Molecular Docking and Molecular Dynamics Studies on Selective Synthesis of α-Amyrin and β-Amyrin by Oxidosqualene Cyclases from Ilex Asprella

 and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Sequence Analysis
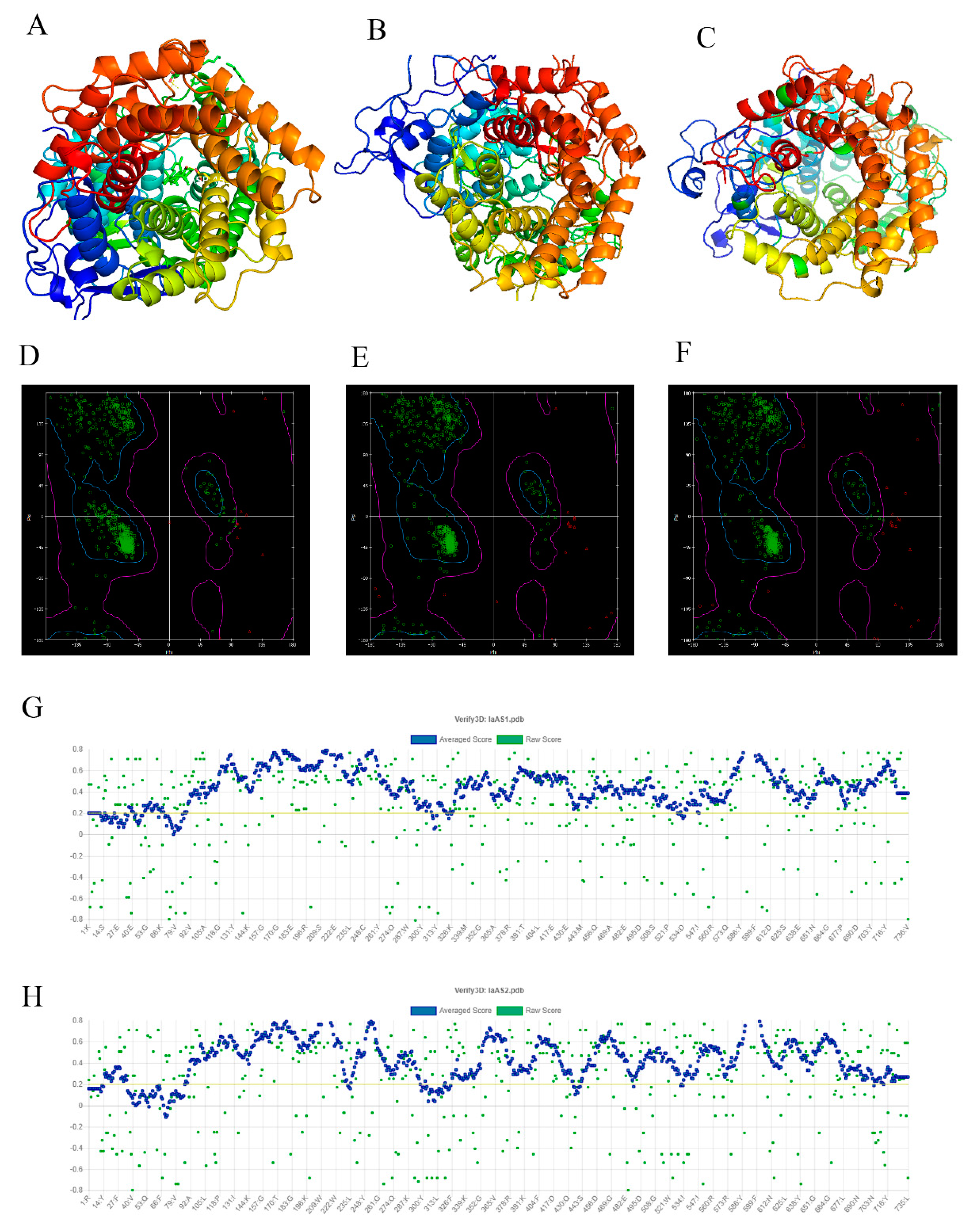
2.2. Verification and Evaluation of the Modeling Structures
2.3. Molecular Docking
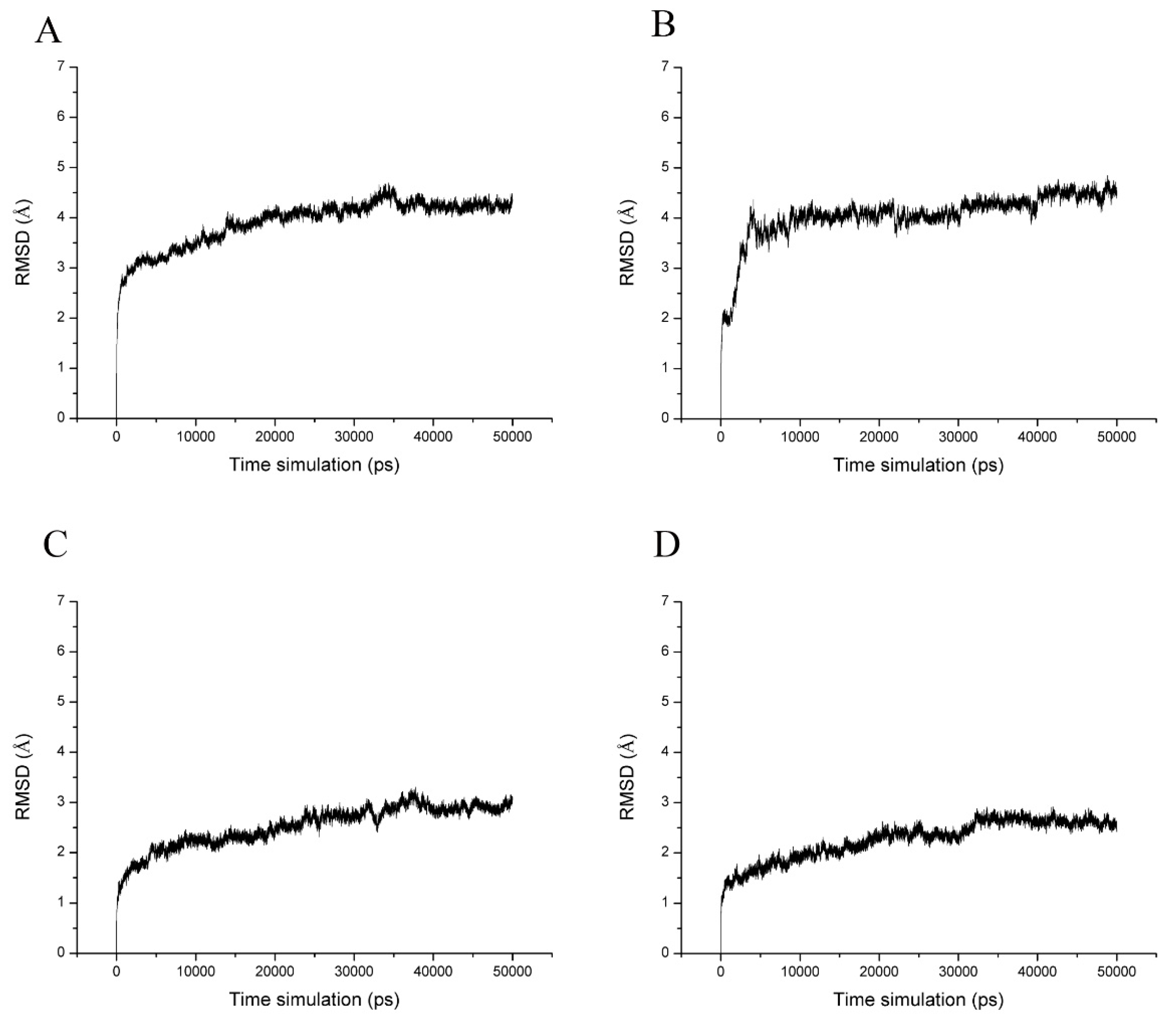
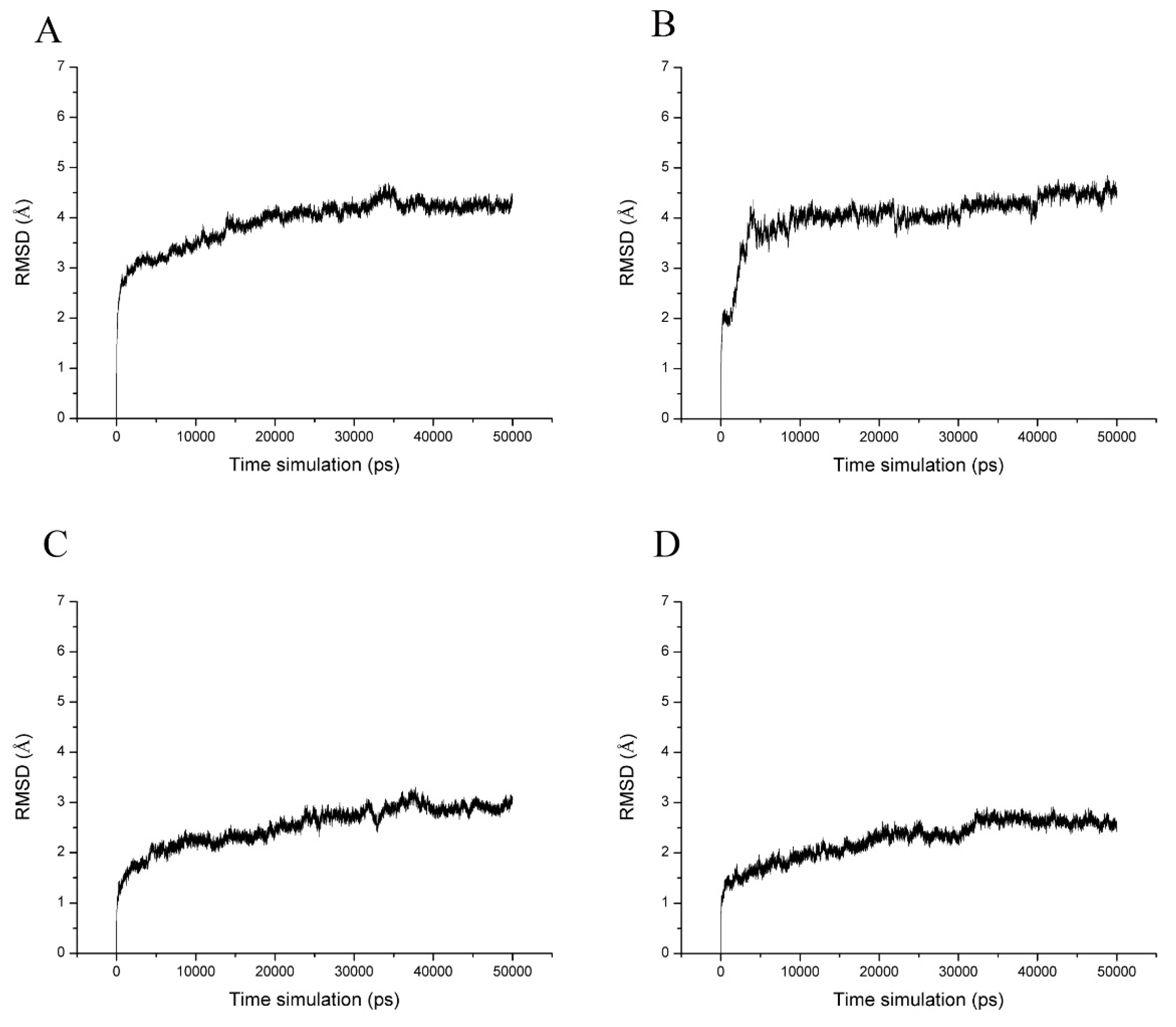
2.4. Molecular Dynamics Simulations and Calculation of Binding Free Energy
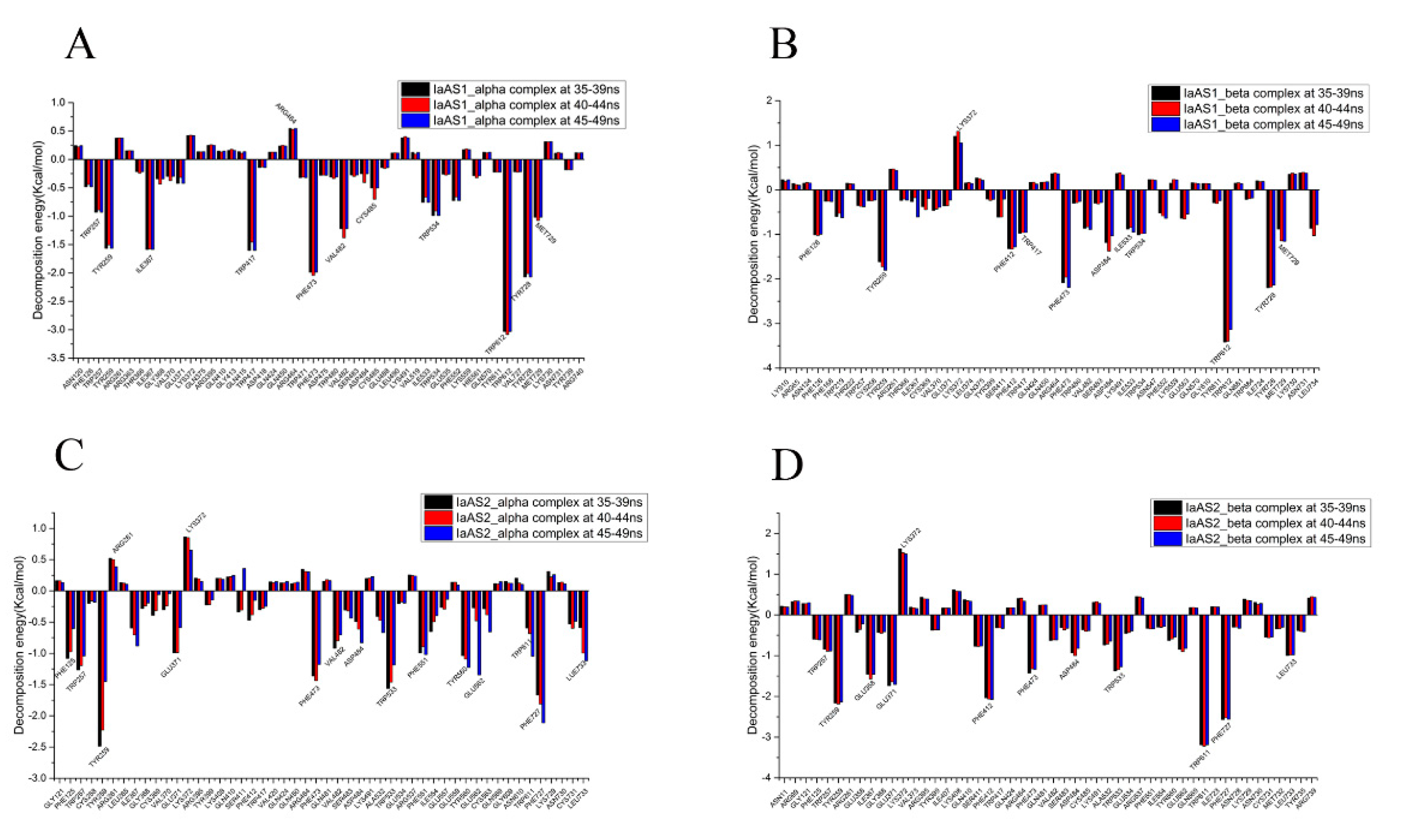
2.5. Decomposition of Binding Free Energy
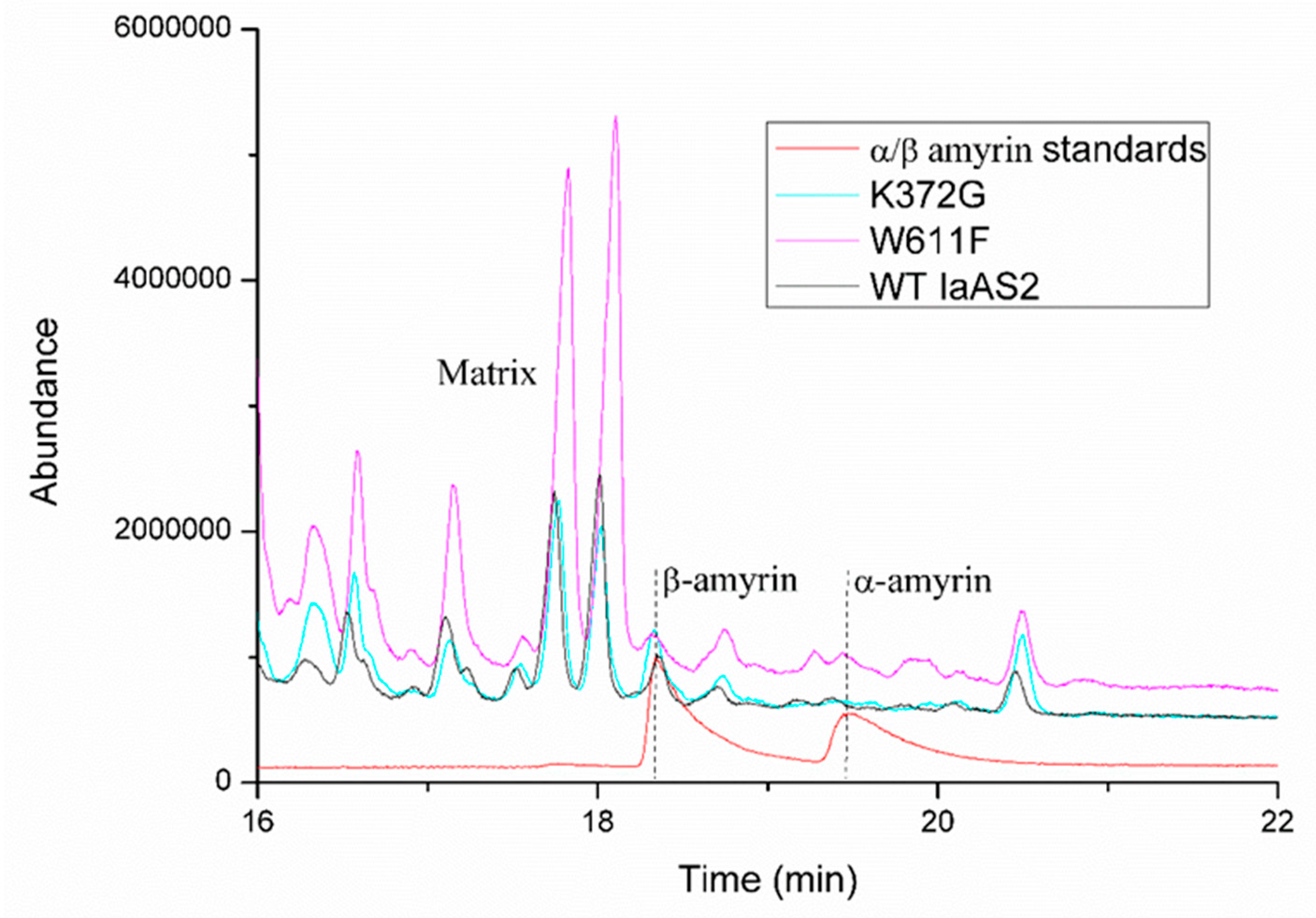
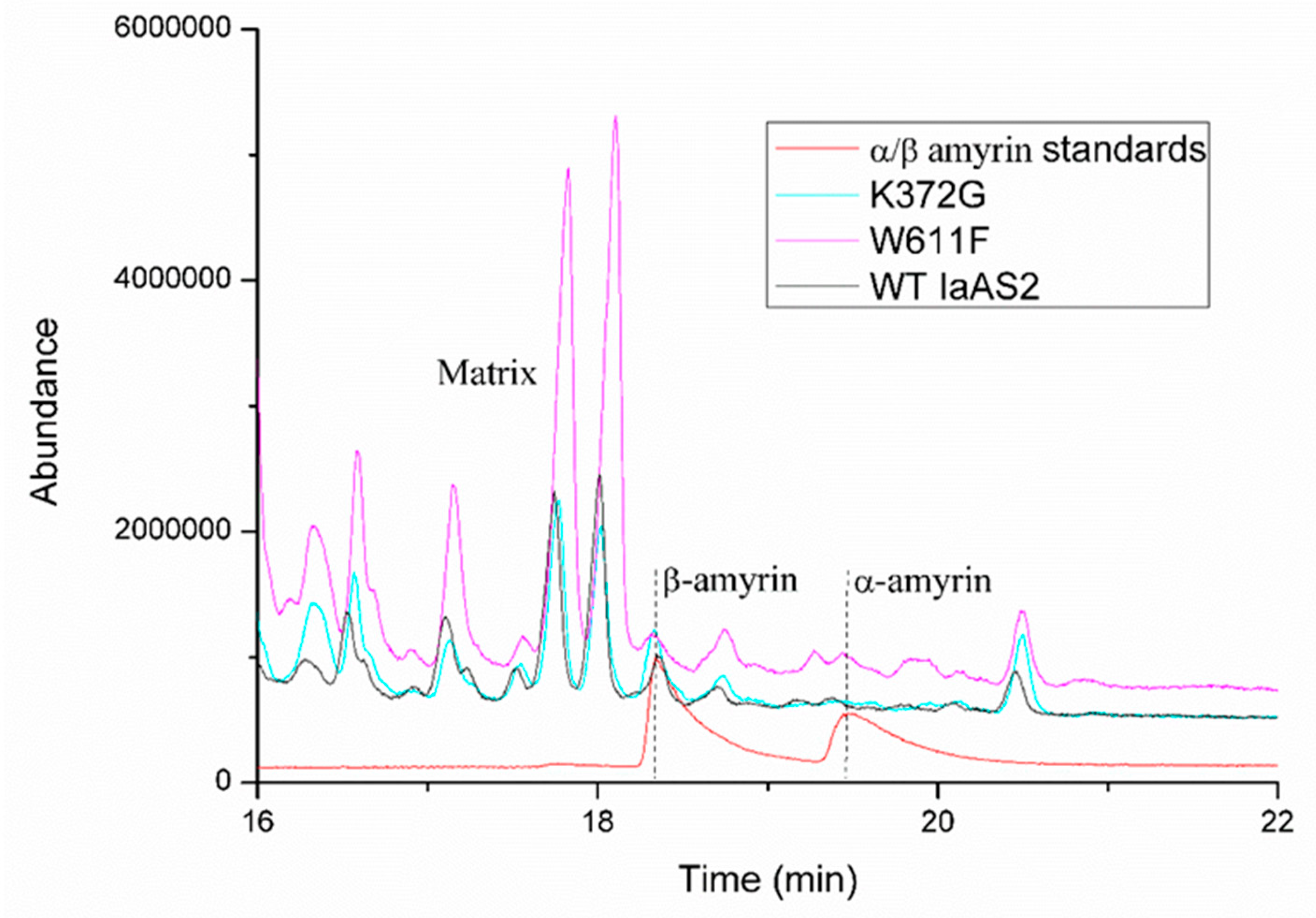
2.6. Quantification of Products Generated by Mutated IaAS1 and IaAS2
3. Materials and Methods
3.1. Homology Modeling to Construct Structures of IaAS1 and IaAS2
3.2. Molecular Docking
3.3. Molecular Dynamics Simulations
3.4. Calculation and Decomposition of Free Energy
3.5. Site-Directed Mutagenesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | amino acid |
| ADT | AutoDocktools v1.5.6 |
| DOPE | Discrete Optimized Protein Energy |
| DS | Discovery Studio v2.5 |
| IaAS1 | amyrin synthase 1 of Ilex asprella |
| IaAS1_alpha | complex of α-amyrin and IaAS1 |
| IaAS1_beta | complex of β-amyrin and IaAS1 |
| IaAS2 | amyrin synthase 2 of Ilex asprella |
| IaAS2_alpha | complex of α-amyrin and IaAS2 |
| IaAS2_beta | complex of β-amyrin and IaAS2 |
| iPCR | inverse PCR |
| LGA | Lamarck’s genetic algorithm |
| MD | molecular dynamics |
| MM/GBSA | Mechanics/Generalized Born Surface Area |
| MM/PBSA | Molecular Mechanics/Poisson Boltzmann Surface Area |
| OSC | Oxidosqualene cyclase |
| OS | 2,3-oxidosqualene |
| Probability density function | |
| SC-U | synthetic complete medium without uracil |
| RMSD | root mean square deviation |
| SHC | squalene-hopene cyclase |
References
- Suzuki, H.; Achnine, L.; Xu, R.; Matsuda, S.P.; Dixon, R.A. A genomics approach to the early stages of triterpene saponin biosynthesis in Medicago truncatula. Plant J. 2002, 32, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Basyuni, M.; Oku, H.; Tsujimoto, E.; Kinjo, K.; Baba, S.; Takara, K. Triterpene synthases from the Okinawan mangrove tribe, Rhizophoraceae. FEBS J. 2007, 274, 5028–5042. [Google Scholar] [CrossRef] [PubMed]
- Abe, I. Enzymatic synthesis of cyclic triterpenes. Nat. Prod. Rep. 2007, 24, 1311–1331. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, J.; Shibuya, M.; Masuda, K.; Ebizuka, Y. Dammaradiene synthase, a squalene cyclase, from Dryopteris crassirhizoma Nakai. Phytochemistry 2008, 69, 2559–2564. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Katsube, Y.; Otsuka, M.; Zhang, H.; Tansakul, P.; Xiang, T.; Ebizuka, Y. Identification of a product specific β-amyrin synthase from Arabidopsis thaliana. Plant Physiol. Biochem. 2009, 47, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cai, Y.; Zhao, Z.; Wang, J.; Li, J.; Xin, W.; Xia, G.; Xiang, F. Cloning and Functional Analysis of a β-amyrin synthase gene associated with oleanolic acid biosynthesis in Gentiana straminea MAXIM. Biol. Pharm. Bull. 2009, 32, 818–824. [Google Scholar] [CrossRef]
- Jin, M.M.; Zhang, W.D.; Jiang, H.H.; Du, Y.F.; Guo, W.; Cao, L.; Xu, H.J. UPLC-Q-TOF-MS/MS-guided dereplication of Pulsatilla chinensis to identify triterpenoid saponins. Phytochem. Anal. 2018, 29, 516–527. [Google Scholar] [CrossRef]
- Nelson, A.T.; Camelio, A.M.; Claussen, K.R.; Cho, J.; Tremmel, L.; DiGiovanni, J.; Siegel, D. Synthesis of oxygenated oleanolic and ursolic acid derivatives with anti-inflammatory properties. Bioorg. Med. Chem. Lett. 2015, 25, 4342–4346. [Google Scholar] [CrossRef] [Green Version]
- Poralla, K.; Hewelt, A.; Prestwich, G.D.; Abe, I.; Reipen, I.; Sprenger, G. A specific amino acid repeat in squalene and oxidosqualene cyclases. Trends Biochem. Sci. 1994, 19, 157–158. [Google Scholar] [CrossRef]
- Abe, I.; Prestwich, G.D. Molecular cloning, characterization, and functional expression of rat oxidosqualene cyclase cDNA. Proc. Natl. Acad. Sci. USA 1995, 92, 9274–9278. [Google Scholar] [CrossRef]
- Dang, T.; Prestwich, G.D. Site-directed mutagenesis of squalene-hopene cyclase: altered substrate specificity and product distribution. Chem. Biol. 2000, 7, 643–649. [Google Scholar] [CrossRef]
- Morita, M.; Shibuya, M.; Kushiro, T.; Masuda, K.; Ebizuka, Y. Molecular cloning and functional expression of triterpene synthases from pea (Pisum sativum) - New alpha-amyrin-producing enzyme is a multifunctional triterpene synthase. Eur. J. Biochem. 2000, 267, 3453–3460. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Fazio, G.C.; Matsuda, S.P. On the origins of triterpenoid skeletal diversity. Phytochemistry 2004, 65, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.R.; Rasbery, J.M.; Bartel, B.; Matsuda, S.P. Biosynthetic diversity in plant triterpene cyclization. Curr. Opin. Plant Biol. 2006, 9, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, J.; Ye, H.; Li, C.; Wang, H.; Liu, B.; Zhang, Y. Molecular characterization of the pentacyclic triterpenoid biosynthetic pathway in Catharanthus roseus. Planta 2012, 236, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Luo, X.; Ye, G.; Chen, Y.; Ji, X.; Wen, L.; Xu, Y.; Xu, H.; Zhan, R.; Chen, W. Characterisation of two oxidosqualene cyclases responsible for triterpenoid biosynthesis in Ilex asprella. Int. J. Mol. Sci. 2015, 16, 3564–3578. [Google Scholar] [CrossRef]
- Basyuni, M.; Oku, H.; Inafuku, M.; Baba, S.; Iwasaki, H.; Oshiro, K.; Okabe, T.; Shibuya, M.; Ebizuka, Y. Molecular cloning and functional expression of a multifunctional triterpene synthase cDNA from a mangrove species Kandelia candel (L.) Druce. Phytochemistry 2006, 67, 2517–2524. [Google Scholar] [CrossRef]
- Salmon, M.; Thimmappa, R.B.; Minto, R.E.; Melton, R.E.; Hughes, R.K.; O’Maille, P.E.; Hemmings, A.M.; Osbourn, A. A conserved amino acid residue critical for product and substrate specificity in plant triterpene synthases. Proc. Natl. Acad. Sci. USA 2016, 113, 4407–4414. [Google Scholar] [CrossRef]
- Wu, T.K.; Griffin, J.H. Conversion of a plant oxidosqualene-cycloartenol synthase to an oxidosqualene-lanosterol cyclase by random mutagenesis. Biochemistry 2002, 41, 8238–8244. [Google Scholar] [CrossRef]
- Takase, S.; Saga, Y.; Kurihara, N.; Naraki, S.; Kuze, K.; Nakata, G.; Araki, T.; Kushiro, T. Control of the 1,2-rearrangement process by oxidosqualene cyclases during triterpene biosynthesis. Org. Biomol. Chem. 2015, 13, 7331–7336. [Google Scholar] [CrossRef]
- Rahier, A.; Karst, F. Plant cyclopropylsterol-cycloisomerase: key amino acids affecting activity and substrate specificity. Biochem. J. 2014, 459, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, M. Investigation of Triterpene Biosynthesis in Arabidopsis thaliana. Ph.D. Thesis, Rice University, Houston, TX, USA, 2008. Available online: https://scholarship.rice.edu/handle/1911/61869 (accessed on 16 February 2017).
- Abe, I.; Naito, K.; Takagi, Y.; Noguchi, H. Molecular cloning, expression, and site-directed mutations of oxidosqualene cyclase from Cephalosporium caerulens. Biochim. Biophys. Acta 2001, 1522, 67–73. [Google Scholar] [CrossRef]
- Hoshino, T.; Nakagawa, K.; Aiba, Y.; Itoh, D.; Nakada, C.; Masukawa, Y. Euphorbia tirucalli β-Amyrin Synthase: Critical Roles of Steric Sizes at Val483 and Met729 and the CH-pi Interaction between Val483 and Trp534 for Catalytic Action. ChemBioChem 2017, 18, 2145–2155. [Google Scholar] [CrossRef] [PubMed]
- Aiba, Y.; Watanabe, T.; Terasawa, Y.; Nakano, C.; Hoshino, T. Strictly Conserved Residues in Euphorbia tirucalli β-Amyrin Cyclase: Trp612 Stabilizes Transient Cation through Cation-pi Interaction and CH-pi Interaction of Tyr736 with Leu734 Confers Robust Local Protein Architecture. ChemBioChem 2018, 19, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Masukawa, Y.; Nakada, C.; Amari, K.; Nakano, C.; Hoshino, T. β-Amyrin synthase from Euphorbia tirucalli. Steric bulk, not the pi-electrons of Phe, at position 474 has a key role in affording the correct folding of the substrate to complete the normal polycyclization cascade. Org. Biomol. Chem. 2014, 12, 3836–3846. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Masukawa, Y.; Hoshino, T. Purification, kinetics, inhibitors and CD for recombinant β-amyrin synthase from Euphorbia tirucalli L and functional analysis of the DCTA motif, which is highly conserved among oxidosqualene cyclases. FEBS J. 2013, 280, 1267–1280. [Google Scholar] [CrossRef]
- Kushiro, T.; Shibuya, M.; Masuda, K.; Ebizuka, Y. Mutational Studies on Triterpene Synthases: Engineering Lupeol Synthase into β-Amyrin Synthase. J. Am. Chem. Soc. 2000, 122, 6816–6824. [Google Scholar] [CrossRef]
- Chang, C.H.; Chen, Y.C.; Tseng, S.W.; Liu, Y.T.; Wen, H.Y.; Li, W.H.; Huang, C.Y.; Ko, C.Y.; Wang, T.T.; Wu, T.K. The cysteine 703 to isoleucine or histidine mutation of the oxidosqualene-lanosterol cyclase from Saccharomyces cerevisiae generates an iridal-type triterpenoid. Biochimie 2012, 94, 2376–2381. [Google Scholar] [CrossRef]
- Wu, T.K.; Li, W.H.; Chang, C.H.; Wen, H.Y.; Liu, Y.T.; Chang, Y.C. Differential Stereocontrolled Formation of Tricyclic Triterpenes by Mutation of Tyrosine 99 of the Oxidosqualene-Lanosterol Cyclase from Saccharomyces cerevisiae. Eur. J. Org. Chem. 2009, 2009, 5731–5737. [Google Scholar] [CrossRef]
- Wu, T.K.; Chang, C.H. Enzymatic formation of multiple triterpenes by mutation of tyrosine 510 of the oxidosqualene-lanosterol cyclase from Saccharomyces cerevisiae. ChemBioChem 2004, 5, 1712–1715. [Google Scholar] [CrossRef]
- Wu, T.K.; Chang, Y.C.; Liu, Y.T.; Chang, C.H.; Wen, H.Y.; Li, W.H.; Shie, W.S. Mutation of isoleucine 705 of the oxidosqualene-lanosterol cyclase from Saccharomyces cerevisiae affects lanosterol’s C/D-ring cyclization and 17alpha/beta-exocyclic side chain stereochemistry. Org. Biomol. Chem. 2011, 9, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.M.; Segura, M.J.; Wilson, W.K.; Matsuda, S.P. Oxidosqualene Cyclase Residues that Promote Formation of Cycloartenol, Lanosterol, and Parkeol. Angew. Chem. Int. Ed. Engl. 2000, 39, 4090–4092. [Google Scholar] [CrossRef]
- Chang, C.H.; Wen, H.Y.; Shie, W.S.; Lu, C.T.; Li, M.E.; Liu, Y.T.; Li, W.H.; Wu, T.K. Protein engineering of oxidosqualene-lanosterol cyclase into triterpene monocyclase. Org. Biomol. Chem. 2013, 11, 4214–4219. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T.; Hu, T.C.; Chang, C.H.; Shie, W.S.; Wu, T.K. Protein engineering of Saccharomyces cerevisiae oxidosqualene-lanosterol cyclase into parkeol synthase. Org. Lett. 2012, 14, 5222–5225. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.K.; Wen, H.Y.; Chang, C.H.; Liu, Y.T. Protein plasticity: A single amino acid substitution in the Saccharomyces cerevisiae oxidosqualene-lanosterol cyclase generates protosta-13(17),24-dien-3beta-ol, a rearrangement product. Org. Lett. 2008, 10, 2529–2532. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.K.; Liu, Y.T.; Chang, C.H.; Yu, M.T.; Wang, H.J. Site-saturated mutagenesis of histidine 234 of Saccharomyces cerevisiae oxidosqualene-lanosterol cyclase demonstrates dual functions in cyclization and rearrangement reactions. J. Am. Chem. Soc. 2006, 128, 6414–6419. [Google Scholar] [CrossRef]
- Lenhart, A.; Reinert, D.J.; Aebi, J.D.; Dehmlow, H.; Morand, O.H.; Schulz, G.E. Binding structures and potencies of oxidosqualene cyclase inhibitors with the homologous squalene-hopene cyclase. J. Med. Chem. 2003, 46, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Reinert, D.J.; Balliano, G.; Schulz, G.E. Conversion of squalene to the pentacarbocyclic hopene. Chem. Biol. 2004, 11, 121–126. [Google Scholar] [CrossRef]
- Lenhart, A.; Weihofen, W.A.; Pleschke, A.E.; Schulz, G.E. Crystal structure of a squalene cyclase in complex with the potential anticholesteremic drug Ro48-8071. Chem. Biol. 2002, 9, 639–645. [Google Scholar] [CrossRef]
- Thoma, R.; Schulz-Gasch, T.; D’Arcy, B.; Benz, J.; Aebi, J.; Dehmlow, H.; Hennig, M.; Stihle, M.; Ruf, A. Insight into steroid scaffold formation from the structure of human oxidosqualene cyclase. Nature 2004, 432, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Nakada, C.; Hoshino, T. β-Amyrin synthase from Euphorbia tirucalli L. functional analyses of the highly conserved aromatic residues Phe413, Tyr259 and Trp257 disclose the importance of the appropriate steric bulk, and cation-pi and CH-pi interactions for the efficient catalytic action of the polyolefin cyclization cascade. Org. Biomol. Chem. 2016, 15, 177–188. [Google Scholar] [PubMed]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowie, J.U.; Luthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham III, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, NC, USA, 2016; Available online: http://ambermd.org/doc12/Amber16.pdf (accessed on 15 May 2017).
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein-protein binding free energies and re-rank binding poses generated by protein-protein docking. Phys. Chem. Chem. Phys. 2016, 18, 22129–22139. [Google Scholar] [CrossRef]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Amir-Hassan, A.; Lee, V.S.; Baharuddin, A.; Othman, S.; Xu, Y.; Huang, M.; Yusof, R.; Rahman, N.A.; Othman, R. Conformational and energy evaluations of novel peptides binding to dengue virus envelope protein. J. Mol. Graph. Model. 2017, 74, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.J.; McLaughlin, W.; Lu, B.; Chen, K.; Wang, W. Prediction of binding affinities between the human amphiphysin-1 SH3 domain and its peptide ligands using homology modeling, molecular dynamics and molecular field analysis. J. Proteome Res. 2006, 5, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Li, N.; Li, Y.; Wang, W. Characterization of domain-peptide interaction interface: prediction of SH3 domain-mediated protein-protein interaction network in yeast by generic structure-based models. J. Proteome Res. 2012, 11, 2982–2995. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.S.; Wong, S.; Peters, J.M.; Almeida, R.; Qi, L.S. Targeted Transcriptional Repression in Bacteria Using CRISPR Interference (CRISPRi). Methods Mol. Biol. 2015, 1311, 349–362. [Google Scholar] [PubMed] [Green Version]
- Gietz, R.D.; Schiestl, R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Conserved Residues |
|---|---|
| 1W6K | LEU229 TRP230 CYS231 HIS232 CYS233 ASN382 GLY383 ASP455 CYS456 THR534 |
| ETAS | MET256 TRP257 CYS258 TYR259 CYS260 PHE413 GLY414 ASP485 CYS486 THR565 |
| IaAS1 | MET256 TRP257 CYS258 TYR259 CYS260 PHE412 GLY413 ASP484 CYS485 THR564 |
| IaAS2 | MET256 TRP257 CYS258 TYR259 CYS260 PHE412 GLY413 ASP484 CYS485 THR564 |
| Complex | MM/GBSA ΔGGBTOT (kcal/moL) | RMSD (mean ± SD) | |||||
|---|---|---|---|---|---|---|---|
| 25–29 ns | 30–34 ns | 35–39 ns | 40–44 ns | 45–49 ns | (mean ± SD) | ||
| IaAS1_alpha | −29.65 | −30.26 | −30.25 | −28.87 | −27.86 | −29.38 ± 1.02 | 4.21 ± 0.13 |
| IaAS1_beta | −26.06 | −25.44 | −24.40 | −25.01 | −24.60 | −25.10 ± 0.67 | 4.92 ± 0.19 |
| IaAS2_alpha | −21.29 | −21.74 | −21.14 | −22.41 | −23.16 | −21.95 ± 0.84 | 3.61 ± 0.19 |
| IaAS2_bet | −32.54 | −32.86 | −31.33 | −30.98 | −29.96 | −31.54 ± 1.18 | 3.34 ± 0.16 |
| Enzyme | Mutant | Primer | Sequence a (5′-3′) | Products Ratio |
|---|---|---|---|---|
| IaAS1 | K372G | K372G-F | GGGAGTTTGCAAATGATGT | No products |
| K372G-R | TTCTACACATCCTATAGTAATGTATCTGCTCT | |||
| W612F | W612F-F | TTCGGAATTTGCTTCCTCTATG | No products | |
| W612F-R | ATAACCATACCATGAACCATCAGG | |||
| IaAS2 | K372G | K372G-F | GGGGTACTATGTATGCTTGCTTG | β:α = 2.2:1 |
| K372G-R | TTCCACACATCCGATGGTG | |||
| W611F | W611F-F | TTCGGTGTGTGTTTCACATATG | β:α = 4.0:1 | |
| W611F-R | GTTTCCATACCATGAACCATCAGAC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Xu, H.; Wang, M.; Zhan, R.; Chen, W.; Zhang, R.; Kuang, Z.; Zhang, F.; Wang, K.; Gu, J. Molecular Docking and Molecular Dynamics Studies on Selective Synthesis of α-Amyrin and β-Amyrin by Oxidosqualene Cyclases from Ilex Asprella. Int. J. Mol. Sci. 2019, 20, 3469. https://doi.org/10.3390/ijms20143469
Wu Z, Xu H, Wang M, Zhan R, Chen W, Zhang R, Kuang Z, Zhang F, Wang K, Gu J. Molecular Docking and Molecular Dynamics Studies on Selective Synthesis of α-Amyrin and β-Amyrin by Oxidosqualene Cyclases from Ilex Asprella. International Journal of Molecular Sciences. 2019; 20(14):3469. https://doi.org/10.3390/ijms20143469
Chicago/Turabian StyleWu, Zhixue, Hui Xu, Meiling Wang, Ruoting Zhan, Weiwen Chen, Ren Zhang, Zaoyuan Kuang, Fengxue Zhang, Kui Wang, and Jiangyong Gu. 2019. "Molecular Docking and Molecular Dynamics Studies on Selective Synthesis of α-Amyrin and β-Amyrin by Oxidosqualene Cyclases from Ilex Asprella" International Journal of Molecular Sciences 20, no. 14: 3469. https://doi.org/10.3390/ijms20143469