MPLs-Pred: Predicting Membrane Protein-Ligand Binding Sites Using Hybrid Sequence-Based Features and Ligand-Specific Models


Abstract

1. Introduction
2. Results and Discussion
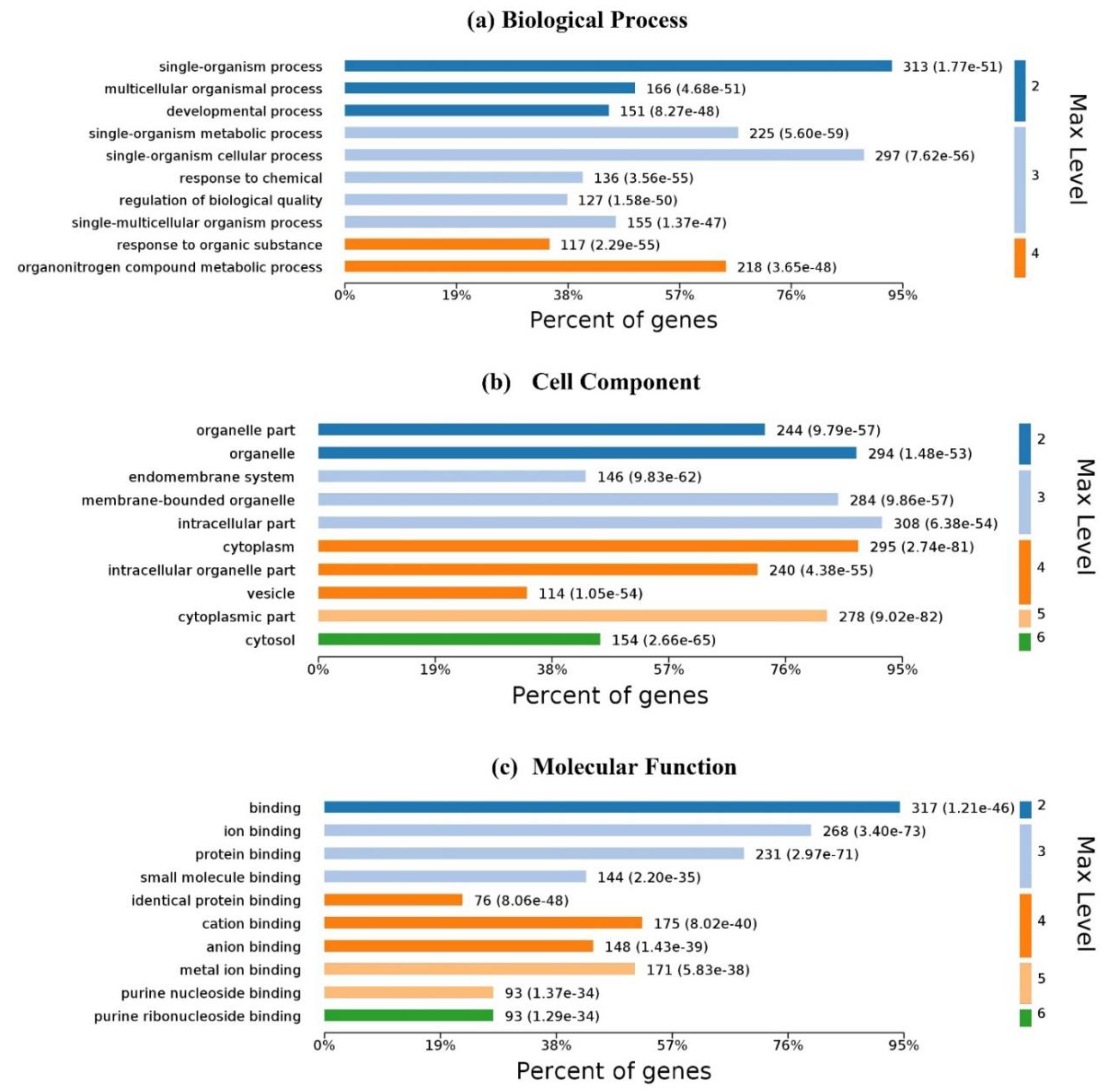
2.1. Characteristics of Ligand-Binding Residues
2.2. The Contribution of Features
2.3. The Random Under-Sampling Scheme Influences Predictor Performance
2.4. Comparison with other Machine Learning Methods Over Cross-Validation
2.5. Performance of MPLs-Pred
2.6. Case Studies
2.7. Comparison with Existing Methods on the Independent Testing Dataset
2.8. Homo’s Membrane Protein-Ligand Interactions
2.9. Performance Evaluation
3. Materials and Methods
3.1. Benchmark Datasets
3.2. Selected Features
3.2.1. Evolutionary Profiles (PSSM)
3.2.2. Topology Structure (TOPO)
3.2.3. Physicochemical Properties (PCP)
3.2.4. Primary Sequence Segment Descriptors (SeqSeg)
3.3. Random Under-Sampling (RUS)
3.4. Random Forest Classifier
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Andreeva, A.; Howorth, D.; Chothia, C.; Kulesha, E.; Murzin, A.G. SCOP2 prototype: A new approach to protein structure mining. Nucleic Acids Res. 2014, 42, D310–D314. [Google Scholar] [CrossRef]
- Almen, M.S.; Nordstrom, K.J.V.; Fredriksson, R.; Schioth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. Opinion—How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Phillips-Jones, M.K. Structural and biophysical characterisation of membrane protein-ligand binding Preface. BBA Biomembr. 2014, 1838, 1–2. [Google Scholar] [CrossRef][Green Version]
- Hernandez, M.; Ghersi, D.; Sanchez, R. SITEHOUND-web: A server for ligand binding site identification in protein structures. Nucleic Acids Res. 2009, 37, W413–W416. [Google Scholar] [CrossRef]
- Wass, M.N.; Kelley, L.A.; Sternberg, M.J. 3DLigandSite: Predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010, 38, W73–W469. [Google Scholar] [CrossRef]
- Roche, D.B.; Tetchner, S.J.; McGuffin, L.J. FunFOLD: An improved automated method for the prediction of ligand binding residues using 3D models of proteins. BMC Bioinform. 2011, 12, 160. [Google Scholar] [CrossRef]
- Honigschmid, P.; Frishman, D. Accurate prediction of helix interactions and residue contacts in membrane proteins. J. Struct. Biol. 2016, 194, 112–123. [Google Scholar] [CrossRef]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef]
- Hu, J.; Li, Y.; Zhang, M.; Yang, X.B.; Shen, H.B.; Yu, D.J. Predicting Protein-DNA Binding Residues by Weightedly Combining Sequence-Based Features and Boosting Multiple SVMs. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 1389–1398. [Google Scholar] [CrossRef]
- Yu, D.J.; Hu, J.; Yang, J.; Shen, H.B.; Tang, J.; Yang, J.Y. Designing template-free predictor for targeting protein-ligand binding sites with classifier ensemble and spatial clustering. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 994–1008. [Google Scholar] [CrossRef]
- Chen, K.; Mizianty, M.J.; Kurgan, L. Prediction and analysis of nucleotide-binding residues using sequence and sequence-derived structural descriptors. Bioinformatics 2012, 28, 331–341. [Google Scholar] [CrossRef]
- Fathima, A.J.; Murugaboopathi, G.; Selvam, P. Pharmacophore Mapping of Ligand Based Virtual Screening, Molecular Docking and Molecular Dynamic Simulation Studies for Finding Potent NS2B/NS3 Protease Inhibitors as Potential Anti-dengue Drug Compounds. Curr. Bioinform. 2018, 13, 606–616. [Google Scholar] [CrossRef]
- Hu, J.; Li, Y.; Zhang, Y.; Yu, D.J. ATPbind: Accurate Protein-ATP Binding Site Prediction by Combining Sequence-Profiling and Structure-Based Comparisons. J. Chem. Inf. Model. 2018, 58, 501–510. [Google Scholar] [CrossRef]
- Capra, J.A.; Laskowski, R.A.; Thornton, J.M.; Singh, M.; Funkhouser, T.A. Predicting Protein Ligand Binding Sites by Combining Evolutionary Sequence Conservation and 3D Structure. PLoS Comput. Biol. 2009, 5, 12. [Google Scholar] [CrossRef]
- Yang, J.Y.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Sun, P.; Ju, H.; Liu, Z.; Ning, Q.; Zhang, J.; Zhao, X.; Huang, Y.; Ma, Z.; Li, Y. Bioinformatics resources and tools for conformational B-cell epitope prediction. Comput. Math. Methods Med. 2013, 2013, 943636. [Google Scholar] [CrossRef]
- Suresh, M.X.; Gromiha, M.M.; Suwa, M. Development of a machine learning method to predict membrane protein-ligand binding residues using basic sequence information. Adv. Bioinform. 2015, 2015, 843030. [Google Scholar] [CrossRef]
- Moraes, I.; Evans, G.; Sanchez-Weatherby, J.; Newstead, S.; Stewart, P.D.S. Membrane protein structure determination the next generation. BBA Biomembr. 2014, 1838, 78–87. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef]
- Hong, H.; Chang, Y.C.; Bowie, J.U. Measuring transmembrane helix interaction strengths in lipid bilayers using steric trapping. Methods Mol. Biol. 2013, 1063, 37–56. [Google Scholar]
- Alonso, M.A.; Millan, J. The role of lipid rafts in signalling and membrane trafficking in T lymphocytes. J. Cell Sci. 2001, 114, 3957–3965. [Google Scholar]
- Maldonado, S.; Lopez, J. Imbalanced data classification using second-order cone programming support vector machines. Pattern Recogn. 2014, 47, 2070–2079. [Google Scholar] [CrossRef]
- O’Brien, R.; Ishwaran, H. A random forests quantile classifier for class imbalanced data. Pattern Recogn. 2019, 90, 232–249. [Google Scholar] [CrossRef]
- Zou, Q.; Li, X.; Jiang, Y.; Zhao, Y.; Wang, G. BinMemPredict: A Web Server and Software for Predicting Membrane Protein Types. Curr. Proteom. 2013, 10, 2–9. [Google Scholar] [CrossRef]
- Zhang, J.; Chai, H.; Gao, B.; Yang, G.; Ma, Z. HEMEsPred: Structure-Based Ligand-Specific Heme Binding Residues Prediction by Using Fast-Adaptive Ensemble Learning Scheme. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 147–156. [Google Scholar] [CrossRef]
- Sodhi, J.S.; Bryson, K.; McGuffin, L.J.; Ward, J.J.; Wernisch, L.; Jones, D.T. Predicting metal-binding site residues in low-resolution structural models. J. Mol. Biol. 2004, 342, 307–320. [Google Scholar] [CrossRef]
- Hu, J.; He, X.; Yu, D.J.; Yang, X.B.; Yang, J.Y.; Shen, H.B. A new supervised over-sampling algorithm with application to protein-nucleotide binding residue prediction. PLoS ONE 2014, 9, e107676. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Zou, Q.; Lin, G.; Jiang, X.; Liu, X.; Zeng, X. Sequence clustering in bioinformatics: An empirical study. Brief. Bioinform. 2019. [Google Scholar] [CrossRef]
- Jeong, J.C.; Lin, X.; Chen, X.W. On position-specific scoring matrix for protein function prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 308–315. [Google Scholar] [CrossRef]
- Zeng, B.; Honigschmid, P.; Frishman, D. Residue co-evolution helps predict interaction sites in alpha-helical membrane proteins. J. Struct. Biol. 2019, 206, 156–169. [Google Scholar] [CrossRef]
- Zangooei, M.H.; Jalili, S. Protein secondary structure prediction using DWKF based on SVR-NSGAII. Neurocomputing 2012, 94, 87–101. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, B.; Chai, H.T.; Ma, Z.Q.; Yang, G.F. Identification of DNA-binding proteins using multi-features fusion and binary firefly optimization algorithm. BMC Bioinform. 2016, 17, 323. [Google Scholar] [CrossRef]
- Qu, K.; Wei, L.; Zou, Q. A Review of DNA-binding Proteins Prediction Methods. Curr. Bioinform. 2019, 14, 246–254. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Von Heijne, G. Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 2006, 7, 909–918. [Google Scholar] [CrossRef]
- Tsirigos, K.D.; Govindarajant, S.; Bassot, C.; Vastermark, A.; Lamb, J.; Shu, N.J.; Elofsson, A. Topology of membrane proteins-predictions, limitations and variations. Curr. Opin. Struct. Biol. 2018, 50, 9–17. [Google Scholar] [CrossRef]
- Fuchs, A.; Kirschner, A.; Frishman, D. Prediction of helix-helix contacts and interacting helices in polytopic membrane proteins using neural networks. Proteins Struct. Funct. Bioinform. 2009, 74, 857–871. [Google Scholar] [CrossRef]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Kall, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef]
- Zhang, J.; Chai, H.T.; Yang, G.F.; Ma, Z.Q. Prediction of bioluminescent proteins by using sequence-derived features and lineage-specific scheme. BMC Bioinform. 2017, 18, 294. [Google Scholar] [CrossRef]
- Chai, H.T.; Zhang, J. Identification of Mammalian Enzymatic Proteins Based on Sequence-Derived Features and Species-Specific Scheme. IEEE Access 2018, 6, 8452–8458. [Google Scholar] [CrossRef]
- Suo, S.B.; Qiu, J.D.; Shi, S.P.; Sun, X.Y.; Huang, S.Y.; Chen, X.; Liang, R.P. Position-Specific Analysis and Prediction for Protein Lysine Acetylation Based on Multiple Features. PLoS ONE 2012, 7, e49108. [Google Scholar] [CrossRef]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2008, 36, D202–D205. [Google Scholar] [CrossRef]
- Wan, S.; Duan, Y.; Zou, Q. HPSLPred: An Ensemble Multi-label Classifier for Human Protein Subcellular Location Prediction with Imbalanced Source. Proteomics 2017, 17, 1700262. [Google Scholar] [CrossRef]
- Song, L.; Li, D.; Zeng, X.; Wu, Y.; Guo, L.; Zou, Q. nDNA-prot: Identification of DNA-binding proteins based on unbalanced classification. BMC Bioinform. 2014, 15, 298. [Google Scholar] [CrossRef]
- BREIMAN, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Su, R.; Liu, X.; Wei, L.; Zou, Q. Deep-Resp-Forest: A deep forest model to predict anti-cancer drug response. Methods 2019. [Google Scholar] [CrossRef]
- Zhao, X.; Zou, Q.; Liu, B.; Liu, X. Exploratory Predicting Protein Folding Model with Random Forest and Hybrid Features. Curr. Proteom. 2014, 11, 289–299. [Google Scholar] [CrossRef]
- Wei, Z.S.; Han, K.; Yang, J.Y.; Shen, H.B.; Yu, D.J. Protein-protein interaction sites prediction by ensembling SVM and sample-weighted random forests. Neurocomputing 2016, 193, 201–212. [Google Scholar] [CrossRef]
- Khan, M.; Hayat, M.; Khan, S.A.; Iqbal, N. Unb-DPC: Identify mycobacterial membrane protein types by incorporating un-biased dipeptide composition into Chou’s general PseAAC. J. Theor. Biol. 2017, 415, 13–19. [Google Scholar] [CrossRef]
- Hu, J.; Li, Y.; Yang, J.Y.; Shen, H.B.; Yu, D.J. GPCR-drug interactions prediction using random forest with drug-association-matrix-based post-processing procedure. Comput. Biol. Chem. 2016, 60, 59–71. [Google Scholar] [CrossRef]
- Liao, Z.; Ju, Y.; Zou, Q. Prediction of G Protein-Coupled Receptors with SVM-Prot Features and Random Forest. Scientifica 2016, 2016, 8309253. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature Combination | ACC | Spe | Sen | MCC |
|---|---|---|---|---|
| PSSM | 0.984 | 0.999 | 0.386 | 0.582 |
| TOPO | 0.548 | 0.309 | 0.788 | 0.113 |
| PCP | 0.973 | 0.998 | 0.216 | 0.413 |
| SeqSeg | 0.973 | 0.999 | 0.189 | 0.389 |
| PSSM+TOPO | 0.98 | 0.998 | 0.415 | 0.603 |
| PSSM+PCP | 0.979 | 0.998 | 0.421 | 0.599 |
| PSSM+ SeqSeg | 0.98 | 0.998 | 0.415 | 0.601 |
| TOPO+PCP | 0.973 | 0.998 | 0.22 | 0.416 |
| TOPO+SeqSeg | 0.904 | 0.981 | 0.139 | 0.201 |
| PCP+SeqSeg | 0.973 | 0.998 | 0.213 | 0.41 |
| PSSM+TOPO+PCP | 0.979 | 0.998 | 0.422 | 0.6 |
| PSSM+TOPO+SeqSeg | 0.98 | 0.998 | 0.416 | 0.603 |
| PSSM+PCP+SeqSeg | 0.979 | 0.998 | 0.421 | 0.601 |
| TOPO+PCP+SeqSeg | 0.973 | 0.998 | 0.216 | 0.414 |
| PSSM+TOPO+PCP+SeqSeg | 0.971 | 0.997 | 0.464 | 0.627 |
| Method | ACC | Spe | Sen | MCC |
|---|---|---|---|---|
| SVM | 0.9578 | 0.98 | 0.774 | 0.347 |
| Naïve Bayes | 0.844 | 0.85 | 0.67 | 0.246 |
| AdaBoost * | 0.974 | 0.995 | 0.334 | 0.472 |
| RF | 0.971 | 0.997 | 0.464 | 0.627 |
| Model | ACC | Spe | Sen | MCC |
|---|---|---|---|---|
| Universal | 0.971 | 0.997 | 0.464 | 0.627 |
| Drug | 0.973 | 1.0 | 0.153 | 0.366 |
| Metal | 0.984 | 0.997 | 0.589 | 0.704 |
| Biomacromolecule | 0.936 | 0.993 | 0.481 | 0.629 |
| Model | ACC | Spe | Sen | MCC |
|---|---|---|---|---|
| Universal | 0.996 | 0.998 | 0.618 | 0.63 |
| Drug | 0.997 | 1.0 | 0.397 | 0.604 |
| Metal | 0.996 | 0.998 | 0.759 | 0.7 |
| Biomacromolecule | 0.997 | 0.999 | 0.596 | 0.692 |
| Method | ACC | Spe | Sen | MCC |
|---|---|---|---|---|
| Tm-lig | 0.857 | 0.857 | 0.769 | 0.126 |
| MPLs-Pred | 0.996 | 0.998 | 0.618 | 0.63 |
| Dataset | Training Dataset | Testing Dataset | ||||
|---|---|---|---|---|---|---|
| No. of Proteins | No. of Residues | Ratio 1 | No. of Protein | No. of Residues | Ratio 1 | |
| Universal | 2500 2 | (10,143, 1,524,372) | 1:150 | 234 | (836, 164,792) | 1:197 |
| Drug | 655 | (1839, 386,979) | 1:210 | 45 | (121, 25,193) | 1:208 |
| Metal | 1375 | (5734, 804,610) | 1:140 | 117 | (503, 85,437) | 1:170 |
| Biomacromolecule | 857 | (2505, 435,298) | 1:174 | 67 | (161, 35,022) | 1:218 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.; Liu, Z.; Zhang, E.; He, F.; Ma, Z.; Wang, H. MPLs-Pred: Predicting Membrane Protein-Ligand Binding Sites Using Hybrid Sequence-Based Features and Ligand-Specific Models. Int. J. Mol. Sci. 2019, 20, 3120. https://doi.org/10.3390/ijms20133120
Lu C, Liu Z, Zhang E, He F, Ma Z, Wang H. MPLs-Pred: Predicting Membrane Protein-Ligand Binding Sites Using Hybrid Sequence-Based Features and Ligand-Specific Models. International Journal of Molecular Sciences. 2019; 20(13):3120. https://doi.org/10.3390/ijms20133120
Chicago/Turabian StyleLu, Chang, Zhe Liu, Enju Zhang, Fei He, Zhiqiang Ma, and Han Wang. 2019. "MPLs-Pred: Predicting Membrane Protein-Ligand Binding Sites Using Hybrid Sequence-Based Features and Ligand-Specific Models" International Journal of Molecular Sciences 20, no. 13: 3120. https://doi.org/10.3390/ijms20133120
APA StyleLu, C., Liu, Z., Zhang, E., He, F., Ma, Z., & Wang, H. (2019). MPLs-Pred: Predicting Membrane Protein-Ligand Binding Sites Using Hybrid Sequence-Based Features and Ligand-Specific Models. International Journal of Molecular Sciences, 20(13), 3120. https://doi.org/10.3390/ijms20133120