The Key Role of Epithelial to Mesenchymal Transition (EMT) in Hypertensive Kidney Disease


Abstract
1. Introduction
2. Tubulointerstitial Damage and Fibrosis in Hypertension
3. Epithelial-to-Mesenchymal Transition (EMT)
4. Markers of EMT
5. EMT as Mediator of TIF
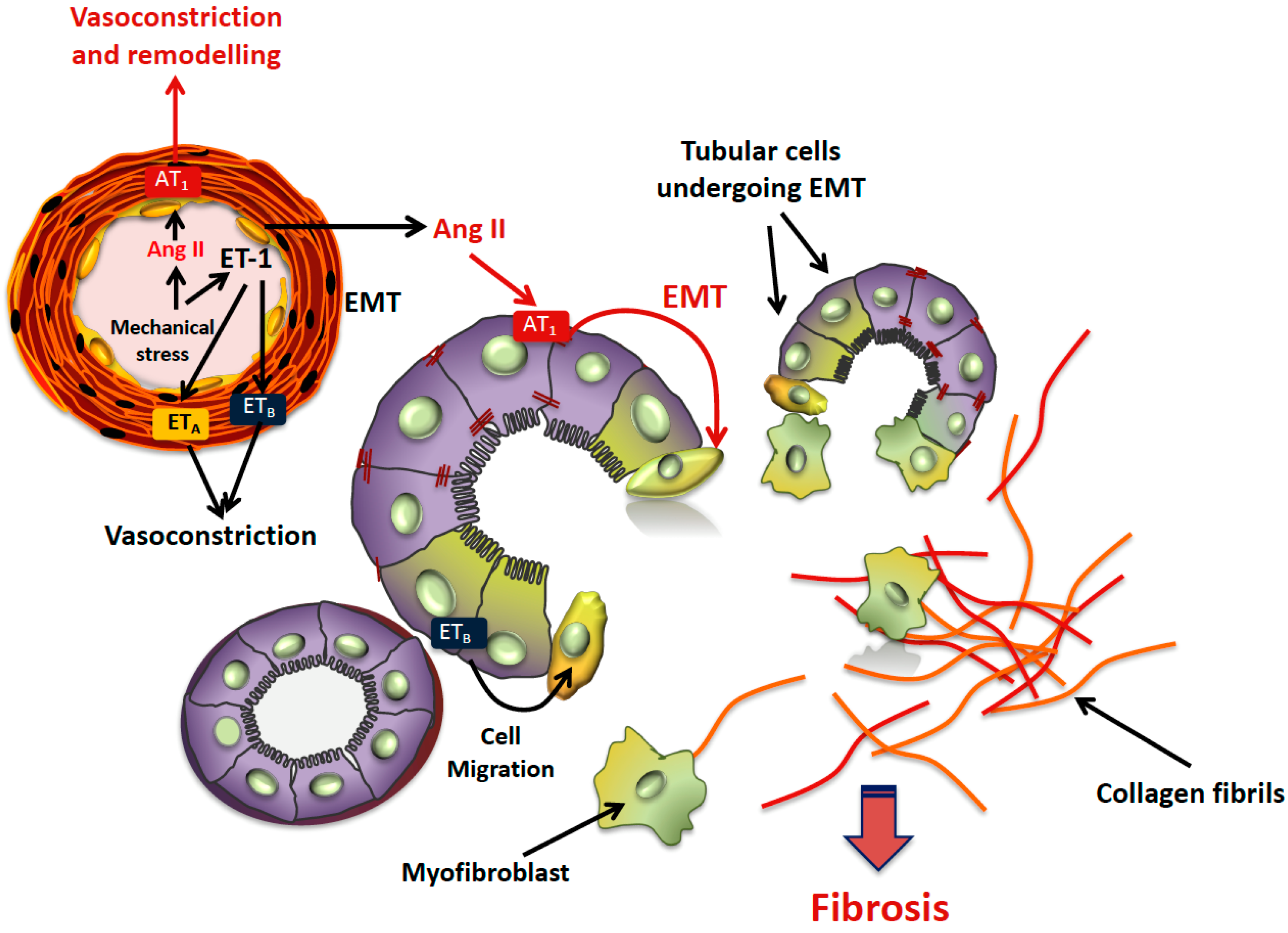
6. EMT in Hypertensive Nephropathy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| αSMA | Alpha smooth muscle actin |
| Ang II | Angiotensin II |
| ACE | Angiotensin-converting enzyme |
| AT1 | Angiotensin II type 1 receptor |
| HT | Arterial hypertension |
| CKD | Chronic kidney disease |
| ET-1 | Endothelin-1 |
| ESRD | End-stage renal disease |
| EMT | Epithelial-to-mesenchymal transition |
| ERA-EDTA | European Renal Association—European Dialysis and Transplant Association |
| FSP-1 | Fibroblast-specific protein–1 |
| MET | Mesenchymal-to-epithelial transition |
| MMP | Metalloproteinases |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| RANTES | Regulated upon Activation Normal T-cell Expressed and Secreted |
| RAAS | Renin angiotensin aldosterone system |
| TGFβR-I | TGF-β receptors type I |
| TLRs | Toll-like receptors |
| TGFβ1 | Transforming growth factor β1 |
| TIF | Tubular-interstitial fibrosis |
References
- Kramer, A.; Pippias, M.; Noordzij, M.; Stel, V.S.; Afentakis, N.; Ambühl, P.M.; Andrusev, A.M.; Fuster, E.A.; Monzón, F.E.A.; Åsberg, A.; et al. The European Renal Association—European Dialysis and Transplant Association (ERA-EDTA) Registry Annual Report 2015: A summary. Clin. Kidney J. 2018, 11, 108–122. [Google Scholar] [CrossRef]
- Levey, A.S.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Rosei, E.A.; Azizi, M.; Burnier, M.; Clement, D.; Coca, A.; De Simone, G.; Dominiczak, A.; et al. 2018 Practice Guidelines for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension. J. Hypertens. 2018, 36, 2284–2309. [Google Scholar] [CrossRef] [PubMed]
- Udani, S.; Lazich, I.; Bakris, G.L. Epidemiology of hypertensive kidney disease. Nat. Rev. Nephrol. 2011, 7, 11–21. [Google Scholar] [CrossRef]
- Ritz, E. The Kidney: Both culprit and victim. Hypertension 2009, 54, 25–26. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hill, G.S.; Heudes, D.; Bariéty, J. Morphometric study of arterioles and glomeruli in the aging kidney suggests focal loss of autoregulation. Kidney Int. 2003, 63, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.S. Hypertensive nephrosclerosis. Curr. Opin. Nephrol. Hypertens. 2008, 17, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Seccia, T.M.; Maniero, C.; Belloni, A.S.; Guidolin, D.; Poulose, P.; Pessina, A.C.; Rossi, G.P. Role of angiotensin II, endothelin-1 and L-type calcium channel in the development of glomerular, tubulointerstitial and perivascular fibrosis. J. Hypertens. 2008, 26, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Seccia, T.M.; Belloni, A.S.; Guidolin, D.; Sticchi, D.; Nussdorfer, G.G.; Pessina, A.C.; Rossi, G.P. The renal antifibrotic effects of angiotensin-converting enzyme inhibition involve bradykinin B2 receptor activation in angiotensin II-dependent hypertension. J. Hypertens. 2006, 24, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Epithelial-mesenchymal transition in morphogenesis, cancer progression and angiogenesis. Exp. Cell Res. 2017, 353, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Seccia, T.M.; Caroccia, B.; Gioco, F.; Piazza, M.; Buccella, V.; Guidolin, D.; Guerzoni, E.; Montini, B.; Petrelli, L.; Pagnin, E.; et al. Endothelin-1 Drives Epithelial-Mesenchymal Transition in Hypertensive Nephroangiosclerosis. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, G.; Rodriguez-Vita, J.; Rodrigues-Díez, R.; Sánchez-López, E.; Rupérez, M.; Cartier, C.; Esteban, V.; Ortiz, A.; Egido, J.; Mezzano, S.A.; et al. Angiotensin II activates the Smad pathway during epithelial mesenchymal transdifferentiation. Kidney Int. 2008, 74, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Macconi, D.; Remuzzi, G.; Benigni, A. Key fibrogenic mediators: Old players. Renin-angiotensin system. Kidney Int. Suppl. 2014, 4, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Fine, L.G.; Ong, A.C.M.; Norman, J.T. Mechanisms of tubulo-interstitiaI injury in progressive renal diseases. Eur. J. Clin. Investig. 1993, 23, 259–265. [Google Scholar] [CrossRef]
- Fine, L.G.; Norman, J.T. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: From hypothesis to novel therapeutics. Kidney Int. 2008, 74, 867–872. [Google Scholar] [CrossRef]
- Choi, Y.J.; Chakraborty, S.; Nguyen, V.; Nguyen, C.; Kim, B.K.; Shim, S.I.; Suki, W.N.; Truong, L.D. Peritubular capillary loss is associated with chronic tubulointerstitial injury in human kidney: Altered expression of vascular endothelial growth factor. Hum. Pathol. 2000, 31, 1491–1497. [Google Scholar] [CrossRef]
- Gross, O.; Schulze-Lohoff, E.; Koepke, M.-L.; Beirowski, B.; Addicks, K.; Bloch, W.; Smyth, N.; Weber, M. Antifibrotic, nephroprotective potential of ACE inhibitors vs AT1 antagonist in a murine model of renal fibrosis. Nephrol. Dial. Transpl. 2004, 19, 1716–1723. [Google Scholar] [CrossRef]
- Hartner, A.; Porst, M.; Klanke, B.; Cordasic, N.; Hilgers, K.F.; Veelken, R. Angiotensin II formation in the kidney and nephrosclerosis in Ren-2 hypertensive rats. Nephrol. Dial. Transpl. 2006, 21, 1778–1785. [Google Scholar] [CrossRef]
- Kriz, W.; Kaissiing, B.; Le Hir, M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: Fact or fantasy? J. Clin. Investig. 2011, 121, 468–474. [Google Scholar] [CrossRef]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Galichon, P.; Finianos, S.; Hertig, A. EMT-MET in renal disease: Should we curb our enthusiasm? Cancer Lett. 2013, 341, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D.; Zuk, A. Transformations between epithelium and mesenchyme: Normal, pathological, and experimentally induced1. Am. J. Kidney Dis. 1995, 26, 678–690. [Google Scholar] [CrossRef]
- Gros, J.; Tabin, C.J. Vertebrate limb bud formation is initiated by localized epithelial-to-mesenchymal transition. Science 2014, 343, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Vainio, S.; Vassileva, G.; McMahon, A.P. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 1994, 372, 679–683. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Neilson, E.G. Mechanisms of Disease: Fibroblasts—A new look at an old problem. Nat. Clin. Pract. Nephrol. 2006, 2, 101–108. [Google Scholar] [CrossRef]
- Strutz, F.; Neilson, E.G. New insights into mechanisms of fibrosis in immune renal injury. Springer Semin. Immunopathol. 2003, 24, 459–476. [Google Scholar] [CrossRef]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.-C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; A De Frutos, C.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef]
- Banerjee, P.; Venkatachalam, S.; Mamidi, M.K.; Bhonde, R.; Shankar, K.; Pal, R. Vitiligo patient-derived keratinocytes exhibit characteristics of normal wound healing via epithelial to mesenchymal transition. Exp. Dermatol. 2015, 24, 391–393. [Google Scholar] [CrossRef]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial–Mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Saitoh, M.; Miyazawa, K. Transcriptional and Post-transcriptional Regulation in TGF–Mediated epithelial-mesenchymal transition. J. Biochem. 2012, 151, 563–571. [Google Scholar] [CrossRef]
- Griggs, L.A.; Hassan, N.T.; Malik, R.S.; Griffin, B.P.; Martinez, B.A.; Elmore, L.W.; Lemmon, C.A. Fibronectin fibrils regulate TGF-β1-induced Epithelial-Mesenchymal Transition. Matrix Biol. 2017, 60–61, 157–175. [Google Scholar] [CrossRef]
- Hewitson, T.D.; Holt, S.G.; Tan, S.J.; Wigg, B.; Samuel, C.S.; Smith, E.R. Epigenetic modifications to H3K9 in renal tubulointerstitial cells after unilateral ureteric obstruction and TGF-β1 stimulation. Front. Pharmacol. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef]
- Inoue, T.; Plieth, D.; Venkov, C.D.; Xu, C.; Neilson, E.G. Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int. 2005, 67, 2488–2493. [Google Scholar] [CrossRef]
- Le Hir, M.; Hegyi, I.; Cueni-Loffing, D.; Loffing, J.; Kaissling, B. Characterization of renal interstitial fibroblast-specific protein 1/S100A4-positive cells in healthy and inflamed rodent kidneys. Histochem. Cell Biol. 2005, 123, 335–346. [Google Scholar] [CrossRef]
- Zhu, M.Q.; De Broe, M.E.; Nouwen, E.J. Vimentin expression and distal tubular damage in the rat kidney. Exp. Nephrol. 1996, 4, 172–183. [Google Scholar]
- Witzgall, R.; Brown, D.; Schwarz, C.; Bonventre, J.V. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J. Clin. Investig. 1994, 93, 2175–2788. [Google Scholar] [CrossRef]
- Gröne, H.J.; Weber, K.; Gröne, E.; Helmchen, U.; Osborn, M. Coexpression of keratin and vimentin in damaged and regenerating tubular epithelia of the kidney. Am. J. Pathol. 1987, 129, 1–8. [Google Scholar]
- Skalli, O.; Pelte, M.F.; Peclet, M.C.; Gabbiani, G.; Gugliotta, P.; Bussolati, G.; Ravazzola, M.; Orci, L. Alpha-smooth muscle actin, a differentiation marker of smooth muscle cells, is present in microfilamentous bundles of pericytes. J. Histochem. Cytochem. 1989, 37, 315–321. [Google Scholar] [CrossRef]
- Gabbiani, G.; Schmid, E.; Winter, S.; Chaponnier, C.; De Ckhastonay, C.; Vandekerckhove, J.; Weber, K.; Franke, W.W. Vascular smooth muscle cells differ from other smooth muscle cells: Predominance of vimentin filaments and a specific alpha-type actin. Proc. Natl. Acad. Sci. USA 1981, 78, 298–302. [Google Scholar] [CrossRef]
- Cheng, S.; Pollock, A.S.; Mahimkar, R.; Olson, J.L.; Lovett, D.H. Matrix metalloproteinase 2 and basement membrane integrity: A unifying mechanism for progressive renal injury. FASEB J. 2006, 20, 1898–1900. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Y. Dissection of Key Events in Tubular Epithelial to Myofibroblast Transition and Its Implications in Renal Interstitial Fibrosis. Am. J. Pathol. 2001, 159, 1465–1475. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Lin, S.-L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef]
- Faulkner, J.L.; Szcykalski, L.M.; Springer, F.; Barnes, J.L. Origin of interstitial fibroblasts in an accelerated model of angiotensin II-induced renal fibrosis. Am. J. Pathol. 2005, 167, 1193–1205. [Google Scholar] [CrossRef]
- Zhu, C.; Mertens, P.R. Epithelial–Mesenchymal transition to be or not to be? Is the answer yes and no at the same time? Int. Urol. Nephrol. 2010, 42, 843–846. [Google Scholar] [CrossRef]
- Huang, S.; Susztak, K. Epithelial Plasticity versus EMT in Kidney Fibrosis. Trends Mol. Med. 2016, 22, 4–6. [Google Scholar] [CrossRef]
- Chen, L.; Liu, B.-C.; Zhang, X.-L.; Zhang, J.-D.; Liu, H.; Li, M.-X. Influence of connective tissue growth factor antisense oligonucleotide on angiotensin II-induced epithelial mesenchymal transition in HK2 cells. Acta Pharmacol. Sin. 2006, 27, 1029–1036. [Google Scholar] [CrossRef]
- Burns, W.C.; Thomas, M.C. Angiotensin II and its role in tubular epithelial to mesenchymal transition associated with chronic kidney disease. Cells Tissues Organs 2010, 193, 74–84. [Google Scholar] [CrossRef]
- Rodrigues-Díez, R.; Carvajal-González, G.; Sánchez-López, E.; Rodríguez-Vita, J.; Rodrigues Díez, R.; Selgas, R.; Ortiz, A.; Egido, J.; Mezzano, S.; Ruiz-Ortega, M. Pharmacological modulation of epithelial mesenchymal transition caused by angiotensin II. Role of ROCK and MAPK pathways. Pharm. Res. 2008, 25, 2447–2461. [Google Scholar] [CrossRef]
- Yang, F.; Huang, X.R.; Chung, A.C.; Hou, C.-C.; Lai, K.N.; Lan, H.Y. Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J. Pathol. 2010, 221, 390–401. [Google Scholar] [CrossRef]
- Kellner, D.; Chen, J.; Richardson, I.; Seshan, S.V.; El Chaar, M.; Vaughan, E.; Poppas, D.; Felsen, D. Angiotensin Receptor Blockade Decreases Fibrosis and Fibroblast Expression in a Rat Model of Unilateral Ureteral Obstruction. J. Urol. 2006, 176, 806–812. [Google Scholar] [CrossRef]
- Hu, H.; Hu, S.; Xu, S.; Gao, Y.; Zeng, F.; Shui, H. miR-29b regulates Ang II-induced EMT of rat renal tubular epithelial cells via targeting PI3K/AKT signaling pathway. Int. J. Mol. Med. 2018, 42, 453–460. [Google Scholar] [CrossRef]


{kind=link}
| Kew Word | Operator | Kew Word | Operator | Kew Word |
|---|---|---|---|---|
| Hypertension | OR | High blood pressure | ||
| AND | ||||
| Nephropathy | OR | Tubular damage | OR | Tubulointerstitial fibrosis |
| AND | ||||
| Epithelial-to-mesenchymal transition | OR | EMT | OR | Epithelial-to-mesenchymal transdifferentiation |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seccia, T.; Caroccia, B.; Piazza, M.; Rossi, G.P. The Key Role of Epithelial to Mesenchymal Transition (EMT) in Hypertensive Kidney Disease. Int. J. Mol. Sci. 2019, 20, 3567. https://doi.org/10.3390/ijms20143567
Seccia T, Caroccia B, Piazza M, Rossi GP. The Key Role of Epithelial to Mesenchymal Transition (EMT) in Hypertensive Kidney Disease. International Journal of Molecular Sciences. 2019; 20(14):3567. https://doi.org/10.3390/ijms20143567
Chicago/Turabian StyleSeccia, Teresa, Brasilina Caroccia, Maria Piazza, and Gian Paolo Rossi. 2019. "The Key Role of Epithelial to Mesenchymal Transition (EMT) in Hypertensive Kidney Disease" International Journal of Molecular Sciences 20, no. 14: 3567. https://doi.org/10.3390/ijms20143567
APA StyleSeccia, T., Caroccia, B., Piazza, M., & Rossi, G. P. (2019). The Key Role of Epithelial to Mesenchymal Transition (EMT) in Hypertensive Kidney Disease. International Journal of Molecular Sciences, 20(14), 3567. https://doi.org/10.3390/ijms20143567