Heart Failure Differentially Modulates Natural (Sinoatrial Node) and Ectopic (Pulmonary Veins) Pacemakers: Mechanism and Therapeutic Implication for Atrial Fibrillation

Abstract
:1. Introduction
2. Distinct Electrophysiological and Structural Characteristics of PVs
2.1. Autonomic Nervous System in PV Electrical Activity
2.2. Calcium Homeostasis in PV Cardiomyocytes
2.3. Role of Renin Angiotensin System in PV Electrical Activity
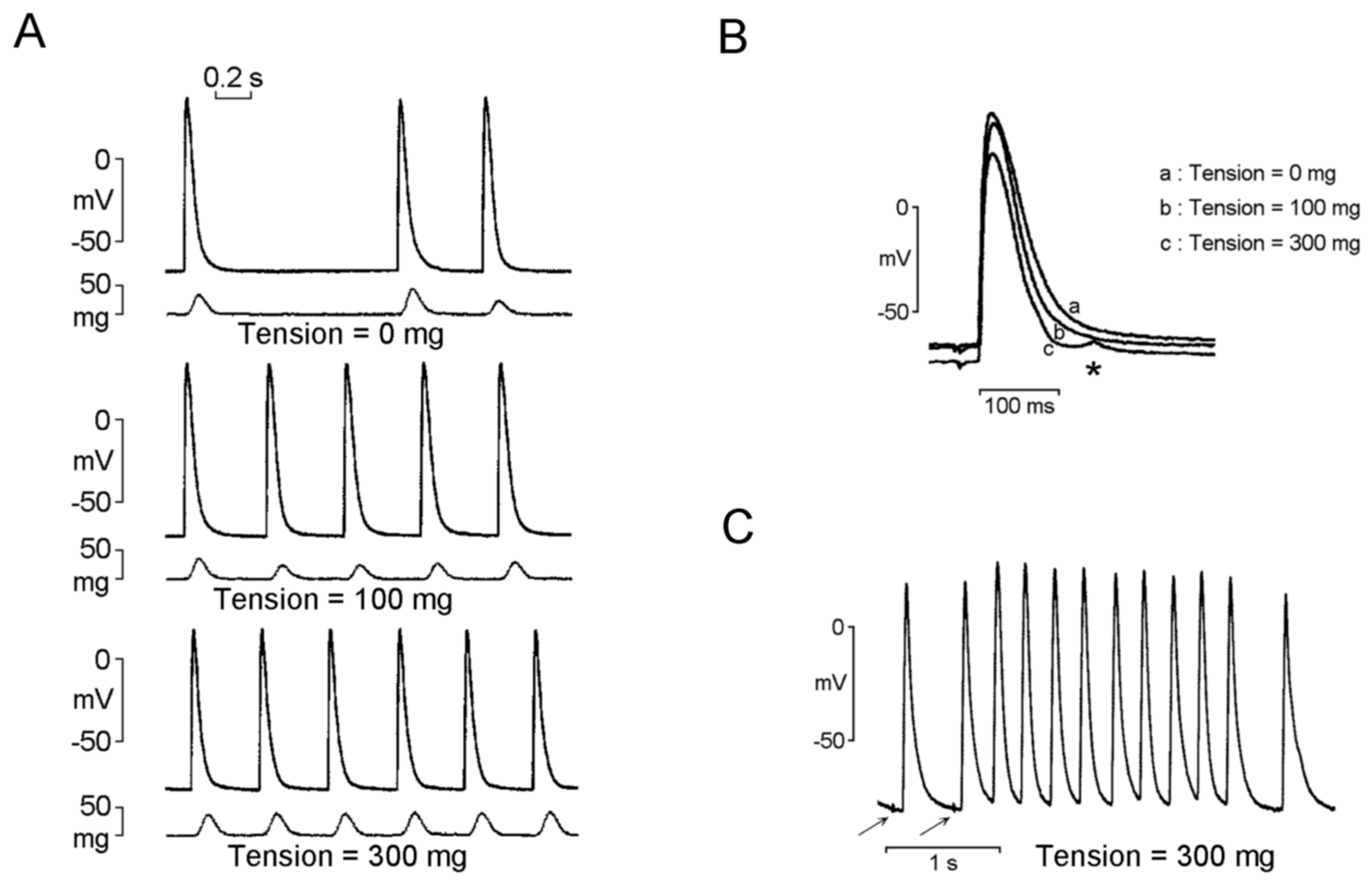
2.4. Mechanoelectrical Feedback on PV Arrhythmogenesis
2.5. Interaction of PV Cardiomyocytes and Fibrosis
3. Distinct Electrophysiological and Structural Characteristics of the SAN
3.1. SAN Dysfunction in AF
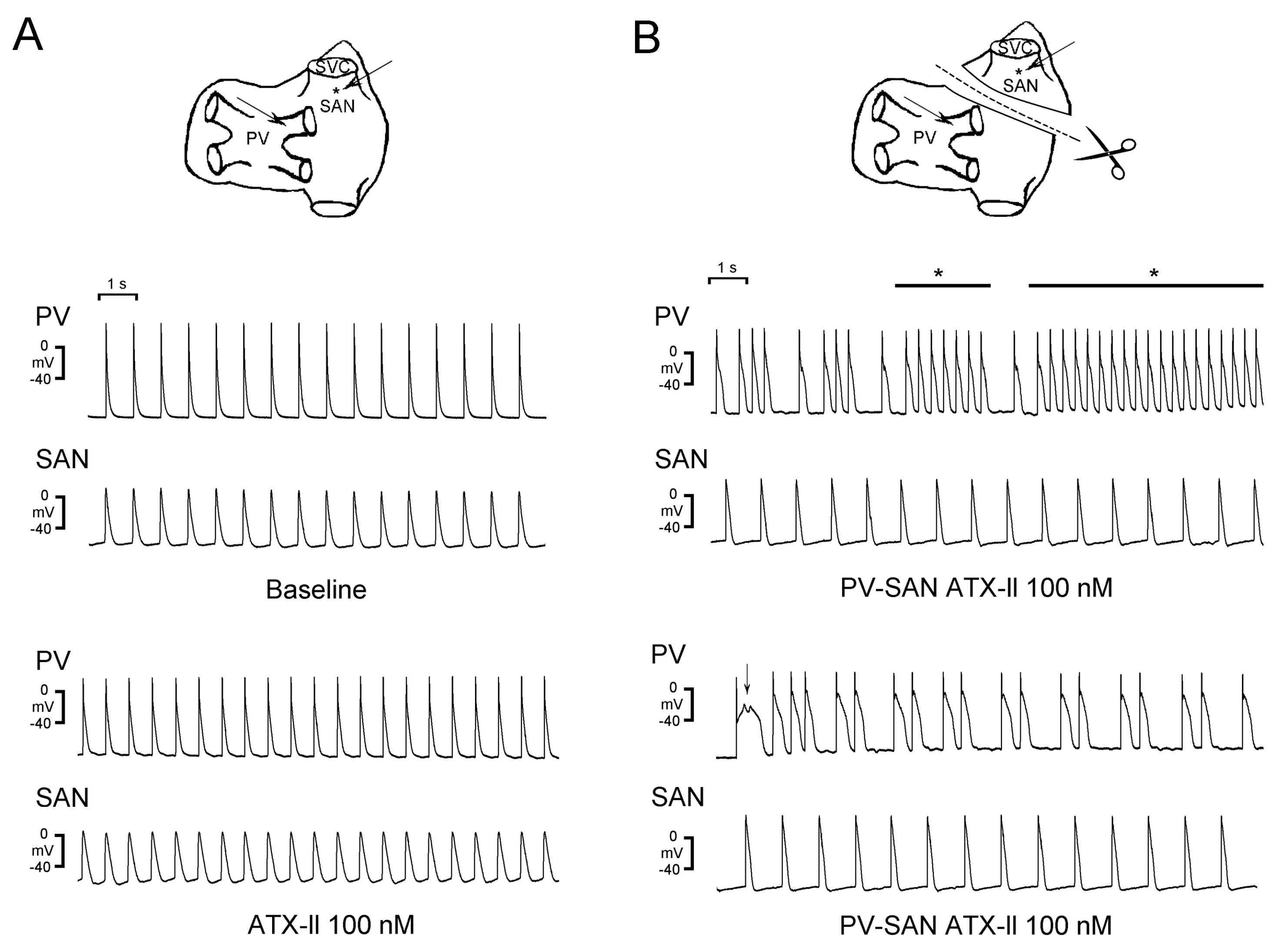
3.2. Crosstalk Between PVs and the SAN
3.3. HF Differentially Modulates Electrical Activity in the SAN and PVs
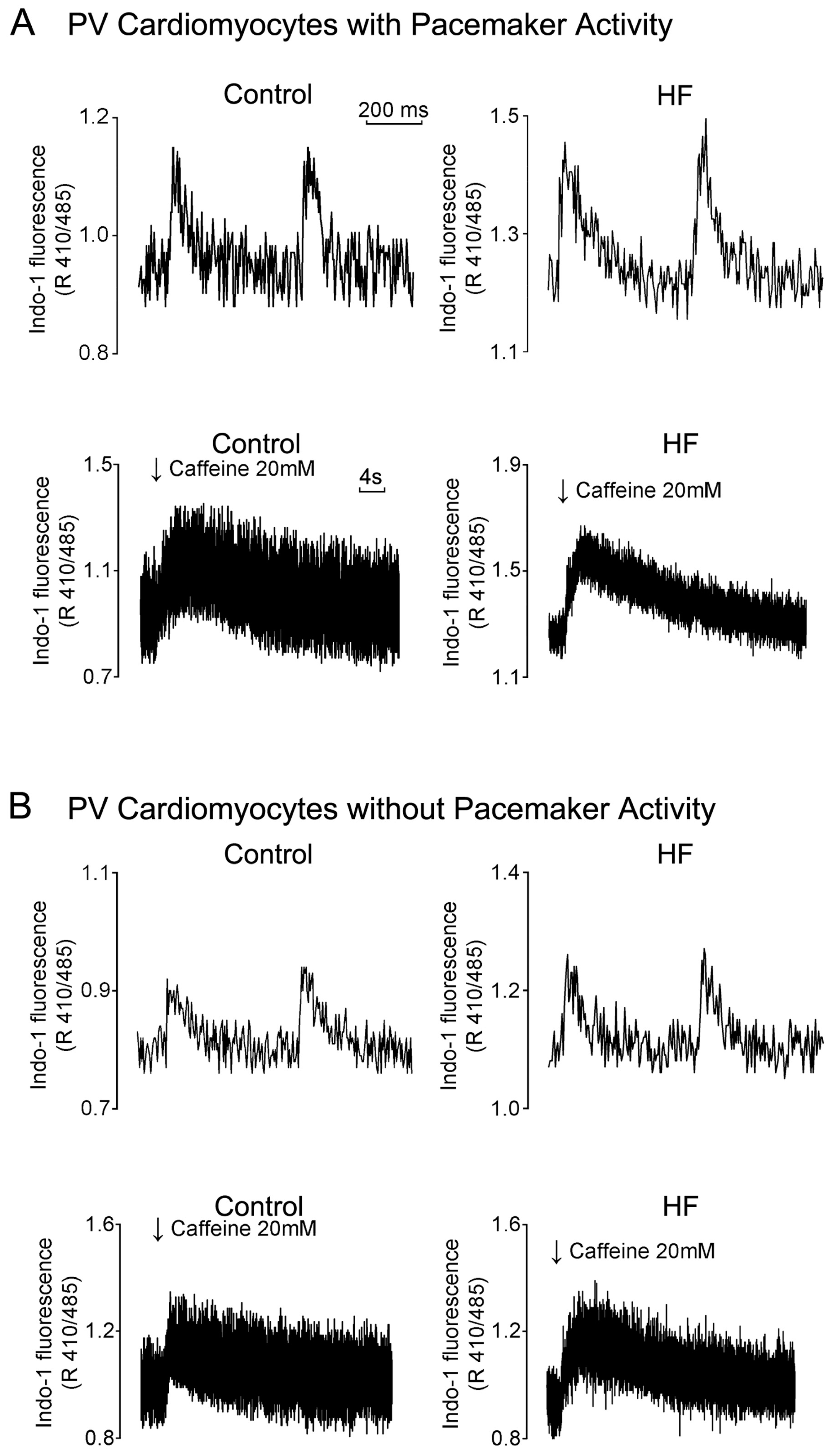
3.4. HF Differentially Induces Calcium Homeostasis Dysregulation in PVs and the SAN
3.5. HF-enhanced Fibrosis and Stretch Differentially Regulates PV and SAN Electrical Activity
3.6. Electrolyte Disturbance Differentially Regulates PV and SAN Electrical Activity
4. Therapeutic Implication for AF
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Braunwald, E. Cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. N. Engl. J. Med. 1997, 337, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Levy, D.; Vaziri, S.M.; D’Agostino, R.B.; Belanger, A.J.; Wolf, P.A. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA 1994, 271, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Mamas, M.A.; Caldwell, J.C.; Chacko, S.; Garratt, C.J.; Fath-Ordoubadi, F.; Neyses, L. A meta-analysis of the prognostic significance of atrial fibrillation in chronic heart failure. Eur. J. Heart Fail. 2009, 11, 676–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.J.; Larson, M.G.; Levy, D.; Vasan, R.S.; Leip, E.P.; Wolf, P.A.; D’Agostino, R.B.; Murabito, J.M.; Kannel, W.B.; Benjamin, E.J. Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: the Framingham Heart Study. Circulation 2003, 107, 2920–2925. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.K.; Nishida, K.; Kato, T.; Nattel, S. Atrial fibrillation pathophysiology: implications for management. Circulation 2011, 124, 2264–2274. [Google Scholar] [CrossRef]
- Wongcharoen, W.; Chen, S.A. The pathophysiology of atrial fibrillation in heart failure. J. Innov. Card. Rhythm Manag. 2012, 3, 865–869. [Google Scholar]
- Kotecha, D.; Piccini, J.P. Atrial fibrillation in heart failure: what should we do? Eur. Heart J. 2015, 36, 3250–3257. [Google Scholar] [CrossRef] [Green Version]
- Køber, L.; Swedberg, K.; McMurray, J.J.; Pfeffer, M.A.; Velazquez, E.J.; Diaz, R.; Maggioni, A.P.; Mareev, V.; Opolski, G.; Van de Werf, F.; et al. Previously known and newly diagnosed atrial fibrillation: a major risk indicator after a myocardial infarction complicated by heart failure or left ventricular dysfunction. Eur. J. Heart Fail. 2006, 8, 591–598. [Google Scholar] [CrossRef]
- Aronow, W.S.; Ahn, C.; Kronzon, I. Prognosis of congestive heart failure after prior myocardial infarction in older person with atrial fibrillation versus sinus rhythm. Am. J. Cardiol. 2001, 87, 224–225. [Google Scholar] [CrossRef]
- Prabhu, S.; Voskoboinik, A.; Kaye, D.M.; Kistler, P.M. Atrial fibrillation and heart failure - cause or effect? Heart Lung Circ. 2017, 26, 967–974. [Google Scholar] [CrossRef]
- Park, K.L.; Anter, E. Atrial fibrillation and heart failure: A review of the intersection of two cardiac epidemics. J. Atr. Fibrillation 2013, 6, 751–759. [Google Scholar]
- Zafrir, B.; Lund, L.H.; Laroche, C.; Ruschitzka, F.; Crespo-Leiro, MG.; Coats, A.J.S.; Anker, S.D.; Filippatos, G.; Seferovic, P.M.; Maggioni, A.P. Prognostic implications of atrial fibrillation in heart failure with reduced, mid-range, and preserved ejection fraction: a report from 14 964 patients in the European Society of Cardiology Heart Failure Long-Term Registry. Eur. Heart J. 2018, 39, 4277–4284. [Google Scholar] [CrossRef] [PubMed]
- Fedele, F.; Mancone, M.; Adamo, F.; Severino, P. Heart failure with preserved, mid-range, and reduced ejection fraction: The misleading definition of the new guidelines. Cardiol. Rev. 2017, 25, 4–5. [Google Scholar] [CrossRef]
- Fedele, F.; Severino, P.; Calcagno, S.; Mancone, M. Heart failure: TNM-like classification. J. Am. Coll. Cardiol. 2014, 63, 1959–1960. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Talajic, M.; Nattel, S.N.; Wyse, D.G.; Dorian, P.; Lee, K.L.; Bourassa, M.G.; Arnold, J.M.; Buxton, A.E.; Camm, A.J.; et al. Rhythm control versus rate control for atrial fibrillation and heart failure. N. Engl. J. Med. 2008, 358, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.C.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Europace 2016, 18, 1609–1678. [Google Scholar] [CrossRef] [PubMed]
- Haïssaguerre, M.; Jaïs, P.; Shah, D.C.; Takahashi, A.; Hocini, M.; Quiniou, G.; Garrigue, S.; Le Mouroux, A.; Le Métayer, P.; Clémenty, J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 1998, 339, 659–666. [Google Scholar] [CrossRef]
- Chen, S.A.; Hsieh, M.H.; Tai, C.T.; Tsai, C.F.; Prakash, V.S.; Yu, W.C.; Hsu, T.L.; Ding, Y.A.; Chang, M.S. Initiation of atrial fibrillation by ectopic beats originating from the pulmonary veins: electrophysiological characteristics, pharmacological responses, and effects of radiofrequency ablation. Circulation 1999, 100, 1879–1886. [Google Scholar] [CrossRef]
- Pappone, C.; Rosanio, S.; Oreto, G.; Tocchi, M.; Gugliotta, F.; Vicedomini, G.; Salvati, A.; Dicandia, C.; Mazzone, P.; Santinelli, V.; et al. Circumferential radiofrequency ablation of pulmonary vein ostia: a new anatomic approach for curing atrial fibrillation. Circulation 2000, 102, 2619–2628. [Google Scholar] [CrossRef]
- Chen, P.S.; Chen, L.S.; Fishbein, M.C.; Lin, S.F.; Nattel, S. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ. Res. 2014, 114, 1500–1515. [Google Scholar] [CrossRef]
- Wongcharoen, W.; Chen, Y.C.; Chen, Y.J.; Chen, S.Y.; Yeh, H.I.; Lin, C.I.; Chen, S.A. Aging increases pulmonary veins arrhythmogenesis and susceptibility to calcium regulation agents. Heart Rhythm 2007, 4, 1338–1349. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Chen, Y.C.; Kao, Y.H.; Lu, Y.Y.; Chen, S.A.; Chen, Y.J. Distinctive sodium and calcium regulation associated with sex differences in atrial electrophysiology of rabbits. Int. J. Cardiol. 2013, 168, 4658–4666. [Google Scholar] [CrossRef] [PubMed]
- Dobrzynski, H.; Boyett, M.R.; Anderson, R.H. New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation 2007, 115, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Birchfield, R.I.; Menefee, E.E.; Bryant, G.D. Disease of the sinoatrial node associated with bradycardia, asystole, syncope, and paroxysmal atrial fibrillation. Circulation 1957, 16, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Lin, Y.J.; Lo, L.W.; Chang, S.L.; Hu, Y.F.; Li, C.H.; Chao, T.F.; Yin, W.H.; Chen, S.A. Sinus node dysfunction in atrial fibrillation patients: the evidence of regional atrial substrate remodelling. Europace 2013, 151, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Lu, Y.Y.; Cheng, C.C.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. Sinoatrial node electrical activity modulates pulmonary vein arrhythmogenesis. Int. J. Cardiol. 2014, 173, 447–452. [Google Scholar] [CrossRef]
- Burch, G.E.; Romney, R.B. Functional anatomy and ‘throttle valve’ action of the pulmonary veins. Am. Heart J. 1954, 47, 58–66. [Google Scholar] [CrossRef]
- Nathan, H.; Eliakim, M. The junction between the left atrium and the pulmonary veins. An anatomic study of human hearts. Circulation 1966, 34, 412–422. [Google Scholar] [CrossRef]
- de Bakker, J.M.; Ho, S.Y.; Hocini, M. Basic and clinical electrophysiology of pulmonary vein ectopy. Cardiovasc. Res. 2002, 54, 287–294. [Google Scholar] [CrossRef]
- Hassink, R.J.; Aretz, H.T.; Ruskin, J.; Keane, D. Morphology of atrial myocardium in human pulmonary veins: a postmortem analysis in patients with and without atrial fibrillation. J. Am. Coll. Cardiol. 2003, 42, 1108–1114. [Google Scholar] [CrossRef]
- Ellis, C.R.; Saavedra, P.; Kanagasundram, A.; Estrada, J.C.; Montgomery, J.; Farrell, M.; Shen, S.; Crossley, G.H.; Michaud, G.; Shoemaker, M.B. Pulmonary vein sleeve length and association with body mass index and sex in atrial fibrillation. JACC Clin. Electrophysiol. 2018, 4, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Baba, M.; Hasebe, H.; Shinoda, Y.; Harunari, T.; Ebine, M.; Uehara, Y.; Watabe, H.; Takeyasu, N.; Horigome, H. Structural relation between the superior vena cava and pulmonary veins in patients with atrial fibrillation. J. Cardiovasc. Electrophysiol. 2017, 28, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chen, S.A.; Chang, M.S.; Lin, C.I. Arrhythmogenic activity of cardiac muscle in pulmonary veins of the dog: implication for the genesis of atrial fibrillation. Cardiovasc. Res. 2000, 48, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.K.; Chen, Y.C.; Chen, S.A.; Chen, Y.J. Ion channel remodeling in pulmonary vein arrhythmogenesis for atrial fibrillation. J. Exp. Clin. Med. 2011, 3, 108–111. [Google Scholar] [CrossRef]
- Chen, Y.C.; Pan, N.H.; Cheng, C.C.; Higa, S.; Chen, Y.J.; Chen, S.A. Heterogeneous expression of potassium currents and pacemaker currents potentially regulates arrhythmogenesis of pulmonary vein cardiomyocytes. J. Cardiovasc. Electrophysiol. 2009, 20, 1039–1045. [Google Scholar] [CrossRef]
- Khan, R. Identifying and understanding the role of pulmonary vein activity in atrial fibrillation. Cardiovasc. Res. 2004, 64, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Chen, S.A.; Chen, Y.J.; Tai, C.T.; Chan, P.; Lin, C.I. T-type calcium current in electrical activity of cardiomyocytes isolated from rabbit pulmonary vein. J. Cardiovasc. Electrophysiol. 2004, 15, 567–571. [Google Scholar] [CrossRef]
- Ehrlich, J.R.; Cha, T.J.; Zhang, L.; Chartier, D.; Melnyk, P.; Hohnloser, S.H.; Nattel, S. Cellular electrophysiology of canine pulmonary vein cardiomyocytes: action potential and ionic current properties. J. Physiol. 2003, 551, 801–813. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chen, S.A.; Chen, Y.C.; Yeh, H.I.; Chan, P.; Chang, M.S.; Lin, C.I. Effects of rapid atrial pacing on the arrhythmogenic activity of single cardiomyocytes from pulmonary veins: Implication in initiation of atrial fibrillation. Circulation 2001, 104, 2849–2854. [Google Scholar] [CrossRef]
- Boyett, M.R.; Inada, S.; Yoo, S.; Li, J.; Liu, J.; Tellez, J.; Greener, I.D.; Honjo, H.; Billeter, R.; Lei, M. Connexins in the sinoatrial and atrioventricular nodes. Adv. Cardiol. 2006, 42, 175–197. [Google Scholar]
- Verheule, S.; Wilson, E.E.; Arora, R.; Engle, S.K.; Scott, L.R.; Olgin, J.E. Tissue structure and connexin expression of canine pulmonary veins. Cardiovasc. Res. 2002, 55, 727–738. [Google Scholar] [CrossRef]
- Armour, J.A.; Murphy, D.A.; Yuan, B.X.; Macdonald, S.; Hopkins, D.A. Gross and microscopic anatomy of the human intrinsic cardiac nervous system. Anat. Rec. 1997, 247, 289–298. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chen, S.A.; Chen, Y.C.; Yeh, H.I.; Chang, M.S.; Lin, C.I. Electrophysiology of single cardiomyocytes isolated from rabbit pulmonary veins: implication in initiation of focal atrial fibrillation. Basic Res. Cardiol. 2002, 97, 26e34. [Google Scholar] [CrossRef]
- Severino, P.; Mariani, M.V.; Maraone, A.; Piro, A.; Ceccacci, A.; Tarsitani, L.; Maestrini, V.; Mancone, M.; Lavalle, C.; Pasquini, M.; et al. Triggers for atrial fibrillation: The role of anxiety. Cardiol. Res. Pract. 2019, 2019, 1208505. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.; Lazzara, R.; Szabo, B.; Liu, H.; Tang, D.; Li, Y.H.; Scherlag, B.J.; Po, S.S. Sodium-calcium exchange initiated by the Ca2+ transient: An arrhythmia trigger within pulmonary veins. J. Am. Coll. Cardiol. 2006, 47, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Wongcharoen, W.; Chen, Y.C.; Chen, Y.J.; Chang, C.M.; Yeh, H.I.; Lin, C.I.; Chen, S.A. Effects of a Na+/Ca2+ exchanger inhibitor on pulmonary vein electrical activity and ouabain-induced arrhythmogenicity. Cardiovasc. Res. 2006, 70, 497e508. [Google Scholar] [CrossRef]
- Honjo, H.; Boyett, M.R.; Niwa, R.; Inada, S.; Yamamoto, M.; Mitsui, K.; Horiuchi, T.; Shibata, N.; Kamiya, K.; Kodama, I. Pacing-induced spontaneous activity in myocardial sleeves of pulmonary veins after treatment with ryanodine. Circulation 2003, 107, 1937e43. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chen, Y.C.; Wongcharoen, W.; Lin, C.I.; Chen, S.A. K201, a novel antiarrhythmic drug on calcium handling and arrhythmogenic activity of pulmonary vein cardiomyocytes. Br. J. Pharmacol. 2008, 153, 915e25. [Google Scholar] [CrossRef]
- Chang, S.H.; Chen, Y.C.; Chiang, S.J.; Higa, S.; Cheng, C.C.; Chen, Y.J.; Chen, S.A. Increased Ca2+ sparks and sarcoplasmic reticulum Ca2+ stores potentially determine the spontaneous activity of pulmonary vein cardiomyocytes. Life Sci. 2008, 83, 284–292. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chen, Y.C.; Tai, C.T.; Yeh, H.I.; Lin, C.I.; Chen, S.A. Angiotensin II and angiotensin II receptor blocker modulate the arrhythmogenic activity of pulmonary veins. Br. J. Pharmacol. 2006, 147, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.F.; Chen, Y.C.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. Electromechanical effects of the direct renin inhibitor (aliskiren) on the pulmonary vein and atrium. Basic Res. Cardiol. 2011, 106, 979–993. [Google Scholar] [CrossRef]
- Kamkin, A.; Kiseleva, I.; Isenberg, G.; Wagner, K.D.; Günther, J.; Theres, H.; Scholz, H. Cardiac fibroblasts and the mechano-electric feedback mechanism in healthy and diseased hearts. Prog. Biophys. Mol. Biol. 2003, 82, 111–120. [Google Scholar] [CrossRef]
- Lin, W.S.; Prakash, V.S.; Tai, C.T.; Hsieh, M.H.; Tsai, C.F.; Yu, W.C.; Lin, Y.K.; Ding, Y.A.; Chang, M.S.; Chen, S.A. Pulmonary vein morphology in patients with paroxysmal atrial fibrillation initiated by ectopic beats originating from the pulmonary veins: implications for catheter ablation. Circulation 2000, 101, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Yamane, T.; Shah, D.C.; Jais, P.; Hocini, M.; Peng, J.T.; Deisenhofer, I.; Clémenty, J.; Haïssaguerre, M. Dilatation as a marker of pulmonary veins initiating atrial fibrillation. J. Interv. Card. Electrophysiol. 2002, 6, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Takase, B.; Nagata, M.; Matsui, T.; Kihara, T.; Kameyama, A.; Hamabe, A.; Noya, K.; Satomura, K.; Ishihara, M.; Kurita, A.; et al. Pulmonary vein dimensions and variation of branching pattern in patients with paroxysmal atrial fibrillation using magnetic resonance angiography. Jpn. Heart J. 2004, 45, 81–92. [Google Scholar] [CrossRef]
- Nattel, S.; Maguy, A.; Le Bouter, S.; Yeh, Y.H. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 2007, 87, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Tsao, H.M.; Yu, W.C.; Cheng, H.C.; Wu, M.H.; Tai, C.T.; Lin, W.S.; Ding, Y.A.; Chang, M.S.; Chen, S.A. Pulmonary vein dilation in patients with atrial fibrillation: detection by magnetic resonance imaging. J. Cardiovasc. Electrophysiol. 2001, 12, 809–813. [Google Scholar] [CrossRef]
- Chang, S.L.; Chen, Y.C.; Chen, Y.J.; Wangcharoen, W.; Lee, S.H.; Lin, C.I.; Chen, S.A. Mechanoelectrical feedback regulates the arrhythmogenic activity of pulmonary veins. Heart 2007, 93, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Kalifa, J.; Jalife, J.; Zaitsev, A.V.; Bagwe, S.; Warren, M.; Moreno, J.; Berenfeld, O.; Nattel, S. Intra-atrial pressure increases rate and organization of waves emanating from the superior pulmonary veins during atrial fibrillation. Circulation 2003, 108, 668–671. [Google Scholar] [CrossRef]
- Tsai, W.C.; Lee, T.I.; Chen, Y.C.; Kao, Y.H.; Lu, Y.Y.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. Testosterone replacement increases aged pulmonary vein and left atrium arrhythmogenesis with enhanced adrenergic activity. Int. J. Cardiol. 2014, 176, 110–118. [Google Scholar] [CrossRef]
- Hamabe, A.; Okuyama, Y.; Miyauchi, Y.; Zhou, S.; Pak, H.N.; Karagueuzian, H.S.; Fishbein, M.C.; Chen, P.S. Correlation between anatomy and electrical activation in canine pulmonary veins. Circulation 2003, 107, 1550–1555. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.Y.; Chen, Y.C.; Kao, Y.H.; Chen, S.A.; Chen, Y.J. Extracellular matrix of collagen modulates arrhythmogenic activity of pulmonary veins through p38 MAPK activation. J. Mol. Cell Cardiol. 2013, 59, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Boyett, M.R.; Honjo, H.; Kodama, I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc. Res. 2000, 47, 658–687. [Google Scholar] [CrossRef]
- Joyner, R.W.; van Capelle, F.J. Propagation through electrically coupled cells. How a small SA node drives a large atrium. Biophys. J. 1986, 50, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Schuessler, R.B.; Boineau, J.P.; Bromberg, B.I. Origin of the sinus impulse. J. Cardiovasc. Electrophysiol. 1996, 7, 263–274. [Google Scholar] [CrossRef] [PubMed]
- John, R.M.; Kumar, S. Sinus node and atrial arrhythmias. Circulation 2016, 133, 1892–1900. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.A.; Kang, P.S.; Matheson, M.; Gough, W.B. Jr.; El-Sherif, N. Coexistence of sick sinus rhythm and atrial flutter-fibrillation. Circulation 1981, 63, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Elvan, A.; Wylie, K.; Zipes, D.P. Pacing-induced chronic atrial fibrillation impairs sinus node function in dogs: electrophysiological remodeling. Circulation 1996, 94, 2953–2960. [Google Scholar] [CrossRef]
- Hadian, D.; Zipes, D.P.; Olgin, J.E.; Miller, J.M. Short-term rapid atrial pacing produces electrical remodeling of sinus node function in humans. J. Cardiovasc. Electrophysiol. 2002, 13, 584–586. [Google Scholar] [CrossRef]
- Yeh, Y.H.; Burstein, B.; Qi, X.Y.; Sakabe, M.; Chartier, D.; Comtois, P.; Wang, Z.; Kuo, C.T.; Nattel, S. Funny current downregulation and sinus node dysfunction associated with atrial tachyarrhythmia: a molecular basis for tachycardia-bradycardia syndrome. Circulation 2009, 119, 1576–1585. [Google Scholar] [CrossRef]
- Morillo, C.A.; Klein, G.J.; Jones, D.L.; Guiraudon, C.M. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation 1995, 91, 1588–1595. [Google Scholar] [CrossRef] [PubMed]
- Kezerashvili, A.; Krumerman, A.K.; Fisher, J.D. Sinus node dysfunction in atrial fibrillation: cause or effect? J. Atr. Fibrillation 2008, 1, 30. [Google Scholar] [PubMed]
- Davies, M.J.; Pomerance, A. Pathology of atrial fibrillation in man. Br. Heart J. 1972, 34, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.S.; Gomes, J.A.; Kelen, G.; El-Sherif, N. Role of Autonomic Regulatory Mechanisms in Sinoatrial Conduction and Sinus Node Automaticity in Sick Sinus Syndrome. Circulation 1981, 64, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, V.V.; Chang, R.; Glukhov, A.V.; Kostecki, G.; Janks, D.; Schuessler, R.B.; Efimov, I.R. Complex interactions between the sinoatrial node and atrium during reentrant arrhythmias in the canine heart. Circulation 2010, 122, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Grundvold, I.; Skretteberg, P.T.; Liestøl, K.; Erikssen, G.; Engeseth, K.; Gjesdal, K.; Kjeldsen, S.E.; Arnesen, H.; Erikssen, J.; Bodegard, J. Low heart rates predict incident atrial fibrillation in healthy middle-aged men. Circ. Arrhythm. Electrophysiol. 2013, 6, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Hai, J.; Chan, P.H.; Tse, H.F.; Siu, C.W. Slow Heart Rate Predicts New Occurrence of Atrial Fibrillation. Heart Lung Circ. 2015, 11, 1087–1093. [Google Scholar] [CrossRef]
- Wu, J.; Fan, X.; Yang, H.; Yan, L.; Xu, X.; Duan, H.; Wang, S.; Chu, Y. Usefulness of a low resting heart rate to predict recurrence of atrial fibrillation after catheter ablation in people ≥65 years of age. Am. J. Cardiol. 2018, 122, 97–101. [Google Scholar] [CrossRef]
- Ferrer, M.I. The sick sinus syndrome in atrial disease. JAMA 1968, 206, 645–646. [Google Scholar] [CrossRef]
- Hocini, M.; Sanders, P.; Deisenhofer, I.; Jais, P.; Hsu, L.F.; Scavee, C.; Weerasoriya, R.; Raybaud, F.; Macle, L.; Shah, D.C.; et al. Reverse remodeling of sinus node function after catheter ablation of atrial fibrillation in patients with prolonged sinus pauses. Circulation 2003, 108, 1172–1175. [Google Scholar] [CrossRef]
- Lamas, G.A.; Lee, K.L.; Sweeney, M.O.; Silverman, R.; Leon, A.; Yee, R.; Marinchak, R.A.; Flaker, G.; Schron, E.; Orav, E.J.; et al. Ventricular pacing or dual-chamber pacing for sinus-node dysfunction. N. Engl. J. Med. 2002, 346, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Amasyali, B.; Kilic, A.; Kilit, C. Sinus node dysfunction and atrial fibrillation: which one dominates? Int. J. Cardiol. 2014, 175, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D.W. Electrical activity of the pulmonary vein and its interaction with the right atrium in the guinea-pig. J. Physiol. 1981, 314, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Cheng, C.C.; Chen, Y.C.; Kao, Y.H.; Chen, S.A.; Chen, Y.J. Gap junction modifiers regulate electrical activities of the sinoatrial node and pulmonary vein: Therapeutic implications in atrial arrhythmogenesis. Int. J. Cardiol. 2016, 221, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.N.; Jaïs, P.; Cummings, J.; Di Biase, L.; Sanders, P.; Martin, D.O.; Kautzner, J.; Hao, S.; Themistoclakis, S.; Fanelli, R.; et al. Pulmonary-vein isolation for atrial fibrillation in patients with heart failure. N. Engl. J. Med. 2008, 359, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.G.; Haldar, S.K.; Hussain, W.; Sharma, R.; Francis, D.P.; Rahman-Haley, S.L.; McDonagh, T.A.; Underwood, S.R.; Markides, V.; Wong, T. A randomized trial to assess catheter ablation versus rate control in the management of persistent atrial fibrillation in heart failure. J. Am. Coll. Cardiol. 2013, 61, 1894–1903. [Google Scholar] [CrossRef]
- Di Biase, L.; Mohanty, P.; Mohanty, S.; Santangeli, P.; Trivedi, C.; Lakkireddy, D.; Reddy, M.; Jais, P.; Themistoclakis, S.; Dello Russo, A.; et al. Ablation versus amiodarone for treatment of persistent atrial fibrillation in patients with congestive heart failure and an implanted device: results from the AATAC multicenter randomized trial. Circulation 2016, 133, 1637–1644. [Google Scholar] [CrossRef]
- Marrouche, N.F.; Brachmann, J.; Andresen, D.; Siebels, J.; Boersma, L.; Jordaens, L.; Merkely, B.; Pokushalov, E.; Sanders, P.; Proff, J.; et al. Catheter ablation for atrial fibrillation with heart failure. N. Engl. J. Med. 2018, 378, 417–427. [Google Scholar] [CrossRef]
- Packer, M. Effect of catheter ablation on pre-existing abnormalities of left atrial systolic, diastolic, and neurohormonal functions in patients with chronic heart failure and atrial fibrillation. Eur. Heart J. 2019, 40, 1873–1879. [Google Scholar] [CrossRef]
- Chan, C.S.; Chen, Y.C.; Chang, S.L.; Lin, Y.K.; Kao, Y.H.; Chen, S.A.; Chen, Y.J. Heart failure differentially modulates the effects of ivabradine on the electrical activity of the sinoatrial node and pulmonary veins. J. Card. Fail. 2018, 24, 763–772. [Google Scholar] [CrossRef]
- Chang, S.L.; Chen, Y.C.; Yeh, Y.H.; Lin, Y.K.; Wu, T.J.; Lin, C.I.; Chen, S.A.; Chen, Y.J. Heart failure enhanced pulmonary vein arrhythmogenesis and dysregulated sodium and calcium homeostasis with increased calcium sparks. J. Cardiovasc. Electrophysiol. 2011, 22, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.L.; Chen, Y.C.; Yeh, Y.H.; Lai, Y.J.; Yeh, H.I.; Lin, C.I.; Lin, Y.K.; Lin, Y.J.; Wu, T.J.; Huang, Y.K.; et al. Heart failure enhances arrhythmogenesis in pulmonary veins. Clin. Exp. Pharmacol. Physiol. 2011, 38, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.; Kistler, P.M.; Morton, J.B.; Spence, S.J.; Kalman, J.M. Remodeling of sinus node function in patients with congestive heart failure: reduction in sinus node reserve. Circulation 2004, 110, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Wilders, R.; Coronel, R.; Ravesloot, J.H.; Verheijck, E.E. Ionic remodeling of sinoatrial node cells by heart failure. Circulation 2003, 108, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Zicha, S.; Fernández-Velasco, M.; Lonardo, G.; L’Heureux, N.; Nattel, S. Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc. Res. 2005, 66, 472–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.L.; Chuang, H.L.; Chen, Y.C.; Kao, Y.H.; Lin, Y.K.; Yeh, Y.H.; Chen, S.A.; Chen, Y.J. Heart failure modulates electropharmacological characteristics of sinoatrial nodes. Exp. Ther. Med. 2017, 13, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, T.; Park, H.W.; Han, S.; Shen, M.J.; Maruyama, M.; Kim, D.; Chen, P.S.; Lin, S.F. Ca2+ clock malfunction in a canine model of pacing-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1805–H1811. [Google Scholar] [CrossRef]
- Yanni, J.; Tellez, J.O.; Maczewski, M.; Mackiewicz, U.; Beresewicz, A.; Billeter, R.; Dobrzynski, H.; Boyett, M.R. Changes in ion channel gene expression underlying heart failure-induced sinoatrial node dysfunction. Circ. Heart Fail. 2011, 4, 496–508. [Google Scholar] [CrossRef]
- Melenovsky, V.; Hwang, S.J.; Redfield, M.M.; Zakeri, R.; Lin, G.; Borlaug, B.A. Left atrial remodeling and function in advanced heart failure with preserved or reduced ejection fraction. Circ. Heart Fail. 2015, 8, 295–303. [Google Scholar] [CrossRef]
- Sridhar, A.; Nishijima, Y.; Terentyev, D.; Khan, M.; Terentyeva, R.; Hamlin, R.L.; Nakayama, T.; Gyorke, S.; Cardounel, A.J.; Carnes, C.A. Chronic heart failure and the substrate for atrial fibrillation. Cardiovasc. Res. 2009, 84, 227–236. [Google Scholar] [CrossRef]
- Lange, G.; Lu, H.H.; Chang, A.; Brooks, C.M. Effect of stretch on the isolated cat sinoatrial node. Am. J. Physiol. 1966, 211, 1192–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, C.M.; Lu, H.H.; Lange, G.; Mangi, R.; Shaw, R.B.; Geoly, K. Effects of localized stretch of the sinoatrial node region of the dog heart. Am. J. Physiol. 1966, 211, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.C.; Amrani, M.; Yacoub, M.H. Heat stress proteins and myocardial protection: experimental model or potential clinical tool? Int. J. Biochem. Cell Biol. 1999, 31, 559–573. [Google Scholar] [CrossRef]
- Lu, Y.Y.; Cheng, C.C.; Chen, Y.C.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. Electrolyte disturbances differentially regulate sinoatrial node and pulmonary vein electrical activity: A contribution to hypokalemia- or hyponatremia-induced atrial fibrillation. Heart Rhythm 2016, 13, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, D.; Holmes, J.; Krum, H.; Altman, D.G.; Manzano, L.; Cleland, J.G.; Lip, G.Y.; Coats, A.J.; Andersson, B.; Kirchhof, P.; et al. Beta-Blockers in Heart Failure Collaborative Group. Efficacy of beta blockers in patients with heart failure plus atrial fibrillation: an individual-patient data meta-analysis. Lancet 2014, 384, 2235–2243. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.; Anker, S.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Di Francesco, D.; Camm, J.A. Heart rate lowering by specific and selective If current inhibition with ivabradine: a new therapeutic perspective in cardiovascular disease. Drugs 2004, 64, 1757–1765. [Google Scholar] [CrossRef]
- Reil, J.C.; Tardif, J.C.; Ford, I.; Lloyd, S.M.; O’Meara, E.; Komajda, M.; Borer, J.S.; Tavazzi, L.; Swedberg, K.; Böhm, M. Selective heart rate reduction with ivabradine unloads the left ventricle in heart failure patients. J. Am. Coll. Cardiol. 2013, 62, 1977–1985. [Google Scholar] [CrossRef]
- Tardif, J.C.; O’Meara, E.; Komajda, M.; B€ohm, M.; Borer, J.S.; Ford, I.; Tavazzi, L.; Swedberg, K.; SHIFT Investigators. Effects of selective heart rate reduction with ivabradine on left ventricular remodeling and function: results from the SHIFT echocardiography sub-study. Eur. Heart J. 2011, 32, 2507–2515. [Google Scholar] [CrossRef]
- Swedberg, K.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L.; SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef]
- Fox, K.; Komajda, M.; Ford, I.; Robertson, M.; Böhm, M.; Borer, J.S.; Steg, P.G.; Tavazzi, L.; Tendera, M.; Ferrari, R.; et al. Effect of ivabradine in patients with left-ventricular systolic dysfunction: a pooled analysis of individual patient data from the BEAUTIFUL and SHIFT trials. Eur. Heart J. 2013, 34, 2263–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, K.; Ford, I.; Steg, P.G.; Tardif, J.C.; Tendera, M.; Ferrari, R.; SIGNIFY investigators. Bradycardia and atrial fibrillation in patients with stable coronary artery disease treated with ivabradine: an analysis from the SIGNIFY study. Eur. Heart J. 2015, 36, 3291–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.I.; Pogoryelova, O.; Koref, M.S.; Bourke, J.P.; Teare, M.D.; Keavney, B.D. Atrial fibrillation associated with ivabradine treatment: meta-analysis of randomized controlled trials. Heart 2014, 100, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Sossalla, S.; Maier, L.S. Role of ranolazine in angina, heart failure, arrhythmias, and diabetes. Pharmacol. Ther. 2010, 133, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Sossalla, S.; Kallmeyer, B.; Wagner, S.; Mazur, M.; Maurer, U.; Toischer, K.; Schmitto, J.D.; Seipelt, R.; Schöndube, F.A.; Hasenfuss, G.; et al. Altered Na+ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 2010, 55, 2330–2342. [Google Scholar] [CrossRef] [PubMed]
- Murray, G.L.; Colomb, J. Ranolazine preserves and improves left ventricular ejection fraction and autonomic measures when added to guideline-driven therapy in chronic heart failure. Heart Int. 2014, 9, 66–73. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calcium Regulation | Pacemaker Current | Connexin | Stretch Channel | Vascular Property | Autonomic Control | |||
|---|---|---|---|---|---|---|---|---|
| 40 | 43 | 45 | ||||||
| PVs | +++ | + | + | ++ | ++ | + | + | + |
| SAN | ++ | ++ | + | - | ++ | + | - | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, C.-S.; Lin, Y.-K.; Chen, Y.-C.; Lu, Y.-Y.; Chen, S.-A.; Chen, Y.-J. Heart Failure Differentially Modulates Natural (Sinoatrial Node) and Ectopic (Pulmonary Veins) Pacemakers: Mechanism and Therapeutic Implication for Atrial Fibrillation. Int. J. Mol. Sci. 2019, 20, 3224. https://doi.org/10.3390/ijms20133224
Chan C-S, Lin Y-K, Chen Y-C, Lu Y-Y, Chen S-A, Chen Y-J. Heart Failure Differentially Modulates Natural (Sinoatrial Node) and Ectopic (Pulmonary Veins) Pacemakers: Mechanism and Therapeutic Implication for Atrial Fibrillation. International Journal of Molecular Sciences. 2019; 20(13):3224. https://doi.org/10.3390/ijms20133224
Chicago/Turabian StyleChan, Chao-Shun, Yung-Kuo Lin, Yao-Chang Chen, Yen-Yu Lu, Shih-Ann Chen, and Yi-Jen Chen. 2019. "Heart Failure Differentially Modulates Natural (Sinoatrial Node) and Ectopic (Pulmonary Veins) Pacemakers: Mechanism and Therapeutic Implication for Atrial Fibrillation" International Journal of Molecular Sciences 20, no. 13: 3224. https://doi.org/10.3390/ijms20133224