Unraveling the Role of Inflammation in the Pathogenesis of Diabetic Kidney Disease

 , ,
, ,
Abstract
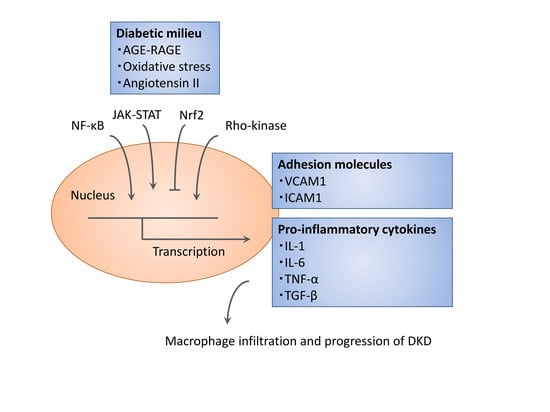
:
{kind=link}
{kind=link}
1. Introduction
2. Inflammatory Mediators in Diabetic Kidney Disease (DKD)
2.1. Macrophages
2.2. Adhesion Molecules and Chemokines
3. Signaling Cascade Governing Inflammatory Reactions
3.1. Nuclear Factor κB (NF-κB) and Activating Protein 1 (AP1)
3.2. Janus Kinase/Signal Transducer and Activator of Transcription (JAK-STAT)
3.3. Nuclear Factor-2 Erythroid Related Factor (Nrf2)-Keep1
3.4. Rho-Kinase Signaling
4. Targeting Renal Inflammation for Prophylaxis and Therapy
4.1. Sodium-Glucose Co-Transporter-2 (SGLT2) Inhibitors
4.2. Glucagon-Like Peptide 1 (GLP-1) Receptor Agonists and Dipeptidyl Peptidase 4 (DPP-4) Inhibitors
4.3. Metformin
4.4. Immunosuppressive Agents
5. Conclusions and Future Perspectives
Author Contributions
Acknowledgements
Conflicts of Interest
References
- Adler, A.I.; Stevens, R.J.; Manley, S.E.; Bilous, R.W.; Cull, C.A.; Holman, R.R.; UKPDS GROUP. Development and progression of nephropathy in type 2 diabetes: The United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int. 2003, 63, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldfine, A.B.; Shoelson, S.E. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Invest. 2017, 127, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Simo-Servat, O.; Simo, R.; Hernandez, C. Circulating Biomarkers of Diabetic Retinopathy: An Overview Based on Physiopathology. J. Diabetes Res. 2016, 2016, 5263798. [Google Scholar] [CrossRef] [PubMed]
- Paeschke, S.; Paeschke, S.; Baum, P.; Toyka, K.V.; Blüher, M.; Koj, S.; Klöting, N.; Bechmann, I.; Thiery, J.; Kosacka, J.; et al. The Role of Iron and Nerve Inflammation in Diabetes Mellitus Type 2-Induced Peripheral Neuropathy. Neuroscience 2019, 406, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lin, X.; Guan, M.; Zeng, Y.; Liu, Y. Verapamil Attenuated Prediabetic Neuropathy in High-Fat Diet-Fed Mice through Inhibiting TXNIP-Mediated Apoptosis and Inflammation. Oxid Med. Cell Longev. 2019, 2019, 1896041. [Google Scholar] [CrossRef]
- Doupis, J.; Lyons, T.E.; Wu, S.; Gnardellis, C.; Dinh, T.; Veves, A. Microvascular reactivity and inflammatory cytokines in painful and painless peripheral diabetic neuropathy. J. Clin. Endocrinol Metab. 2009, 94, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Pop-Busui, R.; Ang, L.; Holmes, C.; Gallagher, K.; Feldman, E.L. Inflammation as a Therapeutic Target for Diabetic Neuropathies. Curr. Diab. Rep. 2016, 16, 29. [Google Scholar] [CrossRef]
- Ganesh Yerra, V.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-kappaB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef]
- Tian, S.; Chen, S.Y. Macrophage polarization in kidney diseases. Macrophage (Houst) 2015, 2, e679. [Google Scholar]
- Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Rabelink, T.J.; Bajema, I.M.; IJpelaar, D.H.T. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol. Dial. Transpl. 2017, 32, 1322–1329. [Google Scholar] [CrossRef]
- Usui, H.K.; Shikata, K.; Sasaki, M.; Okada, S.; Matsuda, M.; Shikata, Y.; Ogawa, D.; Kido, Y.; Nagase, R.; Yozai, K.; et al. Macrophage scavenger receptor-a-deficient mice are resistant against diabetic nephropathy through amelioration of microinflammation. Diabetes 2007, 56, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Landis, R.C.; Quimby, K.R.; Greenidge, A.R. M1/M2 Macrophages in Diabetic Nephropathy: Nrf2/HO-1 as Therapeutic Targets. Curr. Pharm. Des. 2018, 24, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Tobias, P.; Kasinath, B.S.; Ramsamooj, R.; Afify, A.; Jialal, I. Knockout of toll-like receptor-2 attenuates both the proinflammatory state of diabetes and incipient diabetic nephropathy. Arterioscler Thromb. Vasc. Biol. 2011, 31, 1796–1804. [Google Scholar] [CrossRef] [PubMed]
- Clausen, P.; Jacobsen, P.; Rossing, K.; Jensen, J.S.; Parving, H.H.; Feldt-Rasmussen, B. Plasma concentrations of VCAM-1 and ICAM-1 are elevated in patients with Type 1 diabetes mellitus with microalbuminuria and overt nephropathy. Diabet Med. 2000, 17, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Hojs, R.; Ekart, R.; Bevc, S.; Hojs, N. Markers of Inflammation and Oxidative Stress in the Development and Progression of Renal Disease in Diabetic Patients. Nephron 2016, 133, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Yeoh, L.Y.; Sum, C.F.; Tavintharan, S.; Ng, X.W.; Liu, S.; Lee, S.B.; Tang, W.E.; Lim, S.C.; SMART2D study. Vascular cell adhesion molecule-1, but not intercellular adhesion molecule-1, is associated with diabetic kidney disease in Asians with type 2 diabetes. J. Diabetes Complicat. 2015, 29, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.K.; Tesch, G.H. Inflammation in diabetic nephropathy. Mediators Inflamm. 2012, 2012, 146154. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Shikata, K.; Matsuda, M.; Ogawa, D.; Usui, H.; Kido, Y.; Nagase, R.; Wada, J.; Shikata, Y.; Makino, H. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes 2003, 52, 2586–2593. [Google Scholar] [CrossRef]
- Nadkarni, G.N.; Rao, V.; Ismail-Beigi, F.; Fonseca, V.A.; Shah, S.V.; Simonson, M.S.; Cantley, L.; Devarajan, P.; Parikh, C.R.; Coca, S.G. Association of Urinary Biomarkers of Inflammation, Injury, and Fibrosis with Renal Function Decline: The ACCORD Trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 1343–1352. [Google Scholar] [CrossRef] [Green Version]
- Satirapoj, B.; Dispan, R.; Radinahamed, P.; Kitiyakara, C. Urinary epidermal growth factor, monocyte chemoattractant protein-1 or their ratio as predictors for rapid loss of renal function in type 2 diabetic patients with diabetic kidney disease. BMC Nephrol. 2018, 19, 246. [Google Scholar] [CrossRef]
- Niewczas, M.A.; Gohda, T.; Skupien, J.; Smiles, A.M.; Walker, W.H.; Rosetti, F.; Cullere, X.; Eckfeldt, J.H.; Doria, A.; Mayadas, T.N.; et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J. Am. Soc. Nephrol. 2012, 23, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Niewczas, M.A.; Pavkov, M.E.; Skupien, J.; Smiles, A.; Md Dom, Z.I.; Wilson, J.M.; Park, J.; Nair, V.; Schlafly, A.; Saulnier, P.J.; et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med. 2019, 25, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Menne, J.; Eulberg, D.; Beyer, D.; Baumann, M.; Saudek, F.; Valkusz, Z.; Więcek, A.; Haller, H.; Emapticap Study Group. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol. Dial. Transplant. 2017, 32, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Lenda, D.M.; Kikawada, E.; Stanley, E.R.; Kelley, V.R. Reduced macrophage recruitment, proliferation, and activation in colony-stimulating factor-1-deficient mice results in decreased tubular apoptosis during renal inflammation. J. Immunol. 2003, 170, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Yokoyama, H.; Moore, K.J.; Dranoff, G.; Mulligan, R.C.; Kelley, V.R. Macrophage growth factors introduced into the kidney initiate renal injury. Mol. Med. 1996, 2, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Matoba, K.; Kawanami, D.; Tsukamoto, M.; Kinoshita, J.; Ito, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Murai, N.; Matsufuji, S.; et al. Rho-kinase regulation of TNF-alpha-induced nuclear translocation of NF-kappaB RelA/p65 and M-CSF expression via p38 MAPK in mesangial cells. Am. J. Physiol. Renal Physiol. 2014, 307, F571–F580. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.K.; Ma, F.Y.; Nikolic-Paterson, D.J.; Thomas, M.C.; Hurst, L.A.; Tesch, G.H. Antibody blockade of c-fms suppresses the progression of inflammation and injury in early diabetic nephropathy in obese db/db mice. Diabetologia 2009, 52, 1669–1679. [Google Scholar] [CrossRef] [Green Version]
- Pichler, R.; Afkarian, M.; Dieter, B.P.; Tuttle, K.R. Immunity and inflammation in diabetic kidney disease: Translating mechanisms to biomarkers and treatment targets. Am. J. Physiol. Renal Physiol. 2017, 312, F716–F731. [Google Scholar] [CrossRef]
- Sanajou, D.; Ghorbani Haghjo, A.; Argani, H.; Aslani, S. AGE-RAGE axis blockade in diabetic nephropathy: Current status and future directions. Eur. J. Pharmacol. 2018, 833, 158–164. [Google Scholar] [CrossRef]
- Pérez-Morales, R.E.; Del Pino, M.D.; Valdivielso, J.M.; Ortiz, A.; Mora-Fernández, C.; Navarro-González, J.F. Inflammation in Diabetic Kidney Disease. Nephron 2018, 1–5. [Google Scholar] [CrossRef]
- Toth-Manikowski, S.; Atta, M.G. Diabetic Kidney Disease: Pathophysiology and Therapeutic Targets. J. Diabetes Res. 2015, 2015, 697010. [Google Scholar] [CrossRef] [PubMed]
- Weigert, C.; Sauer, U.; Brodbeck, K.; Pfeiffer, A.; Häring, H.U.; Schleicher, E.D. AP-1 proteins mediate hyperglycemia-induced activation of the human TGF-beta1 promoter in mesangial cells. J. Am. Soc. Nephrol. 2000, 11, 2007–2016. [Google Scholar] [PubMed]
- Nicholas, S.B.; Liu, J.; Kim, J.; Ren, Y.; Collins, A.R.; Nguyen, L.; Hsueh, W.A. Critical role for osteopontin in diabetic nephropathy. Kidney Int. 2010, 77, 588–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, K.R.; Brosius III, F.C.; Adler, S.G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: Results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transplant. 2018, 33, 1950–1959. [Google Scholar] [CrossRef] [PubMed]
- Brosius, F.C.; Ju, W. The Promise of Systems Biology for Diabetic Kidney Disease. Adv. Chronic Kidney Dis. 2018, 25, 202–213. [Google Scholar] [CrossRef]
- Zhang, H.; Nair, V.; Saha, J.; Atkins, K.B.; Hodgin, J.B.; Saunders, T.L.; Myers, M.G.Jr.; Werner, T.; Kretzler, M.; Brosius, F.C. Podocyte-specific JAK2 overexpression worsens diabetic kidney disease in mice. Kidney Int. 2017, 92, 909–921. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Lazaro, I.; Lopez-Sanz, L.; Bernal, S.; Oguiza, A.; Recio, C.; Melgar, A.; Jimenez-Castilla, L.; Egido, J.; Madrigal-Matute, J.; Gomez-Guerrero, C. Nrf2 Activation Provides Atheroprotection in Diabetic Mice Through Concerted Upregulation of Antioxidant, Anti-inflammatory, and Autophagy Mechanisms. Front. Pharmacol. 2018, 9, 819. [Google Scholar] [CrossRef]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef]
- Matoba, K.; Kawanami, D.; Okada, R.; Tsukamoto, M.; Kinoshita, J.; Ito, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Murai, N.; et al. Rho-kinase inhibition prevents the progression of diabetic nephropathy by downregulating hypoxia-inducible factor 1alpha. Kidney Int. 2013, 84, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Matoba, K.; Utsunomiya, K. Signaling pathways in diabetic nephropathy. Histol. Histopathol. 2016, 31, 1059–1067. [Google Scholar] [PubMed]
- Kawanami, D.; Matoba, K.; Okada, R.; Tsukamoto, M.; Kinoshita, J.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. Fasudil inhibits ER stress-induced VCAM-1 expression by modulating unfolded protein response in endothelial cells. Biochem. Biophys. Res. Commun. 2013, 435, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Matoba, K.; Kanazawa, Y.; Ishizawa, S.; Yokota, T.; Utsunomiya, K. Thrombin induces MCP-1 expression through Rho-kinase and subsequent p38MAPK/NF-kappaB signaling pathway activation in vascular endothelial cells. Biochem. Biophys. Res. Commun. 2011, 411, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Rajagopalan, L.E. Rho kinase-2 activation in human endothelial cells drives lysophosphatidic acid-mediated expression of cell adhesion molecules via NF-kappaB p65. J. Biol. Chem. 2010, 285, 12536–12542. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Kuhnt-Moore, S.; Zeng, H.; Wu, J.S.; Moyer, M.P.; Pothoulakis, C. Neurotensin stimulates IL-8 expression in human colonic epithelial cells through Rho GTPase-mediated NF-kappa B pathways. Am. J. Physiol. Cell Physiol. 2003, 284, C1397–C1404. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Schwesinger, C.; Dehde, S.; von Ruffer, C.; Gatzemeier, S.; Klug, P.; Wenzel, U.O.; Stahl, R.A.; Thaiss, F.; Meyer, T.N. Rho kinase inhibition attenuates LPS-induced renal failure in mice in part by attenuation of NF-kappaB p65 signaling. Am. J. Physiol. Renal Physiol. 2009, 296, F1088–F1099. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Matoba, K.; Kawanami, D.; Nagai, Y.; Akamine, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. ROCK2 Regulates Monocyte Migration and Cell to Cell Adhesion in Vascular Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 1331. [Google Scholar] [CrossRef]
- Tanaka, T.; Nishimura, D.; Wu, R.C.; Amano, M.; Iso, T.; Kedes, L.; Nishida, H.; Kaibuchi, K.; Hamamori, Y. Nuclear Rho kinase, ROCK2, targets p300 acetyltransferase. J. Biol. Chem. 2006, 281, 15320–15329. [Google Scholar] [CrossRef]
- Thomas, M.C.; Cherney, D.Z.I. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 2018, 61, 2098–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanami, D.; Matoba, K.; Takeda, Y.; Nagai, Y.; Akamine, T.; Yokota, T.; Sango, K.; Utsunomiya, K. SGLT2 Inhibitors as a Therapeutic Option for Diabetic Nephropathy. Int. J. Mol. Sci. 2017, 18, 1083. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef]
- Garvey, W.T.; Van Gaal, L.; Leiter, L.A.; Vijapurkar, U.; List, J.; Cuddihy, R.; Ren, J.; Davies, M.J. Effects of canagliflozin versus glimepiride on adipokines and inflammatory biomarkers in type 2 diabetes. Metabolism 2018, 85, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Tahara, A.; Takasu, T.; Yokono, M.; Imamura, M.; Kurosaki, E. Characterization and comparison of SGLT2 inhibitors: Part 3. Effects on diabetic complications in type 2 diabetic mice. Eur. J. Pharmacol. 2017, 809, 163–171. [Google Scholar] [CrossRef]
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, SC.; Rieg, T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol. Renal Physiol. 2014, 306, F194–F204. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 Inhibition by Empagliflozin Promotes Fat Utilization and Browning and Attenuates Inflammation and Insulin Resistance by Polarizing M2 Macrophages in Diet-induced Obese Mice. EBioMedicine 2017, 20, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Maki, T.; Maeno, S.; Maeda, Y.; Yamato, M.; Sonoda, N.; Ogawa, Y.; Wakisaka, M.; Inoguchi, T. Amelioration of diabetic nephropathy by SGLT2 inhibitors independent of its glucose-lowering effect: A possible role of SGLT2 in mesangial cells. Sci. Rep. 2019, 9, 4703. [Google Scholar] [CrossRef]
- Muskiet, M.H.; Tonneijck, L.; Smits, M.M.; Kramer, M.H.; Diamant, M.; Joles, J.A.; van Raalte, D.H. Acute renal haemodynamic effects of glucagon-like peptide-1 receptor agonist exenatide in healthy overweight men. Diabetes Obes. Metab. 2016, 18, 178–185. [Google Scholar] [CrossRef]
- Kröller-Schön, S.; Knorr, M.; Hausding, M.; Oelze, M.; Schuff, A.; Schell, R.; Sudowe, S.; Scholz, A.; Daub, S.; Karbach, S.; et al. Glucose-independent improvement of vascular dysfunction in experimental sepsis by dipeptidyl-peptidase 4 inhibition. Cardiovasc Res. 2012, 96, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Kanasaki, K. The role of renal dipeptidyl peptidase-4 in kidney disease: Renal effects of dipeptidyl peptidase-4 inhibitors with a focus on linagliptin. Clin. Sci. 2018, 132, 489–507. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, M.; Rizzo, M.R.; Marfella, R.; Boccardi, V.; Esposito, A.; Pansini, A.; Paolisso, G. Decreased carotid atherosclerotic process by control of daily acute glucose fluctuations in diabetic patients treated by DPP-IV inhibitors. Atherosclerosis 2013, 227, 349–354. [Google Scholar] [CrossRef]
- Higashijima, Y.; Tanaka, T.; Yamaguchi, J.; Tanaka, S.; Nangaku, M. Anti-inflammatory role of DPP-4 inhibitors in a nondiabetic model of glomerular injury. Am. J. Physiol. Renal Physiol. 2015, 308, F878–F887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollock, C.; Stefánsson, B.; Reyner, D.; Rossing, P.; Sjöström, C.D.; Wheeler, D.C.; Langkilde, A.M.; Heerspink, H.J.L. Albuminuria-lowering effect of dapagliflozin alone and in combination with saxagliptin and effect of dapagliflozin and saxagliptin on glycaemic control in patients with type 2 diabetes and chronic kidney disease (DELIGHT): A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2019, 7, 429–441. [Google Scholar] [PubMed]
- Rosenstock, J.; Perkovic, V.; Johansen, O.E.; Cooper, M.E.; Kahn, S.E.; Marx, N.; Alexander, J.H.; Pencina, M.; Toto, R.D.; Wanner, C.; et al. Effect of Linagliptin vs Placebo on Major Cardiovascular Events in Adults With Type 2 Diabetes and High Cardiovascular and Renal Risk: The CARMELINA Randomized Clinical Trial. JAMA. 2019, 321, 69–79. [Google Scholar] [CrossRef]
- Kawanami, D.; Matoba, K.; Sango, K.; Utsunomiya, K. Incretin-Based Therapies for Diabetic Complications: Basic Mechanisms and Clinical Evidence. Int. J. Mol. Sci. 2016, 17, 1223. [Google Scholar] [CrossRef]
- Park, C.W.; Kim, H.W.; Ko, S.H.; Lim, J.H.; Ryu, G.R.; Chung, H.W.; Han, S.W.; Shin, S.J.; Bang, B.K.; Breyer, M.D.; et al. Long-term treatment of glucagon-like peptide-1 analog exendin-4 ameliorates diabetic nephropathy through improving metabolic anomalies in db/db mice. J. Am. Soc. Nephrol. 2017, 18, 1227–1238. [Google Scholar] [CrossRef]
- Kodera, R.; Shikata, K.; Kataoka, H.U.; Takatsuka, T.; Miyamoto, S.; Sasaki, M.; Kajitani, N.; Nishishita, S.; Sarai, K.; Hirota, D.; et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia 2011, 54, 965–978. [Google Scholar] [CrossRef] [Green Version]
- Ferdinand, K.C.; White, W.B.; Calhoun, D.A.; Lonn, E.M.; Sager, P.T.; Brunelle, R.; Jiang, H.H.; Threlkeld, R.J.; Robertson, K.E.; Geiger, M.J. Effects of the once-weekly glucagon-like peptide-1 receptor agonist dulaglutide on ambulatory blood pressure and heart rate in patients with type 2 diabetes mellitus. Hypertension 2014, 64, 731–737. [Google Scholar] [CrossRef]
- Fujita, H.; Morii, T.; Fujishima, H.; Sato, T.; Shimizu, T.; Hosoba, M.; Tsukiyama, K.; Narita, T.; Takahashi, T.; Drucker, D.J.; et al. The protective roles of GLP-1R signaling in diabetic nephropathy: Possible mechanism and therapeutic potential. Kidney Int. 2014, 85, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Qin, N.; Ren, H.; Yang, M.; Liu, S.; Wang, Q. Metformin Regulating miR-34a Pathway to Inhibit Egr1 in Rat Mesangial Cells Cultured with High Glucose. Int. J. Endocrinol. 2018, 2018, 6462793. [Google Scholar] [CrossRef] [PubMed]
- Vasamsetti, S.B.; Karnewar, S.; Kanugula, A.K.; Thatipalli, A.R.; Kumar, J.M.; Kotamraju, S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: Potential role in atherosclerosis. Diabetes 2015, 64, 2028–2041. [Google Scholar] [CrossRef]
- Guess, A.; Agrawal, S.; Wei, C.C.; Ransom, R.F.; Benndorf, R.; Smoyer, W.E. Dose- and time-dependent glucocorticoid receptor signaling in podocytes. Am. J. Physiol. Renal Physiol. 2010, 299, F553–F845. [Google Scholar] [CrossRef] [PubMed]
- Utimura, R.; Fujihara, C.K.; Mattar, A.L.; Malheiros, D.M.; Noronha, I.L.; Zatz, R. Mycophenolate mofetil prevents the development of glomerular injury in experimental diabetes. Kidney Int. 2003, 63, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Iturbe, B.; Quiroz, Y.; Shahkarami, A.; Li, Z.; Vaziri, N.D. Mycophenolate mofetil ameliorates nephropathy in the obese Zucker rat. Kidney Int. 2005, 68, 1041–1047. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.W.; Kim, Y.G.; Lee, S.H.; Lee, A.; Kim, D.J.; Jeong, K.H.; Lee, K.H.; Hwang, S.J.; Woo, J.S.; Lim, S.J.; et al. Mycophenolate Mofetil Ameliorates Diabetic Nephropathy in db/db Mice. Biomed. Res. Int. 2015, 2015, 301627. [Google Scholar] [CrossRef]
- Eller, K.; Kirsch, A.; Wolf, A.M.; Sopper, S.; Tagwerker, A.; Stanzl, U.; Wolf, D.; Patsch, W.; Rosenkranz, A.R.; Eller, P. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes 2011, 60, 2954–2962. [Google Scholar] [CrossRef] [PubMed]
- Pena, M.J.; Mischak, H.; Heerspink, H.J. Proteomics for prediction of disease progression and response to therapy in diabetic kidney disease. Diabetologia 2016, 59, 1819–1831. [Google Scholar] [CrossRef] [Green Version]
- Verhave, J.C.; Bouchard, J.; Goupil, R.; Pichette, V.; Brachemi, S.; Madore, F.; Troyanov, S. Clinical value of inflammatory urinary biomarkers in overt diabetic nephropathy: A prospective study. Diabetes Res. Clin. Pract. 2013, 101, 333–340. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Park, A.S.; Mohtat, D.; Thomas, D.B.; Pullman, J.M.; Susztak, K. Transcriptome analysis of human diabetic kidney disease. Diabetes 2011, 60, 2354–2369. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matoba, K.; Takeda, Y.; Nagai, Y.; Kawanami, D.; Utsunomiya, K.; Nishimura, R. Unraveling the Role of Inflammation in the Pathogenesis of Diabetic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 3393. https://doi.org/10.3390/ijms20143393
Matoba K, Takeda Y, Nagai Y, Kawanami D, Utsunomiya K, Nishimura R. Unraveling the Role of Inflammation in the Pathogenesis of Diabetic Kidney Disease. International Journal of Molecular Sciences. 2019; 20(14):3393. https://doi.org/10.3390/ijms20143393
Chicago/Turabian StyleMatoba, Keiichiro, Yusuke Takeda, Yosuke Nagai, Daiji Kawanami, Kazunori Utsunomiya, and Rimei Nishimura. 2019. "Unraveling the Role of Inflammation in the Pathogenesis of Diabetic Kidney Disease" International Journal of Molecular Sciences 20, no. 14: 3393. https://doi.org/10.3390/ijms20143393