Visualization of Bacterial Protein Complexes Labeled with Fluorescent Proteins and Nanobody Binders for STED Microscopy

 ,
,
Abstract
:1. Introduction
2. Results
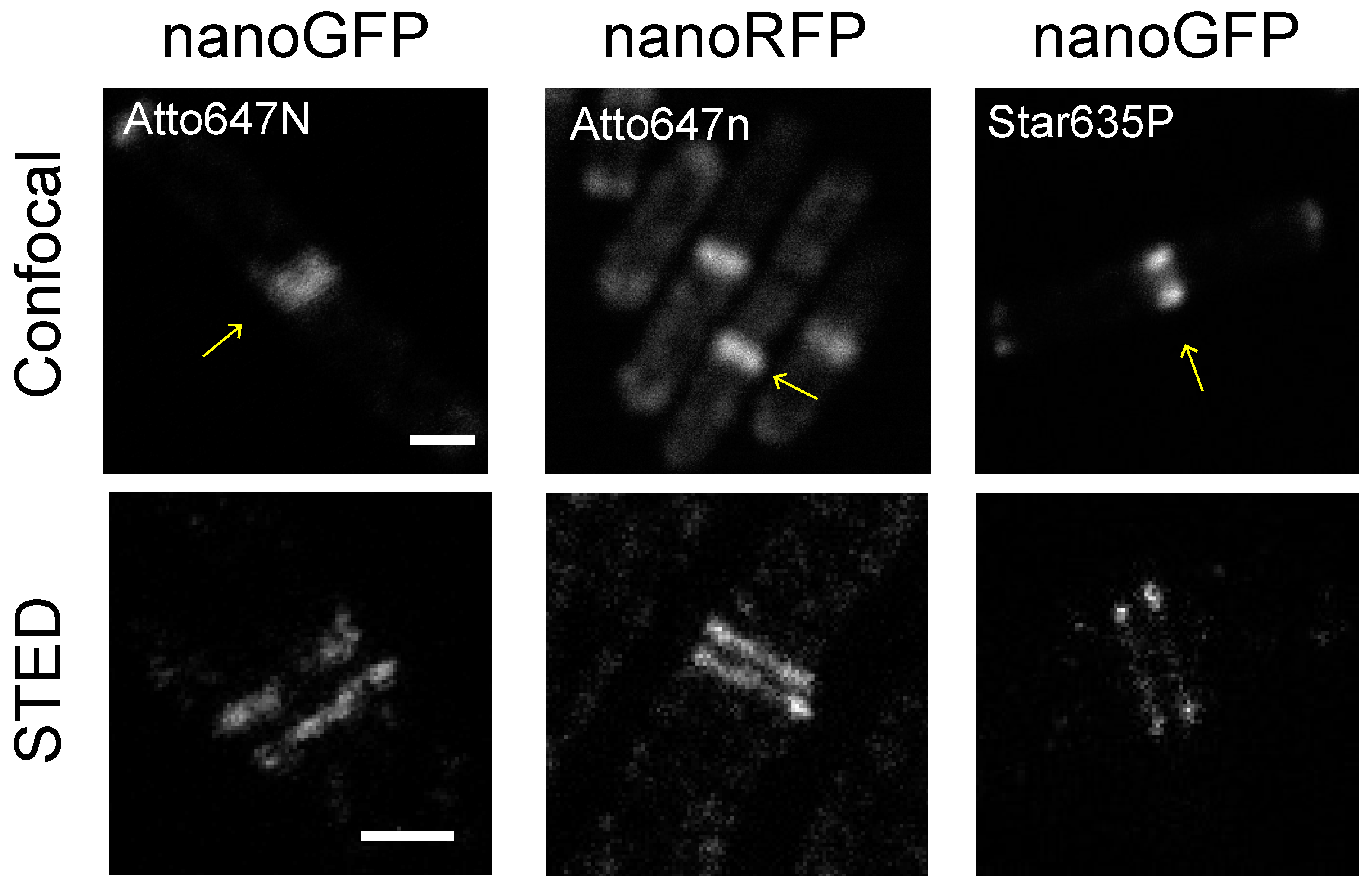
2.1. Nanobodies Recognizing Fluorescent Proteins Enable Visualization of Target Proteins in Bacteria with STED Microscopy
2.2. Evaluation of Unspecific Binding for GFP and RFP Nanobodies in Bacterial Cells
2.3. Evaluation of Fluorescent Dyes for STED Imaging in Bacterial Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Cell Culture
4.2. SIM Imaging
4.2.1. Bacterial Sample Preparation
4.2.2. SIM Data Acquisition and Processing
4.3. STED Imaging
4.3.1. Bacterial Sample Preparation
4.3.2. STED and Confocal Data Acquisition and Processing
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Klar, T.A.; Jakobs, S.; Dyba, M.; Egner, A.; Hell, S.W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. USA 2000, 97, 8206–8210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frohn, J.T.; Knapp, H.F.; Stemmer, A. True optical resolution beyond the Rayleigh limit achieved by standing wave illumination. Proc. Natl. Acad. Sci. USA 2000, 97, 7232–7236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Strauss, S.; Nickels, P.C.; Strauss, M.T.; Jimenez Sabinina, V.; Ellenberg, J.; Carter, J.D.; Gupta, S.; Janjic, N.; Jungmann, R. Modified aptamers enable quantitative sub-10-nm cellular DNA-PAINT imaging. Nat. Methods 2018, 15, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heilemann, M.; van de Linde, S.; Schüttpelz, M.; Kasper, R.; Seefeldt, B.; Mukherjee, A.; Tinnefeld, P.; Sauer, M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 2008, 47, 6172–6176. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef]
- Coltharp, C.; Xiao, J. Superresolution microscopy for microbiology. Cell. Microbiol. 2012, 14, 1808–1818. [Google Scholar] [CrossRef] [Green Version]
- Schoen, I.; Ries, J.; Klotzsch, E.; Ewers, H.; Vogel, V. Binding-activated localization microscopy of DNA structures. Nano Lett. 2011, 11, 4008–4011. [Google Scholar] [CrossRef]
- Schneider, J.P.; Basler, M. Shedding light on biology of bacterial cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holden, S. Probing the mechanistic principles of bacterial cell division with super-resolution microscopy. Curr. Opin. Microbiol. 2018, 43, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Lopez-Garrido, J.; Lee, S.-H.; Diaz-Celis, C.; Fleming, T.; Bustamante, C.; Pogliano, K. Visualization and functional dissection of coaxial paired SpoIIIE channels across the sporulation septum. eLife 2015, 4, 3253. [Google Scholar] [CrossRef] [PubMed]
- Eswaramoorthy, P.; Erb, M.L.; Gregory, J.A.; Silverman, J.; Pogliano, K.; Pogliano, J.; Ramamurthi, K.S. Cellular architecture mediates DivIVA ultrastructure and regulates min activity in Bacillus subtilis. MBio 2011, 2, e00257-11. [Google Scholar] [CrossRef] [PubMed]
- Eswaramoorthy, P.; Winter, P.W.; Wawrzusin, P.; York, A.G.; Shroff, H.; Ramamurthi, K.S. Asymmetric division and differential gene expression during a bacterial developmental program requires DivIVA. PLoS Genet. 2014, 10, e1004526. [Google Scholar] [CrossRef] [PubMed]
- Holden, S.J.; Pengo, T.; Meibom, K.L.; Fernandez Fernandez, C.; Collier, J.; Manley, S. High throughput 3D super-resolution microscopy reveals Caulobacter crescentus in vivo Z-ring organization. Proc. Natl. Acad. Sci. USA 2014, 111, 4566–4571. [Google Scholar] [CrossRef]
- Buss, J.; Coltharp, C.; Shtengel, G.; Yang, X.; Hess, H.; Xiao, J. A multi-layered protein network stabilizes the Escherichia coli FtsZ-ring and modulates constriction dynamics. PLoS Genet. 2015, 11, e1005128. [Google Scholar] [CrossRef]
- Schneider, J.; Klein, T.; Mielich-Süss, B.; Koch, G.; Franke, C.; Kuipers, O.P.; Kovács, Á.T.; Sauer, M.; Lopez, D. Spatio-temporal remodeling of functional membrane microdomains organizes the signaling networks of a bacterium. PLoS Genet. 2015, 11, e1005140. [Google Scholar] [CrossRef]
- Bach, J.N.; Giacomelli, G.; Bramkamp, M. Sample Preparation and Choice of Fluorophores for Single and Dual Color Photo-Activated Localization Microscopy (PALM) with Bacterial Cells. Methods Mol. Biol. 2017, 1563, 129–141. [Google Scholar]
- Dempwolff, F.; Schmidt, F.K.; Hervás, A.B.; Stroh, A.; Rösch, T.C.; Riese, C.N.; Dersch, S.; Heimerl, T.; Lucena, D.; Hülsbusch, N.; et al. Super Resolution Fluorescence Microscopy and Tracking of Bacterial Flotillin (Reggie) Paralogs Provide Evidence for Defined-Sized Protein Microdomains within the Bacterial Membrane but Absence of Clusters Containing Detergent-Resistant Proteins. PLoS Genet. 2016, 12, e1006116. [Google Scholar] [CrossRef]
- Olshausen, P.V.; Defeu Soufo, H.J.; Wicker, K.; Heintzmann, R.; Graumann, P.L.; Rohrbach, A. Superresolution imaging of dynamic MreB filaments in B. subtilis—A multiple-motor-driven transport? Biophys. J. 2013, 105, 1171–1181. [Google Scholar] [PubMed]
- Billaudeau, C.; Yao, Z.; Cornilleau, C.; Carballido-López, R.; Chastanet, A. MreB Forms Subdiffraction Nanofilaments during Active Growth in Bacillus subtilis. MBio 2019, 10, e01879–e01918. [Google Scholar] [PubMed]
- Lesterlin, C.; Ball, G.; Schermelleh, L.; Sherratt, D.J. RecA bundles mediate homology pairing between distant sisters during DNA break repair. Nature 2014, 506, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Söderström, B.; Chan, H.; Shilling, P.J.; Skoglund, U.; Daley, D.O. Spatial separation of FtsZ and FtsN during cell division. Mol. Microbiol. 2018, 107, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Jennings, P.C.; Cox, G.C.; Monahan, L.G.; Harry, E.J. Super-resolution imaging of the bacterial cytokinetic protein FtsZ. Micron 2011, 42, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Strauss, M.P.; Liew, A.T.F.; Turnbull, L.; Whitchurch, C.B.; Monahan, L.G.; Harry, E.J. 3D-SIM super resolution microscopy reveals a bead-like arrangement for FtsZ and the division machinery: Implications for triggering cytokinesis. PLoS Biol. 2012, 10, e1001389. [Google Scholar] [CrossRef] [PubMed]
- Ries, J.; Kaplan, C.; Platonova, E.; Eghlidi, H.; Ewers, H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat. Methods 2012, 9, 582–584. [Google Scholar] [PubMed]
- Deschout, H.; Cella Zanacchi, F.; Mlodzianoski, M.; Diaspro, A.; Bewersdorf, J.; Hess, S.T.; Braeckmans, K. Precisely and accurately localizing single emitters in fluorescence microscopy. Nat. Methods 2014, 11, 253–266. [Google Scholar] [PubMed]
- Bates, M.; Huang, B.; Dempsey, G.T.; Zhuang, X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science 2007, 317, 1749–1753. [Google Scholar] [CrossRef]
- Beghein, E.; Gettemans, J. Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein-Protein Interaction Analysis, and Protein Function Exploration. Front. Immunol. 2017, 8, 771. [Google Scholar] [CrossRef]
- Vedyaykin, A.D.; Vishnyakov, I.E.; Polinovskaya, V.S.; Khodorkovskii, M.A.; Sabantsev, A.V. New insights into FtsZ rearrangements during the cell division of Escherichia coli from single-molecule localization microscopy of fixed cells. Microbiologyopen 2016, 5, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Heilemann, M.; Margeat, E.; Kasper, R.; Sauer, M.; Tinnefeld, P. Carbocyanine dyes as efficient reversible single-molecule optical switch. J. Am. Chem. Soc. 2005, 127, 3801–3806. [Google Scholar] [CrossRef] [PubMed]
- Kasper, R.; Harke, B.; Forthmann, C.; Tinnefeld, P.; Hell, S.W.; Sauer, M. Single-molecule STED microscopy with photostable organic fluorophores. Small 2010, 6, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Stockmar, I.; Feddersen, H.; Cramer, K.; Gruber, S.; Jung, K.; Bramkamp, M.; Shin, J.Y. Optimization of sample preparation and green color imaging using the mNeonGreen fluorescent protein in bacterial cells for photoactivated localization microscopy. Sci. Rep. 2018, 8, 10137. [Google Scholar] [CrossRef] [PubMed]
- Thomaides, H.B.; Freeman, M.; El Karoui, M.; Errington, J. Division site selection protein DivIVA of Bacillus subtilis has a second distinct function in chromosome segregation during sporulation. Genes Dev. 2001, 15, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Oracz, J.; Westphal, V.; Radzewicz, C.; Sahl, S.J.; Hell, S.W. Photobleaching in STED nanoscopy and its dependence on the photon flux applied for reversible silencing of the fluorophore. Sci. Rep. 2017, 7, 11354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurm, C.A.; Kolmakov, K.; Göttfert, F.; Ta, H.; Bossi, M.; Schill, H.; Berning, S.; Jakobs, S.; Donnert, G.; Belov, V.N.; et al. Novel red fluorophores with superior performance in STED microscopy. Opt. Nanoscopy 2012, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- Grotjohann, T.; Testa, I.; Leutenegger, M.; Bock, H.; Urban, N.T.; Lavoie-Cardinal, F.; Willig, K.I.; Eggeling, C.; Jakobs, S.; Hell, S.W. Diffraction-unlimited all-optical imaging and writing with a photochromic GFP. Nature 2011, 478, 204–208. [Google Scholar] [CrossRef]
- Delcanale, P.; Pennacchietti, F.; Maestrini, G.; Rodríguez-Amigo, B.; Bianchini, P.; Diaspro, A.; Iagatti, A.; Patrizi, B.; Foggi, P.; Agut, M.; et al. Subdiffraction localization of a nanostructured photosensitizer in bacterial cells. Sci. Rep. 2015, 5, 15564. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.A.; Ozbay, B.N.; Potcoava, M.; Salcedo, E.; Restrepo, D.; Gibson, E.A. Super-resolution imaging of ciliary microdomains in isolated olfactory sensory neurons using a custom two-color stimulated emission depletion microscope. J. Biomed. Opt. 2016, 21, 66017. [Google Scholar] [CrossRef] [Green Version]
- Winter, F.R.; Loidolt, M.; Westphal, V.; Butkevich, A.N.; Gregor, C.; Sahl, S.J.; Hell, S.W. Multicolour nanoscopy of fixed and living cells with a single STED beam and hyperspectral detection. Sci. Rep. 2017, 7, 46492. [Google Scholar] [CrossRef] [PubMed]
- Vicidomini, G.; Bianchini, P.; Diaspro, A. STED super-resolved microscopy. Nat. Methods 2018, 15, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanobody-Dye | Target | Signal-to-Background Ratio | Cellular Background |
|---|---|---|---|
| NBGFP-Atto647n | GFP | 12 | 11 % |
| NBRFP-Atto647n | mCherry2 | 3 | 36 % |
| NBGFP-Atto594 | GFP | n.a. * | n.a. * |
| NBRFP-Atto594 | mCherry2 | 3 | 38 % |
| NBRFP-Star600 | PAmCherry | 2.4 | 42 % |
| NBGFP-Star635p | GFP | 3 | 34 % |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cramer, K.; Bolender, A.-L.; Stockmar, I.; Jungmann, R.; Kasper, R.; Shin, J.Y. Visualization of Bacterial Protein Complexes Labeled with Fluorescent Proteins and Nanobody Binders for STED Microscopy. Int. J. Mol. Sci. 2019, 20, 3376. https://doi.org/10.3390/ijms20143376
Cramer K, Bolender A-L, Stockmar I, Jungmann R, Kasper R, Shin JY. Visualization of Bacterial Protein Complexes Labeled with Fluorescent Proteins and Nanobody Binders for STED Microscopy. International Journal of Molecular Sciences. 2019; 20(14):3376. https://doi.org/10.3390/ijms20143376
Chicago/Turabian StyleCramer, Kimberly, Anna-Lena Bolender, Iris Stockmar, Ralf Jungmann, Robert Kasper, and Jae Yen Shin. 2019. "Visualization of Bacterial Protein Complexes Labeled with Fluorescent Proteins and Nanobody Binders for STED Microscopy" International Journal of Molecular Sciences 20, no. 14: 3376. https://doi.org/10.3390/ijms20143376