Effects of the Pentapeptide P33 on Memory and Synaptic Plasticity in APP/PS1 Transgenic Mice: A Novel Mechanism Presenting the Protein Fe65 as a Target

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
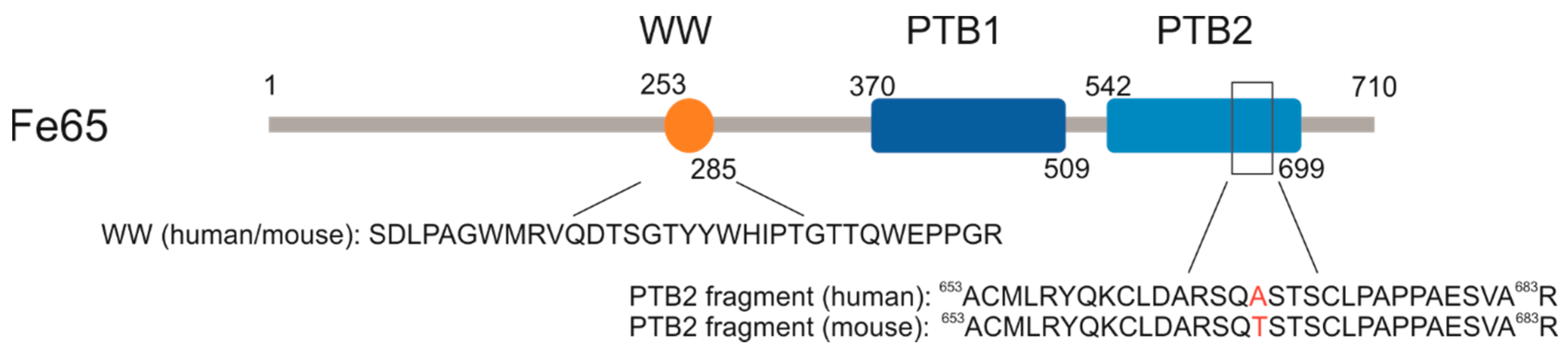
2.1. P33 Binds to Fe65-WW but Not to Pin1-WW In Vitro
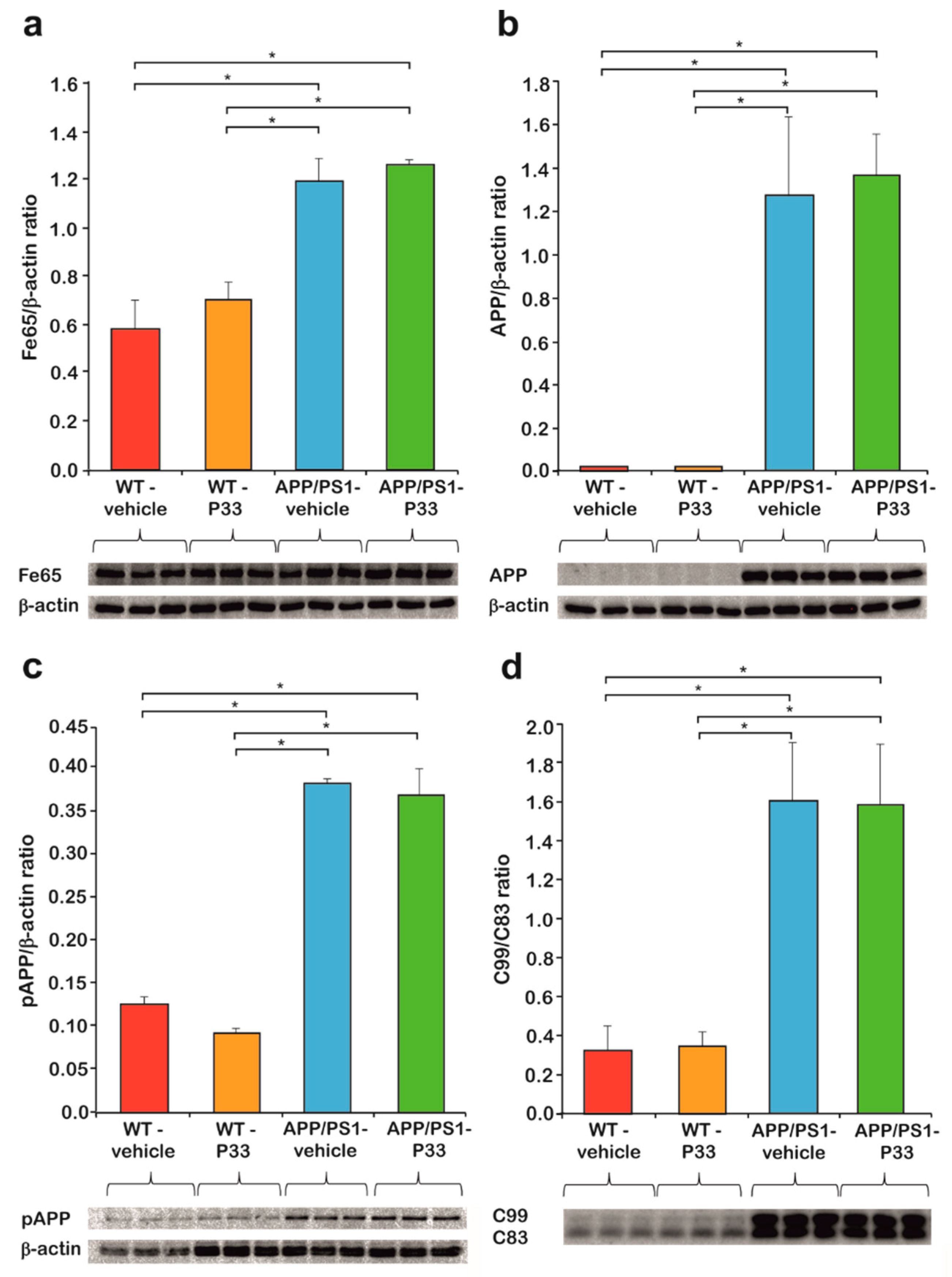
2.2. P33 Treatment Does Not Influence Key Protein Levels Involved in the Fe65/APP Route in the Transgenic Mice
2.3. P33 Restores the Pathologically Reduced Spine Density and Protects the Synapses
2.4. P33 Significantly Reduces the Amounts of Both Soluble and Deposited Aβ Forms in the APP/PS1 Animals
2.5. P33 Hinders Inflammatory Processes in the Mouse Brain
2.6. P33 Exerts a Positive Effect on the Learning Ability and Memory Functions in an MWM Paradigm
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis
4.3. Purification
4.4. ITC
4.5. Animals
4.6. MWM
4.7. ELISA
4.8. WB
4.9. Quantification of Spine Density
4.10. Immunohistochemistry
4.11. Quantification of the Immunohistochemical Data
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RIP | regulated intramembrane proteolysis |
| APP | amyloid precursor protein |
| AICD | intracellular domain of APP |
| AD | Alzheimer’s disease |
| Aβ | beta amyloid |
| HEK293 | human embryonic kidney cells 293 |
| H4 | human H4 neuroglioma cells |
| MDCK | Madin–Darby Canine Kidney cells |
| ITC | isothermal titration calorimetry |
| APP/PS1 | APPswe/PS1ΔE9 double transgenic |
| WT | C57BL/6 wild type |
| MWM | Morris water maze |
| GFAP | glial fibrillary acidic protein |
| Iba1 | ionized calcium-binding adapter molecule 1 |
| DCC | N,N′-dicyclohexyl-carbodiimide |
| HOBt | 1-hydroxybenzotriazole |
| HATU | 1-[bis(dimethylamino)-methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxid hexafluorophos-phate |
| MBHA x HCl | 4-methylbenz-hydryl-amine hydrochloride |
| DIPEA | N,N-diisopropylethylamine |
| TFA | trifluoroacetic acid |
| ACN | acetonitrile |
| RT | room temperature |
| HC | hippocampus |
| CTX | cerebral cortex |
| WB | Western blot |
| TBS | Tris-buffered saline |
| BSA | bovine serum albumin |
| DAB | 3,3′-diaminobenzidine |
| DPX | dibutyl phthalate xylene |
| ROI | regions of interest |
| CNS | central nervous system |
| mFe65 | mouse Fe65 |
| hAPP | human APP |
| ELISA | enzyme-linked immunosorbent assay |
| PSD95 | postsynaptic density protein 95 |
| SYN | synaptophysin |
| NFT | neurofibrillary tangles |
References
- Borquez, D.A.; Gonzalez-Billault, C. The amyloid precursor protein intracellular domain-Fe65 multiprotein complexes: A challenge to the amyloid hypothesis for Alzheimer’s disease? Int. J. Alzheimer’s Dis. 2012, 353145, 10. [Google Scholar] [CrossRef] [PubMed]
- Borg, J.-P.; Yang, Y.; De Taddeo-Borg, M.; Margolis, B.; Turner, R.S. The X11α protein slows cellular amyloid precursor protein processing and reduces Aβ40 and Aβ42 secretion. J. Biol. Chem. 1998, 273, 14761–14766. [Google Scholar] [CrossRef] [PubMed]
- Dunning, C.J.R.; Black, H.L.; Andrews, K.L.; Davenport, E.C.; Conboy, M.; Chawla, S.; Dowle, A.A.; Ashford, D.; Thomas, J.R.; Evans, G.J.O. Multisite tyrosine phosphorylation of the N-terminus of Mint1/X11α by Src kinase regulates the trafficking of amyloid precursor protein. J. Neurochem. 2016, 137, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.C.J.; McLoughlin, D.M.; Lau, K.-F.; Tennant, M.E.; Rogelj, B. The X11 proteins, Aβ production and Alzheimer’s disease. Trends Neurosci. 2006, 29, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Tamayev, R.; Zhou, D.; D’Adamio, L. The interactome of the amyloid β precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol. Neurodegener. 2009, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Hoe, H.-S.; Tran, T.S.; Matsuoka, Y.; Howell, B.W.; Rebeck, G.W. DAB1 and Reelin Effects on Amyloid Precursor Protein and ApoE Receptor 2 Trafficking and Processing. J. Biol. Chem. 2006, 281, 35176–35185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, C.; Dolcini, V.; Salis, S.; Venezia, V.; Zambrano, N.; Russo, T.; Schettini, G. Signal Transduction through Tyrosine-phosphorylated C-terminal Fragments of Amyloid Precursor Protein via an Enhanced Interaction with Shc/Grb2 Adaptor Proteins in Reactive Astrocytes of Alzheimer’s Disease Brain. J. Biol. Chem. 2002, 277, 35282–35288. [Google Scholar] [CrossRef]
- Tarr, P.E.; Roncarati, R.; Pelicci, G.; Pelicci, P.G.; D’Adamio, L. Tyrosine phosphorylation of the β-amyloid precursor protein cytoplasmic tail promotes interaction with Shc. J. Biol. Chem. 2002, 277, 16798–16804. [Google Scholar] [CrossRef]
- Bukhari, H.; Glotzbach, A.; Kolbe, K.; Leonhardt, G.; Loosse, C.; Mueller, T. Small things matter: Implications of APP intracellular domain AICD nuclear signaling in the progression and pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2017, 156, 189–213. [Google Scholar] [CrossRef]
- Chang, K.-A.; Kim, H.-S.; Ha, T.-Y.; Ha, J.-W.; Shin, K.Y.; Jeong, Y.H.; Lee, J.-P.; Park, C.-H.; Kim, S.; Baik, T.-K.; et al. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol. Cell. Biol. 2006, 26, 4327–4338. [Google Scholar] [CrossRef]
- Ando, K.; Iijima, K.-I.; Elliott, J.I.; Kirino, Y.; Suzuki, T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of β-amyloid. J. Biol. Chem. 2001, 276, 40353–40361. [Google Scholar] [CrossRef] [PubMed]
- Penke, B.; Bogar, F.; Paragi, G.; Gera, J.; Fulop, L. Key Peptides and Proteins in Alzheimer’s Disease. Curr. Protein Pept. Sci. 2019, 20, 577–599. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, D.M.; Miller, C.C.J. The FE65 proteins and Alzheimer’s disease. J. Neurosci. Res. 2008, 86, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Sabo, S.L.; Ikin, A.F.; Buxbaum, J.D.; Greengard, P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J. Cell Biol. 2001, 153, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Sabo, S.L.; Ikin, A.F.; Buxbaum, J.D.; Greengard, P. The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J. Neurosci. 2003, 23, 5407–5415. [Google Scholar] [CrossRef] [PubMed]
- Minopoli, G.; Gargiulo, A.; Parisi, S.; Russo, T. Fe65 matters: New light on an old molecule. IUBMB Life 2012, 64, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Santiard-Baron, D.; Langui, D.; Delehedde, M.; Delatour, B.; Schombert, B.; Touchet, N.; Tremp, G.; Paul, M.-F.; Blanchard, V.; Sergeant, N.; et al. Expression of human FE65 in amyloid precursor protein transgenic mice is associated with a reduction in β-amyloid load. J. Neurochem. 2005, 93, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Tesco, G.; Jeong, W.J.; Lindsley, L.; Eckman, E.A.; Eckman, C.B.; Tanzi, R.E.; Guenette, S.Y. Generation of the β-Amyloid Peptide and the Amyloid Precursor Protein C-terminal Fragment γ Are Potentiated by FE65L1. J. Biol. Chem. 2003, 278, 51100–51107. [Google Scholar] [CrossRef]
- Guenette, S.Y.; Chen, J.; Ferland, A.; Haass, C.; Capell, A.; Tanzi, R.E. hFE65L influences amyloid precursor protein maturation and secretion. J. Neurochem. 1999, 73, 985–993. [Google Scholar] [CrossRef]
- Sabo, S.L.; Lanier, L.M.; Ikin, A.F.; Khorkova, O.; Sahasrabudhe, S.; Greengard, P.; Buxbaum, J.D. Regulation of β-amyloid secretion by FE65, an amyloid protein precursor-binding protein. J. Biol. Chem. 1999, 274, 7952–7957. [Google Scholar] [CrossRef]
- Tanahashi, H.; Tabira, T. Characterization of an amyloid precursor protein-binding protein Fe65L2 and its novel isoforms lacking phosphotyrosine-interaction domains. Biochem. J. 2002, 367, 687–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Dong, Y.; Maeda, U.; Xia, W.; Tanzi, R.E. RNA Interference Silencing of the Adaptor Molecules ShcC and Fe65 Differentially Affect Amyloid Precursor Protein Processing and Aβ Generation. J. Biol. Chem. 2007, 282, 4318–4325. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Lyckman, A.; Wang, L.; Eckman, E.A.; Guenette, S.Y. FE65 proteins regulate NMDA receptor activation-induced amyloid precursor protein processing. J. Neurochem. 2011, 119, 377–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenette, S.; Chang, Y.; Hiesberger, T.; Richardson, J.A.; Eckman, C.B.; Eckman, E.A.; Hammer, R.E.; Herz, J. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO J. 2006, 25, 420–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Hu, Q.; Hearn, M.G.; Shimizu, K.; Ware, C.B.; Liggitt, D.H.; Jin, L.-W.; Cool, B.H.; Storm, D.R.; Martin, G.M. Isoform-specific knockout of FE65 leads to impaired learning and memory. J. Neurosci. Res. 2004, 75, 12–24. [Google Scholar] [CrossRef]
- Macias, M.J.; Hyvoenen, M.; Baraldi, E.; Schultz, J.; Sudol, M.; Saraste, M.; Oschkinat, H. Structure of the WW domain of a kinase-associated protein complexed with a proline-rich peptide. Nature 1996, 382, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M.; Sliwa, K.; Russo, T. Functions of WW domains in the nucleus. FEBS Lett. 2001, 490, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Staub, O.; Rotin, D. WW domains. Structure 1996, 4, 495–499. [Google Scholar] [CrossRef] [Green Version]
- Sudol, M.; Chen, H.I.; Bougeret, C.; Einbond, A.; Bork, P. Characterization of a novel protein-binding module—The WW domain. FEBS Lett. 1995, 369, 67–71. [Google Scholar] [CrossRef]
- Kato, Y.; Ito, M.; Kawai, K.; Nagata, K.; Tanokura, M. Determinants of ligand specificity in groups I and IV WW domains as studied by surface plasmon resonance and model building. J. Biol. Chem. 2002, 277, 10173–10177. [Google Scholar] [CrossRef]
- Lambrechts, A.; Kwiatkowski, A.V.; Lanier, L.M.; Bear, J.E.; Vandekerckhove, J.; Ampe, C.; Gertler, F.B. cAMP-dependent protein kinase phosphorylation of EVL, a Mena/VASP relative, regulates its interaction with actin and SH3 domains. J. Biol. Chem. 2000, 275, 36143–36151. [Google Scholar] [CrossRef] [PubMed]
- Masin, M.; Kerschensteiner, D.; Duemke, K.; Rubio, M.E.; Soto, F. Fe65 Interacts with P2X2 Subunits at Excitatory Synapses and Modulates Receptor Function. J. Biol. Chem. 2006, 281, 4100–4108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meiyappan, M.; Birrane, G.; Ladias, J.A.A. Structural Basis for Polyproline Recognition by the FE65 WW Domain. J. Mol. Biol. 2007, 372, 970–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telese, F.; Bruni, P.; Donizetti, A.; Gianni, D.; D’Ambrosio, C.; Scaloni, A.; Zambrano, N.; Rosenfeld, M.G.; Russo, T. Transcription regulation by the adaptor protein Fe65 and the nucleosome assembly factor SET. EMBO Rep. 2005, 6, 77–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermekova, K.S.; Zambrano, N.; Linn, H.; Minopoli, G.; Gertler, F.; Russo, T.; Sudol, M. The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. J. Biol. Chem. 1997, 272, 32869–32877. [Google Scholar] [CrossRef]
- Vargas, L.M.; Cerpa, W.; Munoz, F.J.; Zanlungo, S.; Alvarez, A.R. Amyloid-β oligomers synaptotoxicity: The emerging role of EphA4/c-Abl signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1148–1159. [Google Scholar] [CrossRef]
- Pastorino, L.; Sun, A.; Lu, P.-J.; Zhou, X.Z.; Balastik, M.; Finn, G.; Wulf, G.; Lim, J.; Li, S.-H.; Li, X.; et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production. Nature 2006, 440, 528–534. [Google Scholar] [CrossRef]
- Liou, Y.-C.; Sun, A.; Ryo, A.; Zhou, X.Z.; Yu, Z.-X.; Huang, H.-K.; Uchida, T.; Bronson, R.; Bing, G.; Li, X.; et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 2003, 424, 556–561. [Google Scholar] [CrossRef]
- Maudsley, S.; Mattson, M.P. Protein twists and turns in Alzheimer disease. Nat. Med. 2006, 12, 392–393. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: Evidence for augmentation of a 42-specific γ secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef]
- Lee, M.-S.; Kao, S.-C.; Lemere, C.A.; Xia, W.; Tseng, H.-C.; Zhou, Y.; Neve, R.; Ahlijanian, M.K.; Tsai, L.-H. APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 2003, 163, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Vanderstichele, H.; Van, K.E.; Hesse, C.; Davidsson, P.; Buyse, M.A.; Andreasen, N.; Minthon, L.; Wallin, A.; Blennow, K.; Vanmechelen, E. Standardization of measurement of β-amyloid1–42 in cerebrospinal fluid and plasma. Amyloid 2000, 7, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Masliah, E.; Yu, G.-Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Ohgi, K.A.; Rose, D.W.; Koo, E.H.; Glass, C.K.; Rosenfeld, M.G. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-κB and β-amyloid precursor protein. Cell 2002, 110, 55–67. [Google Scholar] [CrossRef]
- Delatour, B.; Mercken, L.; El Hachimi, K.H.; Colle, M.-A.; Pradier, L.; Duyckaerts, C. FE65 in Alzheimer’s disease: Neuronal distribution and association with neurofibrillary tangles. Am. J. Pathol. 2001, 158, 1585–1591. [Google Scholar] [CrossRef]
- Chen, D.; Liu, S.; Zhang, W.; Sun, L. Rational design of YAP WW1 domain-binding peptides to target TGFβ/BMP/Smad-YAP interaction in heterotopic ossification. J. Pept. Sci. 2015, 21, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Rubini, C.; Ruzza, P.; Spaller, M.R.; Siligardi, G.; Hussain, R.; Udugamasooriya, D.G.; Bellanda, M.; Mammi, S.; Borgogno, A.; Calderan, A.; et al. Recognition of lysine-rich peptide ligands by murine cortactin SH3 domain: CD, ITC, and NMR studies. Biopolymers 2010, 94, 298–306. [Google Scholar] [CrossRef]
- Sudol, M.; Hunter, T. New wrinkles for an old domain. Cell 2000, 103, 1001–1004. [Google Scholar] [CrossRef]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid β oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef]
- Wei, W.; Nguyen, L.N.; Kessels, H.W.; Hagiwara, H.; Sisodia, S.; Malinow, R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 2010, 13, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Suedhof, T.C. Dissection of Amyloid-β Precursor Protein-dependent Transcriptional Transactivation. J. Biol. Chem. 2004, 279, 24601–24611. [Google Scholar] [CrossRef] [PubMed]
- Feilen, L.P.; Haubrich, K.; Stier, G.; Sinning, I.; Wild, K.; Haubrich, K.; Simon, B.; Strecker, P.; Eggert, S.; Kins, S.; et al. Fe65-PTB2 Dimerization Mimics Fe65-APP Interaction. Front. Mol. Neurosci. 2017, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Borbely, E.; Horvath, J.; Furdan, S.; Bozso, Z.; Penke, B.; Fulop, L. Simultaneous changes of spatial memory and spine density after intrahippocampal administration of fibrillar Aβ1-42 to the rat brain. BioMed Res. Int. 2014, 2014, 345305. [Google Scholar] [CrossRef] [PubMed]
- Nagy, D.; Kocsis, K.; Fuzik, J.; Marosi, M.; Kis, Z.; Teichberg, V.I.; Toldi, J.; Farkas, T. Kainate postconditioning restores LTP in ischemic hippocampal CA1: Onset-dependent second pathophysiological stress. Neuropharmacology 2011, 61, 1026–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szögi, T.; Schuster, I.; Borbély, E.; Gyebrovszki, A.; Bozsó, Z.; Gera, J.; Rajkó, R.; Sántha, M.; Penke, B.; Fülöp, L. Effects of the Pentapeptide P33 on Memory and Synaptic Plasticity in APP/PS1 Transgenic Mice: A Novel Mechanism Presenting the Protein Fe65 as a Target. Int. J. Mol. Sci. 2019, 20, 3050. https://doi.org/10.3390/ijms20123050
Szögi T, Schuster I, Borbély E, Gyebrovszki A, Bozsó Z, Gera J, Rajkó R, Sántha M, Penke B, Fülöp L. Effects of the Pentapeptide P33 on Memory and Synaptic Plasticity in APP/PS1 Transgenic Mice: A Novel Mechanism Presenting the Protein Fe65 as a Target. International Journal of Molecular Sciences. 2019; 20(12):3050. https://doi.org/10.3390/ijms20123050
Chicago/Turabian StyleSzögi, Titanilla, Ildikó Schuster, Emőke Borbély, Andrea Gyebrovszki, Zsolt Bozsó, János Gera, Róbert Rajkó, Miklós Sántha, Botond Penke, and Lívia Fülöp. 2019. "Effects of the Pentapeptide P33 on Memory and Synaptic Plasticity in APP/PS1 Transgenic Mice: A Novel Mechanism Presenting the Protein Fe65 as a Target" International Journal of Molecular Sciences 20, no. 12: 3050. https://doi.org/10.3390/ijms20123050