Crystal Structure of Phosphoserine BlaC from Mycobacterium tuberculosis Inactivated by Bis(Benzoyl) Phosphate

 and
and
Abstract
:
1. Introduction
2. Results
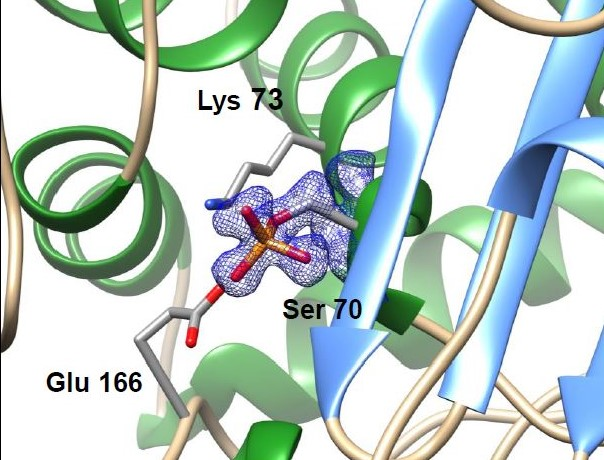
2.1. Crystal Structures of Phosphoserine BlaC and Inactivation by Bis(Benzoyl) Phosphate
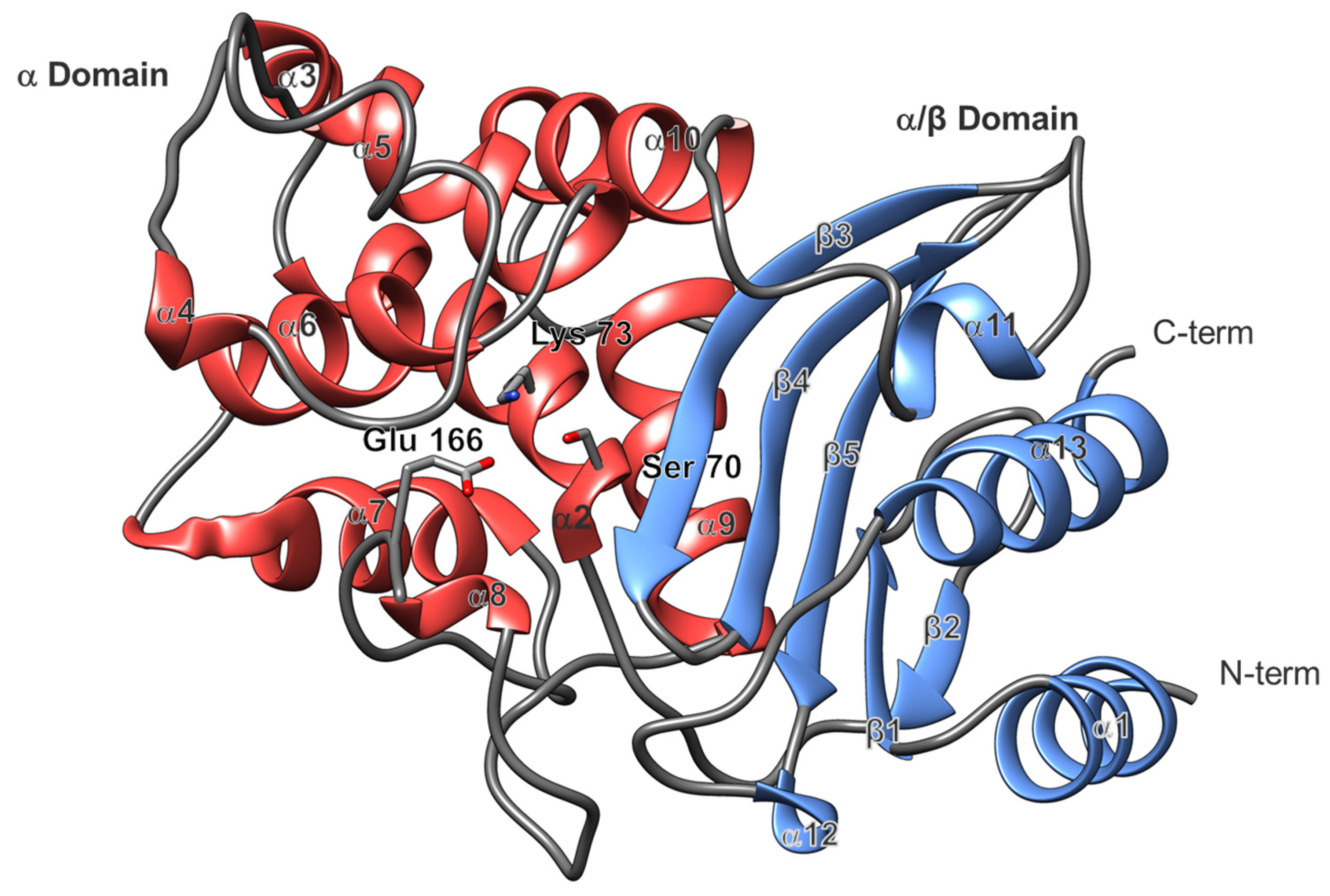
2.2. Overall Structure and Active Site
3. Discussion
4. Materials and Methods
4.1. Expression and Purification
4.2. Synthesis of the Bis(Benzoyl) Phosphate
4.3. Time-Dependent Inhibition of BlaC
4.4. Protein Crystallization and Structure Determination
4.5. Sequence Alignment
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Laura Anderson, A.B.; Dean, A.; Dias, H.M.; Falzon, D.; Floyd, K.; Baena, I.G.; Gebreselassie, N.; Gilpin, C.; Glaziou, P.; Hamada, Y.; et al. Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2017; Available online: https://www.who.int/tb/en/ (accessed on 26 March 2018).
- Hugonnet, J.-E.; Tremblay, L.W.; Boshoff, H.I.; Barry, C.E.; Blanchard, J.S. Meropenem-Clavulanate Is Effective Against Extensively Drug-Resistant Mycobacterium tuberculosis. Science 2009, 323, 1215–1218. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.M.; Miggiano, R.; Rossi, F.; Rizzi, M. Mycobacterium tuberculosis Molecular Determinants of Infection, Survival Strategies, and Vulnerable Targets. Pathogens 2018, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- J Libardo, M.D.; Boshoff, H.I.M.; Barry, C.E. The present state of the tuberculosis drug development pipeline. Curr. Opin. Pharmacol. 2018, 42, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Prigozhin, D.M.; Krieger, I.V.; Huizar, J.P.; Mavrici, D.; Waldo, G.S.; Hung, L.-W.; Sacchettini, J.C.; Terwilliger, T.C.; Alber, T. Subfamily-Specific Adaptations in the Structures of Two Penicillin-Binding Proteins from Mycobacterium tuberculosis. PloS ONE 2014, 9, e116249. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G.; Barlow, M. Evolution of the serine β-lactamases: Past, present and future. Drug Resist. Updates 2004, 7, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Cassidy, C.; Sacchettini, J.C. Crystal Structure and Activity Studies of the Mycobacterium tuberculosis β-Lactamase Reveal Its Critical Role in Resistance to β-Lactam Antibiotics. Antimicrob. Agents Chemother. 2006, 50, 2762–2771. [Google Scholar] [CrossRef]
- Kurz, S.G.; Wolff, K.A.; Hazra, S.; Bethel, C.R.; Hujer, A.M.; Smith, K.M.; Xu, Y.; Tremblay, L.W.; Blanchard, J.S.; Nguyen, L.; et al. Can inhibitor-resistant substitutions in the Mycobacterium tuberculosis β-Lactamase BlaC lead to clavulanate resistance?: A biochemical rationale for the use of β-lactam-β-lactamase inhibitor combinations. Antimicrob. Agents Chemother. 2013, 57, 6085–6096. [Google Scholar] [CrossRef]
- Xu, H.; Hazra, S.; Blanchard, J.S. NXL104 irreversibly inhibits the beta-lactamase from Mycobacterium tuberculosis. Biochemistry 2012, 51, 4551–4557. [Google Scholar] [CrossRef]
- Chambers, H.F.; Kocagöz, T.; Sipit, T.; Turner, J.; Hopewell, P.C. Activity of Amoxicillin/Clavulanate in Patients with Tuberculosis. Clin. Infect. Dis. 1998, 26, 874–877. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.M.; Clasman, J.R.; June, C.M.; Kaitany, K.-C.J.; LaFleur, J.R.; Taracila, M.A.; Klinger, N.V.; Bonomo, R.A.; Wymore, T.; Szarecka, A.; et al. The structural basis of activity against aztreonam and extended spectrum cephalosporins for two carbepenem-hydrolyzing class D b-lactamases from Acinetobacter baumannii. Biochemistry 2015, 54, 1976–1987. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, C.; De Luca, F.; Benvenuti, M.; Poirel, L.; Nordmann, P.; Rossolini, G.M.; Mangani, S.; Docquier, J.-D. Crystal Structure of the Pseudomonas aeruginosa BEL-1 Extended-Spectrum β-Lactamase and Its Complexes with Moxalactam and Imipenem. Antimicrob. Agents Chemother. 2016, 60, 7189–7199. [Google Scholar] [PubMed]
- Maveyraud, L.; Pratt, R.F.; Samama, J.-P. Crystal Structure of an Acylation Transition-State Analog of the TEM-1 β-Lactamase. Mechanistic Implications for Class A β-Lactamases. Biochemistry 1998, 37, 2622–2628. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Pratt, R.F. Inhibition of Serine β-Lactamases by Acyl Phosph(on)ates: A New Source of Inert Acyl [and Phosphyl] Enzymes. J. Am. Chem. Soc. 1998, 120, 4264–4268. [Google Scholar] [CrossRef]
- Hazra, S.; Kurz, S.G.; Wolff, K.; Nguyen, L.; Bonomo, R.A.; Blanchard, J.S. Kinetic and Structural Characterization of the Interaction of 6-Methylidene Penem 2 with the beta-Lactamase from Mycobacterium tuberculosis. Biochemistry 2015, 54, 5657–5664. [Google Scholar] [CrossRef] [PubMed]
- Elings, W.; Tassoni, R.; van der Schoot, S.A.; Luu, W.; Kynast, J.P.; Dai, L.; Blok, A.J.; Timmer, M.; Florea, B.I.; Pannu, N.S.; et al. Phosphate Promotes the Recovery of Mycobacterium tuberculosis beta-Lactamase from Clavulanic Acid Inhibition. Biochemistry 2017, 56, 6257–6267. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Delmas, J.; Sirot, J.; Shoichet, B.; Bonnet, R. Atomic Resolution Structures of CTX-M β-Lactamases: Extended Spectrum Activities from Increased Mobility and Decreased Stability. J. Mol. Biol. 2005, 348, 349–362. [Google Scholar] [CrossRef]
- Kuzin, A.P.; Nukaga, M.; Nukaga, Y.; Hujer, A.M.; Bonomo, R.A.; Knox, J.R. Structure of the SHV-1 β-Lactamase. Biochemistry 1999, 38, 5720–5727. [Google Scholar] [CrossRef]
- Stec, B.; Holtz, K.M.; Wojciechowski, C.L.; Kantrowitz, E.R. Structure of the wild-type TEM-1 [beta]-lactamase at 1.55 A and the mutant enzyme Ser70Ala at 2.1 A suggest the mode of noncovalent catalysis for the mutant enzyme. Acta Crystallogr. Sect. D 2005, 61, 1072–1079. [Google Scholar] [CrossRef]
- Ambler, R.P.; Coulson, A.F.; Frere, J.M.; Ghuysen, J.M.; Joris, B.; Forsman, M.; Levesque, R.C.; Tiraby, G.; Waley, S.G. A standard numbering scheme for the class A beta-lactamases. Biochem. J. 1991, 276, 269–270. [Google Scholar] [CrossRef]
- Vandavasi, V.G.; Weiss, K.L.; Cooper, J.B.; Erskine, P.T.; Tomanicek, S.J.; Ostermann, A.; Schrader, T.E.; Ginell, S.L.; Coates, L. Exploring the Mechanism of beta-Lactam Ring Protonation in the Class A beta-lactamase Acylation Mechanism Using Neutron and X-ray Crystallography. J. Med. Chem. 2016, 59, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, L.W.; Fan, F.; Blanchard, J.S. Biochemical and Structural Characterization of Mycobacterium tuberculosis β-Lactamase with the Carbapenems Ertapenem and Doripenem. Biochemistry 2010, 49, 3766–3773. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; He, Y.; Lei, J.; Huang, X.; Zhao, Y. Crystallographic Snapshots of Class A beta-Lactamase Catalysis Reveal Structural Changes That Facilitate beta-Lactam Hydrolysis. J. Biol. Chem. 2017, 292, 4022–4033. [Google Scholar] [CrossRef] [PubMed]
- Pratt, R.F. Inhibition of a class C beta-lactamase by a specific phosphonate monoester. Science 1989, 246, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Rahil, J.; Pratt, R.F. Phosphonate monoester inhibitors of class A beta-lactamases. Biochem. J. 1991, 275, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.J.; Li, N.; Pratt, R.F. Inverse acyl phosph(on)ates: Substrates or inhibitors of beta-lactam-recognizing enzymes? Bioorganic Chem. 2001, 29, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Pratt, R.F. Inhibition of Class A and C β-Lactamases by Diaroyl Phosphates. Biochemistry 2009, 48, 8285–8292. [Google Scholar] [CrossRef]
- Chen, C.C.; Rahil, J.; Pratt, R.F.; Herzberg, O. Structure of a phosphonate-inhibited beta-lactamase. An analog of the tetrahedral transition state/intermediate of beta-lactam hydrolysis. J. Mol. Biol. 1993, 234, 165–178. [Google Scholar] [CrossRef]
- Aldridge, W.N. Some properties of specific cholinesterase with particular reference to the mechanism of inhibition by diethyl p-nitrophenyl thiophosphate (E 605) and analogues. Biochem. J. 1950, 46, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Carletti, E.; Colletier, J.-P.; Schopfer, L.M.; Santoni, G.; Masson, P.; Lockridge, O.; Nachon, F.; Weik, M. Inhibition Pathways of the Potent Organophosphate CBDP with Cholinesterases Revealed by X-ray Crystallographic Snapshots and Mass Spectrometry. Chem. Res. Toxicol. 2013, 26, 280–289. [Google Scholar] [CrossRef]
- Sanson, B.; Nachon, F.; Colletier, J.-P.; Froment, M.-T.; Toker, L.; Greenblatt, H.M.; Sussman, J.L.; Ashani, Y.; Masson, P.; Silman, I.; et al. Crystallographic Snapshots of Nonaged and Aged Conjugates of Soman with Acetylcholinesterase, and of a Ternary Complex of the Aged Conjugate with Pralidoxime. J. Med. Chem. 2009, 52, 7593–7603. [Google Scholar] [CrossRef] [PubMed]
- Bharathi, S.; Wong, P.T.; Desai, A.; Lykhytska, O.; Choe, V.; Kim, H.; Thomas, T.P.; Baker, J.R.; Choi, S.K. Design and mechanistic investigation of oxime-conjugated PAMAM dendrimers as the catalytic scavenger of reactive organophosphate. J. Mater. Chem. B 2014, 2, 1068–1078. [Google Scholar] [CrossRef]
- Tassoni, R.; Blok, A.; Pannu, N.S.; Ubbink, M. New Conformations of Acylation Adducts of Inhibitors of β-Lactamase from Mycobacterium tuberculosis. Biochemistry 2019, 58, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Toia, R.F.; Casida, J.E. Phosphorylation, “aging” and possible alkylation reactions of saligenin cyclic phosphorus esters with α-chymotrypsin. Biochem. Pharmacol. 1979, 28, 211–216. [Google Scholar] [CrossRef]
- Carletti, E.; Schopfer, L.M.; Colletier, J.-P.; Froment, M.-T.; Nachon, F.; Weik, M.; Lockridge, O.; Masson, P. Reaction of Cresyl Saligenin Phosphate, the Organophosphorus Agent Implicated in Aerotoxic Syndrome, with Human Cholinesterases: Mechanistic Studies Employing Kinetics, Mass Spectrometry, and X-ray Structure Analysis. Chem. Res. Toxicol. 2011, 24, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, T.R. Mechanism of Action of Organophosphorus and Carbamate Insecticides. Environ. Health Perspect. 1990, 87, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1997; Vol. 276, pp. 307–326. [Google Scholar]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput Chem 2004, 25, 605–1612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Naas, T.; Oueslati, S.; Bonnin, R.A.; Dabos, M.L.; Zavala, A.; Dortet, L.; Retailleau, P.; Iorga, B.I. Beta-lactamase database (BLDB) – structure and function. J. Enzym. Inhib. Med. Chem. 2017, 32, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT; Nucleic acids symposium series, 1999; Oxford University Press: Oxford, UK, 1999; pp. 95–98. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data collection | BlaC - phosphoserine |
| PDB ID | 6N14 |
| Space group | P212121 |
| Cell dimensions | |
| (a, b, c) (Å) | 49.84, 68.04, 75.45 |
| Molecules per asymetric unit | 1 |
| Resolution (Å) | 28.10−1.52 |
| Wavelength (Å) | 1.00 |
| Rsym | 0.099 (0.816) |
| I/σ | 17.6 (1.92) |
| CC1/2 | 0.992 (0.623) |
| Completeness | 99.75 (97.88) |
| Redundancy | 6.8 (4.7) |
| Refinement | |
| Resolution (Å) | 28.10−1.52 |
| Unique reflections | 39,978 (3,879) |
| Rwork/Rfree | 0.155/0.169 |
| RMSD | |
| RMSD bonds (Å) | 0.003 |
| RMSD angles (Å) | 0.745 |
| Ramachandrans (%) | |
| Favored | 98 |
| Outliers | 0 |
| Number of atoms | 2,322 |
| Protein and ligand | 1,999 |
| Water | 323 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moural, T.W.; White, D.S.-D.; Choy, C.J.; Kang, C.; Berkman, C.E. Crystal Structure of Phosphoserine BlaC from Mycobacterium tuberculosis Inactivated by Bis(Benzoyl) Phosphate. Int. J. Mol. Sci. 2019, 20, 3247. https://doi.org/10.3390/ijms20133247
Moural TW, White DS-D, Choy CJ, Kang C, Berkman CE. Crystal Structure of Phosphoserine BlaC from Mycobacterium tuberculosis Inactivated by Bis(Benzoyl) Phosphate. International Journal of Molecular Sciences. 2019; 20(13):3247. https://doi.org/10.3390/ijms20133247
Chicago/Turabian StyleMoural, Timothy W., Dawanna Shar-Day White, Cindy J. Choy, Chulhee Kang, and Clifford E. Berkman. 2019. "Crystal Structure of Phosphoserine BlaC from Mycobacterium tuberculosis Inactivated by Bis(Benzoyl) Phosphate" International Journal of Molecular Sciences 20, no. 13: 3247. https://doi.org/10.3390/ijms20133247