.; Mann, B.; Skott, O.; Kurtz, A. Differential regulation of renal prostaglandin receptor mRNAs by dietary salt intake in the rat. Kidney Int. 1999, 56, 528–537. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stitt-Cavanagh, E.M.; Faour, W.H.; Takami, K.; Carter, A.; Vanderhyden, B.; Guan, Y.; Schneider, A.; Breyer, M.D.; Kennedy, C.R. A maladaptive role for EP4 receptors in podocytes. J. Am. Soc. Nephrol. 2010, 21, 1678–1690. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Izawa, T.; Juniantito, V.; Kuwamura, M.; Sugiura, K.; Takeuchi, T.; Yamate, J. Involvement of endogenous prostaglandin E2 in tubular epithelial regeneration through inhibition of apoptosis and epithelial-mesenchymal transition in cisplatin-induced rat renal lesions. Histol. Histopathol. 2010, 25, 995–1007. [Google Scholar] [CrossRef]
- Vukicevic, S.; Simic, P.; Borovecki, F.; Grgurevic, L.; Rogic, D.; Orlic, I.; Grasser, W.A.; Thompson, D.D.; Paralkar, V.M. Role of EP2 and EP4 receptor-selective agonists of prostaglandin E(2) in acute and chronic kidney failure. Kidney Int. 2006, 70, 1099–1106. [Google Scholar] [CrossRef][Green Version]
- Mohamed, R.; Jayakumar, C.; Ramesh, G. Chronic administration of EP4-selective agonist exacerbates albuminuria and fibrosis of the kidney in streptozotocin-induced diabetic mice through IL-6. Lab. Investig. 2013, 93, 933–945. [Google Scholar] [CrossRef]
- Chang, J.; Blazek, E.; Kreft, A.F.; Lewis, A.J. Inhibition of platelet and neutrophil phospholipase A2 by hydroxyeicosatetraenoic acids (HETES). A novel pharmacological mechanism for regulating free fatty acid release. Biochem. Pharmacol. 1985, 34, 1571–1575. [Google Scholar] [CrossRef]
- Johnson, E.N.; Brass, L.F.; Funk, C.D. Increased platelet sensitivity to ADP in mice lacking platelet-type 12-lipoxygenase. Proc. Natl. Acad. Sci. USA 1998, 95, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Coulon, L.; Calzada, C.; Moulin, P.; Vericel, E.; Lagarde, M. Activation of p38 mitogen-activated protein kinase/cytosolic phospholipase A2 cascade in hydroperoxide-stressed platelets. Free Radic. Biol. Med. 2003, 35, 616–625. [Google Scholar] [CrossRef]
- Jansen, M.P.; Emal, D.; Teske, G.J.; Dessing, M.C.; Florquin, S.; Roelofs, J.J. Release of extracellular DNA influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps. Kidney Int. 2017, 91, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Auboeuf, D.; Raspe, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.P.; Akiyama, T.E.; Meinke, P.T. PPARs: Therapeutic targets for metabolic disease. Trends Pharmacol. Sci. 2005, 26, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Platt, C.; Coward, R.J. Peroxisome proliferator activating receptor-gamma and the podocyte. Nephrol. Dial. Transplant. 2017, 32, 423–433. [Google Scholar] [CrossRef]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef]
- Huang, J.T.; Welch, J.S.; Ricote, M.; Binder, C.J.; Willson, T.M.; Kelly, C.; Witztum, J.L.; Funk, C.D.; Conrad, D.; Glass, C.K. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 1999, 400, 378–382. [Google Scholar] [CrossRef]
- Okamura, D.M.; Lopez-Guisa, J.M.; Koelsch, K.; Collins, S.; Eddy, A.A. Atherogenic scavenger receptor modulation in the tubulointerstitium in response to chronic renal injury. Am. J. Physiol. Ren. Physiol. 2007, 293, F575–F585. [Google Scholar] [CrossRef]
- Fruchart, J.C. Peroxisome proliferator-activated receptor-alpha (PPARalpha): At the crossroads of obesity, diabetes and cardiovascular disease. Atherosclerosis 2009, 205, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kume, S.; Araki, S.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Sugimoto, T.; Koya, D.; Haneda, M.; Kashiwagi, A.; et al. Fenofibrate, a PPARalpha agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int. 2011, 79, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr. Diabetes Rep. 2015, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Muroya, Y.; Roman, R.J. Cytochrome P450 eicosanoids in hypertension and renal disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Croft, K.D.; McGiff, J.C.; Sanchez-Mendoza, A.; Carroll, M.A. Angiotensin II releases 20-HETE from rat renal microvessels. Am. J. Physiol. Ren. Physiol. 2000, 279, F544–F551. [Google Scholar] [CrossRef] [PubMed]
- Parmentier, J.H.; Muthalif, M.M.; Saeed, A.E.; Malik, K.U. Phospholipase D activation by norepinephrine is mediated by 12(s)-, 15(s)-, and 20-hydroxyeicosatetraenoic acids generated by stimulation of cytosolic phospholipase a2. Tyrosine phosphorylation of phospholipase d2 in response to norepinephrine. J. Biol. Chem. 2001, 276, 15704–15711. [Google Scholar] [CrossRef] [PubMed]
- Escalante, B.; Erlij, D.; Falck, J.R.; McGiff, J.C. Effect of cytochrome P450 arachidonate metabolites on ion transport in rabbit kidney loop of Henle. Science 1991, 251, 799–802. [Google Scholar] [CrossRef]
- Capdevila, J.H.; Falck, J.R. The CYP P450 arachidonic acid monooxygenases: From cell signaling to blood pressure regulation. Biochem. Biophys. Res. Commun. 2001, 285, 571–576. [Google Scholar] [CrossRef]
- Williams, J.M.; Fan, F.; Murphy, S.; Schreck, C.; Lazar, J.; Jacob, H.J.; Roman, R.J. Role of 20-HETE in the antihypertensive effect of transfer of chromosome 5 from Brown Norway to Dahl salt-sensitive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R1209–R1218. [Google Scholar] [CrossRef][Green Version]
- Williams, J.M.; Sarkis, A.; Hoagland, K.M.; Fredrich, K.; Ryan, R.P.; Moreno, C.; Lopez, B.; Lazar, J.; Fenoy, F.J.; Sharma, M.; et al. Transfer of the CYP4A region of chromosome 5 from Lewis to Dahl S rats attenuates renal injury. Am. J. Physiol. Ren. Physiol. 2008, 295, F1764–F1777. [Google Scholar] [CrossRef]
- Dahly-Vernon, A.J.; Sharma, M.; McCarthy, E.T.; Savin, V.J.; Ledbetter, S.R.; Roman, R.J. Transforming growth factor-beta, 20-HETE interaction, and glomerular injury in Dahl salt-sensitive rats. Hypertension 2005, 45, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Roman, R.J.; Ma, Y.H.; Frohlich, B.; Markham, B. Clofibrate prevents the development of hypertension in Dahl salt-sensitive rats. Hypertension 1993, 21, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.H.; Schwartzman, M.L.; Roman, R.J. Altered renal P-450 metabolism of arachidonic acid in Dahl salt-sensitive rats. Am. J. Physiol. 1994, 267, R579–R589. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Maier, K.G.; Hoagland, K.M.; Yu, M.; Roman, R.J. Abnormal pressure-natriuresis in hypertension: Role of cytochrome P450 metabolites of arachidonic acid. Am. J. Hypertens. 2001, 14, 90S–97S. [Google Scholar] [CrossRef]
- Alonso-Galicia, M.; Maier, K.G.; Greene, A.S.; Cowley, A.W., Jr.; Roman, R.J. Role of 20-hydroxyeicosatetraenoic acid in the renal and vasoconstrictor actions of angiotensin II. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R60–R68. [Google Scholar] [CrossRef] [PubMed]
- McGiff, J.C.; Quilley, J. 20-HETE and the kidney: Resolution of old problems and new beginnings. Am. J. Physiol. 1999, 277, R607–R623. [Google Scholar] [CrossRef] [PubMed]
- Roman, R.J. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 2002, 82, 131–185. [Google Scholar] [CrossRef]
- Williams, J.M.; Sarkis, A.; Lopez, B.; Ryan, R.P.; Flasch, A.K.; Roman, R.J. Elevations in renal interstitial hydrostatic pressure and 20-hydroxyeicosatetraenoic acid contribute to pressure natriuresis. Hypertension 2007, 49, 687–694. [Google Scholar] [CrossRef]
- Williams, J.M.; Murphy, S.; Burke, M.; Roman, R.J. 20-hydroxyeicosatetraeonic acid: A new target for the treatment of hypertension. J. Cardiovasc. Pharmacol. 2010, 56, 336–344. [Google Scholar] [CrossRef]
- Nilakantan, V.; Maenpaa, C.; Jia, G.; Roman, R.J.; Park, F. 20-HETE-mediated cytotoxicity and apoptosis in ischemic kidney epithelial cells. Am. J. Physiol. Ren. Physiol. 2008, 294, F562–F570. [Google Scholar] [CrossRef]
- Lameire, N.; Vanholder, R. Pathophysiologic features and prevention of human and experimental acute tubular necrosis. J. Am. Soc. Nephrol. 2001, 12 (Suppl. 17), S20–S32. [Google Scholar]
- Fan, F.; Ge, Y.; Lv, W.; Elliott, M.R.; Muroya, Y.; Hirata, T.; Booz, G.W.; Roman, R.J. Molecular mechanisms and cell signaling of 20-hydroxyeicosatetraenoic acid in vascular pathophysiology. Front. Biosci. 2016, 21, 1427–1463. [Google Scholar]
- Regner, K.R.; Zuk, A.; Van Why, S.K.; Shames, B.D.; Ryan, R.P.; Falck, J.R.; Manthati, V.L.; McMullen, M.E.; Ledbetter, S.R.; Roman, R.J. Protective effect of 20-HETE analogues in experimental renal ischemia reperfusion injury. Kidney Int. 2009, 75, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Roman, R.J.; Akbulut, T.; Park, F.; Regner, K.R. 20-HETE in acute kidney injury. Kidney Int. 2011, 79, 10–13. [Google Scholar] [CrossRef]
- Eid, A.A.; Gorin, Y.; Fagg, B.M.; Maalouf, R.; Barnes, J.L.; Block, K.; Abboud, H.E. Mechanisms of podocyte injury in diabetes: Role of cytochrome P450 and NADPH oxidases. Diabetes 2009, 58, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Eid, S.; Maalouf, R.; Jaffa, A.A.; Nassif, J.; Hamdy, A.; Rashid, A.; Ziyadeh, F.N.; Eid, A.A. 20-HETE and EETs in diabetic nephropathy: A novel mechanistic pathway. PLoS ONE 2013, 8, e70029. [Google Scholar] [CrossRef] [PubMed]
- Roshanravan, H.; Kim, E.Y.; Dryer, S.E. 20-Hydroxyeicosatetraenoic Acid (20-HETE) Modulates Canonical Transient Receptor Potential-6 (TRPC6) Channels in Podocytes. Front. Physiol. 2016, 7, 351. [Google Scholar] [CrossRef]
- Kriz, W.; Lemley, K.V. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J. Am. Soc. Nephrol. 2015, 26, 258–269. [Google Scholar] [CrossRef]
- Omata, K.; Tsutsumi, E.; Sheu, H.L.; Utsumi, Y.; Abe, K. Effect of aging on renal cytochrome P450-dependent arachidonic acid metabolism in Dahl rats. J. Lipid Mediat. 1993, 6, 369–373. [Google Scholar] [PubMed]
- Moore, K.P.; Wood, J.; Gove, C.; Tan, K.C.; Eason, J.; Taylor, G.W.; Williams, R. Synthesis and metabolism of cysteinyl leukotrienes by the isolated pig kidney. Adv. Prostaglandin Thromboxane Leukot. Res. 1991, 21B, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Klausner, J.M.; Paterson, I.S.; Goldman, G.; Kobzik, L.; Rodzen, C.; Lawrence, R.; Valeri, C.R.; Shepro, D.; Hechtman, H.B. Postischemic renal injury is mediated by neutrophils and leukotrienes. Am. J. Physiol. 1989, 256, F794–F802. [Google Scholar] [CrossRef] [PubMed]
- Sener, G.; Sakarcan, A.; Sehirli, O.; Eksioglu-Demiralp, E.; Sener, E.; Ercan, F.; Gedik, N.; Yegen, B.C. Chronic renal failure-induced multiple-organ injury in rats is alleviated by the selective CysLT1 receptor antagonist montelukast. Prostaglandins Other Lipid Mediat. 2007, 83, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Noiri, E.; Yokomizo, T.; Nakao, A.; Izumi, T.; Fujita, T.; Kimura, S.; Shimizu, T. An in vivo approach showing the chemotactic activity of leukotriene B(4) in acute renal ischemic-reperfusion injury. Proc. Natl. Acad. Sci. USA 2000, 97, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, S.S.; Silva, L.S.; Peruchetti, D.B.; Sirtoli, G.M.; Moraes-Santos, F.; Portella, V.G.; Silva-Filho, J.L.; Pinheiro, C.S.; Abreu, T.P.; Takiya, C.M.; et al. 5-Lypoxygenase products are involved in renal tubulointerstitial injury induced by albumin overload in proximal tubules in mice. PLoS ONE 2014, 9, e107549. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, G.A.; McIntyre, T.M. Neutrophil adherence to human endothelium in vitro occurs by CDw18 (Mo1, MAC-1/LFA-1/GP 150,95) glycoprotein-dependent and -independent mechanisms. J. Clin. Investig. 1988, 81, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Yared, A.; Albrightson-Winslow, C.; Griswold, D.; Takahashi, K.; Fogo, A.; Badr, K.F. Functional significance of leukotriene B4 in normal and glomerulonephritic kidneys. J. Am. Soc. Nephrol. 1991, 2, 45–56. [Google Scholar] [PubMed]
- Zedan, M.M.; El-Refaey, A.; Zaghloul, H.; Abdelrahim, M.E.; Osman, A.; Zedan, M.M.; Eltantawy, N. Montelukast as an add-on treatment in steroid dependant nephrotic syndrome, randomised-controlled trial. J. Nephrol. 2016, 29, 585–592. [Google Scholar] [CrossRef]
- Planaguma, A.; Kazani, S.; Marigowda, G.; Haworth, O.; Mariani, T.J.; Israel, E.; Bleecker, E.R.; Curran-Everett, D.; Erzurum, S.C.; Calhoun, W.J.; et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. Am. J. Respir. Crit. Care Med. 2008, 178, 574–582. [Google Scholar] [CrossRef]
- Cheng, X.; He, S.; Yuan, J.; Miao, S.; Gao, H.; Zhang, J.; Li, Y.; Peng, W.; Wu, P. Lipoxin A4 attenuates LPS-induced mouse acute lung injury via Nrf2-mediated E-cadherin expression in airway epithelial cells. Free Radic. Biol. Med. 2016, 93, 52–66. [Google Scholar] [CrossRef]
- Kieran, N.E.; Maderna, P.; Godson, C. Lipoxins: Potential anti-inflammatory, proresolution, and antifibrotic mediators in renal disease. Kidney Int. 2004, 65, 1145–1154. [Google Scholar] [CrossRef]
- Christie, P.E.; Spur, B.W.; Lee, T.H. The effects of lipoxin A4 on airway responses in asthmatic subjects. Am. Rev. Respir. Dis. 1992, 145, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Dahlen, S.E.; Franzen, L.; Raud, J.; Serhan, C.N.; Westlund, P.; Wikstrom, E.; Bjorck, T.; Matsuda, H.; Webber, S.E.; Veale, C.A.; et al. Actions of lipoxin A4 and related compounds in smooth muscle preparations and on the microcirculation in vivo. Adv. Exp. Med. Biol. 1988, 229, 107–130. [Google Scholar] [PubMed]
- Wu, S.H.; Liao, P.Y.; Yin, P.L.; Zhang, Y.M.; Dong, L. Elevated expressions of 15-lipoxygenase and lipoxin A4 in children with acute poststreptococcal glomerulonephritis. Am. J. Pathol. 2009, 174, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.; McMahon, B.; Mitchell, D.; Sadlier, D.; Godson, C. Lipoxin A4 modifies platelet-derived growth factor-induced pro-fibrotic gene expression in human renal mesangial cells. Am. J. Pathol. 2005, 167, 683–694. [Google Scholar] [CrossRef]
- O’Meara, Y.M.; Brady, H.R. Lipoxins, leukocyte recruitment and the resolution phase of acute glomerulonephritis. Kidney Int. Suppl. 1997, 58, S56–S61. [Google Scholar] [PubMed]
- Munger, K.A.; Montero, A.; Fukunaga, M.; Uda, S.; Yura, T.; Imai, E.; Kaneda, Y.; Valdivielso, J.M.; Badr, K.F. Transfection of rat kidney with human 15-lipoxygenase suppresses inflammation and preserves function in experimental glomerulonephritis. Proc. Natl. Acad. Sci. USA 1999, 96, 13375–13380. [Google Scholar] [CrossRef] [PubMed]
- Davin, J.C.; Coppo, R. Henoch-Schonlein purpura nephritis in children. Nat. Rev. Nephrol. 2014, 10, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Borgeson, E.; Docherty, N.G.; Murphy, M.; Rodgers, K.; Ryan, A.; O’Sullivan, T.P.; Guiry, P.J.; Goldschmeding, R.; Higgins, D.F.; Godson, C. Lipoxin A(4) and benzo-lipoxin A(4) attenuate experimental renal fibrosis. FASEB J. 2011, 25, 2967–2979. [Google Scholar] [CrossRef]
- Borgeson, E.; Johnson, A.M.; Lee, Y.S.; Till, A.; Syed, G.H.; Ali-Shah, S.T.; Guiry, P.J.; Dalli, J.; Colas, R.A.; Serhan, C.N.; et al. Lipoxin A4 Attenuates Obesity-Induced Adipose Inflammation and Associated Liver and Kidney Disease. Cell Metab. 2015, 22, 125–137. [Google Scholar] [CrossRef]
- Brennan, E.P.; Mohan, M.; McClelland, A.; Tikellis, C.; Ziemann, M.; Kaspi, A.; Gray, S.P.; Pickering, R.; Tan, S.M.; Ali-Shah, S.T.; et al. Lipoxins Regulate the Early Growth Response-1 Network and Reverse Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 1437–1448. [Google Scholar] [CrossRef]
- Sun, S.; Ning, X.; Zhai, Y.; Du, R.; Lu, Y.; He, L.; Li, R.; Wu, W.; Sun, W.; Wang, H. Egr-1 mediates chronic hypoxia-induced renal interstitial fibrosis via the PKC/ERK pathway. Am. J. Nephrol. 2014, 39, 436–448. [Google Scholar] [CrossRef] [PubMed]
- McMahon, B.; Stenson, C.; McPhillips, F.; Fanning, A.; Brady, H.R.; Godson, C. Lipoxin A4 antagonizes the mitogenic effects of leukotriene D4 in human renal mesangial cells. Differential activation of MAP kinases through distinct receptors. J. Biol. Chem. 2000, 275, 27566–27575. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.C.; Sung, J.M.; Shen, Y.T.; Jheng, H.F.; Chen, S.H.; Tsai, P.J.; Tsai, Y.S. Egr-1 deficiency protects from renal inflammation and fibrosis. J. Mol. Med. 2016, 94, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, M.M.; Hye Khan, M.A.; Chesnik, M.A.; Sharma, A.; Paudyal, M.P.; Falck, J.R.; Imig, J.D. The epoxyeicosatrienoic acid analog PVPA ameliorates cyclosporine-induced hypertension and renal injury in rats. Am. J. Physiol. Ren. Physiol. 2016, 311, F576–F585. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Imig, J.D.; Yang, J.; Hammock, B.D.; Padanilam, B.J. Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am. J. Physiol. Ren. Physiol. 2014, 307, F971–F980. [Google Scholar] [CrossRef]
- Manhiani, M.; Quigley, J.E.; Knight, S.F.; Tasoobshirazi, S.; Moore, T.; Brands, M.W.; Hammock, B.D.; Imig, J.D. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am. J. Physiol. Ren. Physiol. 2009, 297, F740–F748. [Google Scholar] [CrossRef] [PubMed]
- Roche, C.; Guerrot, D.; Harouki, N.; Duflot, T.; Besnier, M.; Remy-Jouet, I.; Renet, S.; Dumesnil, A.; Lejeune, A.; Morisseau, C.; et al. Impact of soluble epoxide hydrolase inhibition on early kidney damage in hyperglycemic overweight mice. Prostaglandins Other Lipid Mediat. 2015, 120, 148–154. [Google Scholar] [CrossRef]
- Zhu, Y.; Blum, M.; Hoff, U.; Wesser, T.; Fechner, M.; Westphal, C.; Gurgen, D.; Catar, R.A.; Philippe, A.; Wu, K.; et al. Renal Ischemia/Reperfusion Injury in Soluble Epoxide Hydrolase-Deficient Mice. PLoS ONE 2016, 11, e0145645. [Google Scholar] [CrossRef][Green Version]
- Hercule, H.C.; Schunck, W.H.; Gross, V.; Seringer, J.; Leung, F.P.; Weldon, S.M.; da Costa Goncalves, A.; Huang, Y.; Luft, F.C.; Gollasch, M. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 54–60. [Google Scholar] [CrossRef]
- Deng, B.Q.; Luo, Y.; Kang, X.; Li, C.B.; Morisseau, C.; Yang, J.; Lee, K.S.S.; Huang, J.; Hu, D.Y.; Wu, M.Y.; et al. Epoxide metabolites of arachidonate and docosahexaenoate function conversely in acute kidney injury involved in GSK3beta signaling. Proc. Natl. Acad. Sci. USA 2017, 114, 12608–12613. [Google Scholar] [CrossRef]
- Wang, D.; Borrego-Conde, L.J.; Falck, J.R.; Sharma, K.K.; Wilcox, C.S.; Umans, J.G. Contributions of nitric oxide, EDHF, and EETs to endothelium-dependent relaxation in renal afferent arterioles. Kidney Int. 2003, 63, 2187–2193. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.K.; Wang, Y.; Chen, J.; Zhang, M.Z.; Harris, R.C.; Chen, J.K. Overexpression of G-protein-coupled receptor 40 enhances the mitogenic response to epoxyeicosatrienoic acids. PLoS ONE 2015, 10, e0113130. [Google Scholar] [CrossRef] [PubMed]
- Pomposiello, S.I.; Quilley, J.; Carroll, M.A.; Falck, J.R.; McGiff, J.C. 5,6-epoxyeicosatrienoic acid mediates the enhanced renal vasodilation to arachidonic acid in the SHR. Hypertension 2003, 42, 548–554. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheng, M.K.; Doumad, A.B.; Jiang, H.; Falck, J.R.; McGiff, J.C.; Carroll, M.A. Epoxyeicosatrienoic acids mediate adenosine-induced vasodilation in rat preglomerular microvessels (PGMV) via A2A receptors. Br. J. Pharmacol. 2004, 141, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.A.; Doumad, A.B.; Li, J.; Cheng, M.K.; Falck, J.R.; McGiff, J.C. Adenosine2A receptor vasodilation of rat preglomerular microvessels is mediated by EETs that activate the cAMP/PKA pathway. Am. J. Physiol. Ren. Physiol. 2006, 291, F155–F161. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D.; Dimitropoulou, C.; Reddy, D.S.; White, R.E.; Falck, J.R. Afferent arteriolar dilation to 11, 12-EET analogs involves PP2A activity and Ca2+-activated K+ Channels. Microcirculation 2008, 15, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Zhang, C.; Lin, D.H.; Wang, L.; Graves, J.P.; Zeldin, D.C.; Capdevila, J.H. Cyp2c44 epoxygenase in the collecting duct is essential for the high K+ intake-induced antihypertensive effect. Am. J. Physiol. Ren. Physiol. 2014, 307, F453–F460. [Google Scholar] [CrossRef]
- Sharma, M.; McCarthy, E.T.; Reddy, D.S.; Patel, P.K.; Savin, V.J.; Medhora, M.; Falck, J.R. 8,9-Epoxyeicosatrienoic acid protects the glomerular filtration barrier. Prostaglandins Other Lipid Mediat. 2009, 89, 43–51. [Google Scholar] [CrossRef]
- Beck, L.H., Jr.; Fervenza, F.C.; Beck, D.M.; Bonegio, R.G.; Malik, F.A.; Erickson, S.B.; Cosio, F.G.; Cattran, D.C.; Salant, D.J. Rituximab-induced depletion of anti-PLA2R autoantibodies predicts response in membranous nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1543–1550. [Google Scholar] [CrossRef]
- Thokhonelidze, I.; Maglakelidze, N.; Sarishvili, N.; Kasradze, T.; Dalakishvili, K. Association of anti-phospholipasea2-receptor antibodies with clinical course of idiopathic membranous nephropathy. Georgian Med News 2015, 49–53. [Google Scholar]
- Ramachandran, R.; Hn, H.K.; Kumar, V.; Nada, R.; Yadav, A.K.; Goyal, A.; Kumar, V.; Rathi, M.; Jha, V.; Gupta, K.L.; et al. Tacrolimus combined with corticosteroids versus Modified Ponticelli regimen in treatment of idiopathic membranous nephropathy: Randomized control trial. Nephrology 2016, 21, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.Y.; Wang, Y.X.; Li, J.S.; Zhao, S.L.; Diao, T.T.; Wang, Y.; Wang, C.; Qin, Y.; Cao, Y.; Wei, Q.; et al. Serum Anti-PLA2R Antibody Predicts Treatment Outcome in Idiopathic Membranous Nephropathy. Am. J. Nephrol. 2016, 43, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Bech, A.P.; Hofstra, J.M.; Brenchley, P.E.; Wetzels, J.F. Association of anti-PLA(2)R antibodies with outcomes after immunosuppressive therapy in idiopathic membranous nephropathy. Clin. J. Am. Soc. Nephrol. 2014, 9, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, J.; Cattran, D.C. The KDIGO practice guideline on glomerulonephritis: Reading between the (guide)lines--application to the individual patient. Kidney Int. 2012, 82, 840–856. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo-Gil, E.; Garcia-Herrero, C.; Toldos, O.; Usategui, A.; Criado, G.; Perez-Yague, S.; Barber, D.F.; Pablos, J.L.; Galindo, M. Microthrombotic Renal Vascular Lesions Are Associated to Increased Renal Inflammatory Infiltration in Murine Lupus Nephritis. Front. Immunol. 2018, 9, 1948. [Google Scholar] [CrossRef] [PubMed]
- Gerrah, R.; Ehrlich, S.; Tshori, S.; Sahar, G. Beneficial effect of aspirin on renal function in patients with renal insufficiency postcardiac surgery. J. Cardiovasc. Surg. 2004, 45, 545–550. [Google Scholar]
- Garg, A.X.; Kurz, A.; Sessler, D.I.; Cuerden, M.; Robinson, A.; Mrkobrada, M.; Parikh, C.R.; Mizera, R.; Jones, P.M.; Tiboni, M.; et al. Perioperative aspirin and clonidine and risk of acute kidney injury: A randomized clinical trial. JAMA 2014, 312, 2254–2264. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Chiarugi, A.; Boner, A.; Montini, G.; De’ Angelis, G.L. Working towards an Appropriate Use of Ibuprofen in Children: An Evidence-Based Appraisal. Drugs 2017, 77, 1295–1311. [Google Scholar] [CrossRef]
- Lipman, G.S.; Shea, K.; Christensen, M.; Phillips, C.; Burns, P.; Higbee, R.; Koskenoja, V.; Eifling, K.; Krabak, B.J. Ibuprofen versus placebo effect on acute kidney injury in ultramarathons: A randomised controlled trial. Emerg. Med. J. 2017, 34, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Kaojarern, S.; Chennavasin, P.; Anderson, S.; Brater, D.C. Nephron site of effect of nonsteroidal anti-inflammatory drugs on solute excretion in humans. Am. J. Physiol. 1983, 244, F134–F139. [Google Scholar] [CrossRef]
- Wu, Y.J.; Xue, M.; Chen, H. Licofelone inhibits interleukin-18-induced pro-inflammatory cytokine release and cellular proliferation in human mesangial cells. Basic Clin. Pharmacol. Toxicol. 2012, 111, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, J.; Pye, C.; Al-Shabrawey, M.; Elmarakby, A.A. Inhibition of 12/15-Lipoxygenase Reduces Renal Inflammation and Injury in Streptozotocin-Induced Diabetic Mice. J. Diabetes Metab. 2015, 6. [Google Scholar] [CrossRef]
- Hofstra, J.M.; Beck, L.H., Jr.; Beck, D.M.; Wetzels, J.F.; Salant, D.J. Anti-phospholipase A(2) receptor antibodies correlate with clinical status in idiopathic membranous nephropathy. Clin. J. Am. Soc. Nephrol. 2011, 6, 1286–1291. [Google Scholar] [CrossRef] [PubMed]
- Debiec, H.; Ronco, P. PLA2R autoantibodies and PLA2R glomerular deposits in membranous nephropathy. N. Engl. J. Med. 2011, 364, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Pourcine, F.; Dahan, K.; Mihout, F.; Cachanado, M.; Brocheriou, I.; Debiec, H.; Ronco, P. Prognostic value of PLA2R autoimmunity detected by measurement of anti-PLA2R antibodies combined with detection of PLA2R antigen in membranous nephropathy: A single-centre study over 14 years. PLoS ONE 2017, 12, e0173201. [Google Scholar] [CrossRef] [PubMed]
- Jullien, P.; Seitz Polski, B.; Maillard, N.; Thibaudin, D.; Laurent, B.; Ollier, E.; Alamartine, E.; Lambeau, G.; Mariat, C. Anti-phospholipase A2 receptor antibody levels at diagnosis predicts spontaneous remission of idiopathic membranous nephropathy. Clin. Kidney J. 2017, 10, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Buysen, J.G.; Houthoff, H.J.; Krediet, R.T.; Arisz, L. Acute interstitial nephritis: A clinical and morphological study in 27 patients. Nephrol. Dial. Transplant. 1990, 5, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Eddy, A.A. Drug-induced tubulointerstitial nephritis: Hypersensitivity and necroinflammatory pathways. Pediatric Nephrol. 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; Giroud, J.P.; Willoughby, D.A. Studies on the mediators of the acute inflammatory response induced in rats in different sites by carrageenan and turpentine. J. Pathol. 1971, 104, 15–29. [Google Scholar] [CrossRef] [PubMed]
- De Gaetano, G.; Bucchi, F.; Gambino, M.C.; Cerletti, C. Does oral aspirin spare the kidney? Lancet 1986, 1, 736. [Google Scholar] [CrossRef]
- Berger, J.S.; Brown, D.L.; Becker, R.C. Low-dose aspirin in patients with stable cardiovascular disease: A meta-analysis. Am. J. Med. 2008, 121, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Antithrombotic Trialists, C.; Baigent, C.; Blackwell, L.; Collins, R.; Emberson, J.; Godwin, J.; Peto, R.; Buring, J.; Hennekens, C.; Kearney, P.; et al. Aspirin in the primary and secondary prevention of vascular disease: Collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009, 373, 1849–1860. [Google Scholar] [CrossRef]
- Vandvik, P.O.; Lincoff, A.M.; Gore, J.M.; Gutterman, D.D.; Sonnenberg, F.A.; Alonso-Coello, P.; Akl, E.A.; Lansberg, M.G.; Guyatt, G.H.; Spencer, F.A. Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141, e637S–e668S. [Google Scholar] [CrossRef] [PubMed]
- Karzai, W.; Priebe, H.J. Aspirin and mortality from coronary bypass surgery. N. Engl. J. Med. 2003, 348, 1057–1059. [Google Scholar] [PubMed]
- Ali, H.; Shaaban, A.; Murtaza, A.; Howell, L.E.; Ahmed, A. Effect of Long-Term, Low-Dose Aspirin Therapy on Renal Graft Function. Exp. Clin. Transplant. 2017, 15, 400–404. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rainsford, K.D. Ibuprofen: Pharmacology, efficacy and safety. Inflammopharmacology 2009, 17, 275–342. [Google Scholar] [CrossRef] [PubMed]
- Van Overmeire, B.; Allegaert, K.; Casaer, A.; Debauche, C.; Decaluwe, W.; Jespers, A.; Weyler, J.; Harrewijn, I.; Langhendries, J.P. Prophylactic ibuprofen in premature infants: A multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2004, 364, 1945–1949. [Google Scholar] [CrossRef]
- Haas, M.; Spargo, B.H.; Wit, E.J.; Meehan, S.M. Etiologies and outcome of acute renal insufficiency in older adults: A renal biopsy study of 259 cases. Am. J. Kidney Dis. 2000, 35, 433–447. [Google Scholar] [CrossRef]
- Martinez Lopez, A.B.; Alvarez Blanco, O.; De Pablos, A.L.; San-Jose, M.D.M.; De La Blanca, A.R.S. Ibuprofen-induced acute interstitial nephritis in the paediatric population. Nefrologia 2016, 36, 69–71. [Google Scholar] [CrossRef]
- Steinhauslin, F.; Munafo, A.; Buclin, T.; Macciocchi, A.; Biollaz, J. Renal effects of nimesulide in furosemide-treated subjects. Drugs 1993, 46 (Suppl. 1), 257–262. [Google Scholar] [CrossRef]
- Warrington, S.J.; Ravic, M.; Dawnay, A. Renal and general tolerability of repeated doses of nimesulide in normal subjects. Drugs 1993, 46 (Suppl. 1), 263–269. [Google Scholar] [CrossRef]
- Ceserani, R.; Casciarri, I.; Cavalletti, E.; Cazzulani, P. Action of Nimesulide on Rat Gastric Prostaglandins and Renal Function. Drug Investig. 1991, 3, 14–21. [Google Scholar] [CrossRef]
- Forget, P.; Machiels, J.P.; Coulie, P.G.; Berliere, M.; Poncelet, A.J.; Tombal, B.; Stainier, A.; Legrand, C.; Canon, J.L.; Kremer, Y.; et al. Neutrophil: Lymphocyte ratio and intraoperative use of ketorolac or diclofenac are prognostic factors in different cohorts of patients undergoing breast, lung, and kidney cancer surgery. Ann. Surg. Oncol. 2013, 20 (Suppl. 3), S650–S660. [Google Scholar] [CrossRef]
- Yakar, I.; Melamed, R.; Shakhar, G.; Shakhar, K.; Rosenne, E.; Abudarham, N.; Page, G.G.; Ben-Eliyahu, S. Prostaglandin e(2) suppresses NK activity in vivo and promotes postoperative tumor metastasis in rats. Ann. Surg. Oncol. 2003, 10, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Ungprasert, P.; Cheungpasitporn, W.; Crowson, C.S.; Matteson, E.L. Individual non-steroidal anti-inflammatory drugs and risk of acute kidney injury: A systematic review and meta-analysis of observational studies. Eur. J. Intern. Med. 2015, 26, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Colebatch, A.N.; Marks, J.L.; van der Heijde, D.M.; Edwards, C.J. Safety of nonsteroidal antiinflammatory drugs and/or paracetamol in people receiving methotrexate for inflammatory arthritis: A Cochrane systematic review. J. Rheumatol. Suppl. 2012, 90, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, F.A.; Larsen, C.P.; Troxell, M.L. Membranous nephropathy and nonsteroidal anti-inflammatory agents. Am. J. Kidney Dis. 2013, 62, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Catella-Lawson, F.; McAdam, B.; Morrison, B.W.; Kapoor, S.; Kujubu, D.; Antes, L.; Lasseter, K.C.; Quan, H.; Gertz, B.J.; FitzGerald, G.A. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J. Pharmacol. Exp. Ther. 1999, 289, 735–741. [Google Scholar] [PubMed]
- Takahashi, K.; Patel, A.K.; Nagai, S.; Safwan, M.; Putchakayala, K.G.; Kane, W.J.; Malinzak, L.E.; Denny, J.E.; Yoshida, A.; Kim, D.Y. Perioperative Ketorolac Use: A Potential Risk Factor for Renal Dysfunction After Live-Donor Nephrectomy. Ann. Transplant. 2017, 22, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.R. High-dose non-steroidal anti-inflammatories: Painful choices. Lancet 2013, 382, 746–748. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, E.L.; Song, Y. Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: Meta-analysis of randomized trials. JAMA 2006, 296, 1619–1632. [Google Scholar] [CrossRef] [PubMed]
- Gounaris, E.; Heiferman, M.J.; Heiferman, J.R.; Shrivastav, M.; Vitello, D.; Blatner, N.R.; Knab, L.M.; Phillips, J.D.; Cheon, E.C.; Grippo, P.J.; et al. Zileuton, 5-lipoxygenase inhibitor, acts as a chemopreventive agent in intestinal polyposis, by modulating polyp and systemic inflammation. PLoS ONE 2015, 10, e0121402. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.M.M.; Ribeiro, D.; Silva, A.M.S.; Fernandes, E. 2,3-Diarylxanthones as Potential Inhibitors of Arachidonic Acid Metabolic Pathways. Inflammation 2017, 40, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.N.; Theuer, J.; Shagdarsuren, E.; Kaergel, E.; Honeck, H.; Park, J.K.; Markovic, M.; Barbosa-Sicard, E.; Dechend, R.; Wellner, M.; et al. A peroxisome proliferator-activated receptor-alpha activator induces renal CYP2C23 activity and protects from angiotensin II-induced renal injury. Am. J. Pathol. 2004, 164, 521–532. [Google Scholar] [CrossRef]
- Gai, Z.; Visentin, M.; Gui, T.; Zhao, L.; Thasler, W.E.; Hausler, S.; Hartling, I.; Cremonesi, A.; Hiller, C.; Kullak-Ublick, G.A. Effects of Farnesoid X Receptor Activation on Arachidonic Acid Metabolism, NF-kB Signaling, and Hepatic Inflammation. Mol. Pharmacol. 2018, 94, 802–811. [Google Scholar] [CrossRef]
- Shlipak, M.G.; Fried, L.F.; Cushman, M.; Manolio, T.A.; Peterson, D.; Stehman-Breen, C.; Bleyer, A.; Newman, A.; Siscovick, D.; Psaty, B. Cardiovascular mortality risk in chronic kidney disease: Comparison of traditional and novel risk factors. JAMA 2005, 293, 1737–1745. [Google Scholar] [CrossRef]
- Muntner, P.; He, J.; Astor, B.C.; Folsom, A.R.; Coresh, J. Traditional and nontraditional risk factors predict coronary heart disease in chronic kidney disease: Results from the atherosclerosis risk in communities study. J. Am. Soc. Nephrol. 2005, 16, 529–538. [Google Scholar] [CrossRef]
- Garg, A.; Grundy, S.M. Management of dyslipidemia in NIDDM. Diabetes Care 1990, 13, 153–169. [Google Scholar] [CrossRef]
- Garg, A.; Grundy, S.M. Gemfibrozil alone and in combination with lovastatin for treatment of hypertriglyceridemia in NIDDM. Diabetes 1989, 38, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Siavash, M.; Amini, M. Vitamin C may have similar beneficial effects to Gemfibrozil on serum high-density lipoprotein-cholesterol in type 2 diabetic patients. J. Res. Pharm. Pract. 2014, 3, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Martin-Navarro, J.A.; Petkov-Stoyanov, V.; Gutierrez-Sanchez, M.J.; Pedraza-Cezon, L. Acute renal failure secondary to interstitial acute nephritis and Fanconi syndrome for metamizole and gemfibrozil. Nefrologia 2016, 36, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Liu, J.; Kumar, G.; Skapek, S.X.; Falck, J.R.; Imig, J.D. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J. 2013, 27, 2946–2956. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Farzamikia, N.; Sakhinia, E.; Afrasiabirad, A. Pharmacogenetics-Based Warfarin Dosing in Patients with Cardiac Valve Replacement: The Effects of CYP2C9 and VKORC1 Gene Polymorphisms. Lab. Med. 2017, 49, 25–34. [Google Scholar] [CrossRef]
- Pei, L.; Tian, X.; Long, Y.; Nan, W.; Jia, M.; Qiao, R.; Zhang, J. Establishment of a Han Chinese-specific pharmacogenetic-guided warfarin dosing algorithm. Medicine 2018, 97, e12178. [Google Scholar] [CrossRef]
- Spatzenegger, M.; Jaeger, W. Clinical importance of hepatic cytochrome P450 in drug metabolism. Drug Metab. Rev. 1995, 27, 397–417. [Google Scholar] [CrossRef]
- Yanagita, M. Gas6, warfarin, and kidney diseases. Clin. Exp. Nephrol. 2004, 8, 304–309. [Google Scholar] [CrossRef]
- Zeldin, D.C.; Wei, S.; Falck, J.R.; Hammock, B.D.; Snapper, J.R.; Capdevila, J.H. Metabolism of epoxyeicosatrienoic acids by cytosolic epoxide hydrolase: Substrate structural determinants of asymmetric catalysis. Arch. Biochem. Biophys. 1995, 316, 443–451. [Google Scholar] [CrossRef]
- Imig, J.D.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 2009, 8, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.C.; Hammock, B.D. Discovery of inhibitors of soluble epoxide hydrolase: A target with multiple potential therapeutic indications. J. Med. Chem. 2012, 55, 1789–1808. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compounds | Species | Targets | Kidney Disease | Outcome | Reference |
|---|---|---|---|---|---|
| Cyclosporine+ methylprednisolone | Human | Calcineurin and corticosteroid hormone receptor | IMN | Proteinuria ↓ PLA2R Ab ↓ Infiltration of defense cells ↓ | [189] |
| Tacrolimus+corticosteroids, corticosteroids+cyclophosphamide, or corticosteroids alone. | Human | Peptidyl-prolyl isomerase and glucocorticoid receptors | IMN | Proteinuria ↓ Serum albumin ↑ Glomerular PLA2R ↓ Serum PLA2R-Ab ↓ Infiltration of defense cells ↑ | [190] |
| Rituximab | Human | Pan-B-cell marker CD20 | IMN/IgA Nephritis | Proteinuria ↓ GFR ↑ Serum albumin ↑ PLA2R Ab ↓ Infiltration of defense cells ↓ | [191,192,193] |
| Prednisolone | Human | glucocorticoid receptors | IMN | GFR ↑ Proteinuria ↓ Serum albumin ↑ PLA2R Ab ↓ Infiltration of defense cells ↓ | [194] |
| Cyclophosphamide or +corticosteroids | Human | glucocorticoid receptors (for corticosteroids) | IMN/IgA Nephritis | GFR ↑ Proteinuria ↓ Serum albumin ↑ PLA2R Ab ↓ Infiltration of defense cells ↓ | [193,194] |
| Aspirin | Mouse and Human | COX-1/COX-2 | AKI | GFR ↑ Serum creatinine ↓ Urinary output ↑ Proteinuria ↓ | [195,196] |
| Ibuprofen | Human | COX-1/COX-2 | ATIN | Pain control ↓ | [197,198] |
| Nimesulide | Rats and Human | COX-1/COX-2 | ATIN | Plasma renin activity ↓ Aldosterone level ↓ Urinary PTE2 level ↓ | [199] |
| Indomethacin | Human | COX-1/COX-2 | Renal failure | IL-6 ↓ IL-10 ↓ | [199] |
| Carprofen | Human | COX-2 | Renal failure | IL-1β ↓ | [199] |
| Diclofenac acid | Rats and Human | COX1/COX2 | Renal cancer | PGE2 level ↓ | [200] |
| Zileuton | Human Mesangial Cells | LOX/COX-2 | Renal cancer | Serum creatinine ↓ Interstitial fibrosis ↓ | [201] |
| Licofelone | Mouse and Human | 5-LOX/COX | Glomerulonephritis | IL-18 ↓ PGE2 ↓ | [201] |
| Baicalein | Mouse | 12/15-LOX | Diabetic nephropathy | 12-HETE ↓ IL-6 ↓ Proteinuria ↓ | [202] |
| PVPA | Rats | CYP450 | Acute and chronic glomerulonephritis | Proteinuria ↓ Apoptosis in tubular epithelial cells ↓ Generation of reactive oxygen species ↓ | [174] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Fu, X.; Chen, Q.; Patra, J.K.; Wang, D.; Wang, Z.; Gai, Z. Arachidonic Acid Metabolism and Kidney Inflammation. Int. J. Mol. Sci. 2019, 20, 3683. https://doi.org/10.3390/ijms20153683
Wang T, Fu X, Chen Q, Patra JK, Wang D, Wang Z, Gai Z. Arachidonic Acid Metabolism and Kidney Inflammation. International Journal of Molecular Sciences. 2019; 20(15):3683. https://doi.org/10.3390/ijms20153683
Chicago/Turabian StyleWang, Tianqi, Xianjun Fu, Qingfa Chen, Jayanta Kumar Patra, Dongdong Wang, Zhenguo Wang, and Zhibo Gai. 2019. "Arachidonic Acid Metabolism and Kidney Inflammation" International Journal of Molecular Sciences 20, no. 15: 3683. https://doi.org/10.3390/ijms20153683
APA StyleWang, T., Fu, X., Chen, Q., Patra, J. K., Wang, D., Wang, Z., & Gai, Z. (2019). Arachidonic Acid Metabolism and Kidney Inflammation. International Journal of Molecular Sciences, 20(15), 3683. https://doi.org/10.3390/ijms20153683