The Complete Chloroplast Genomes of Punica granatum and a Comparison with Other Species in Lythraceae

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
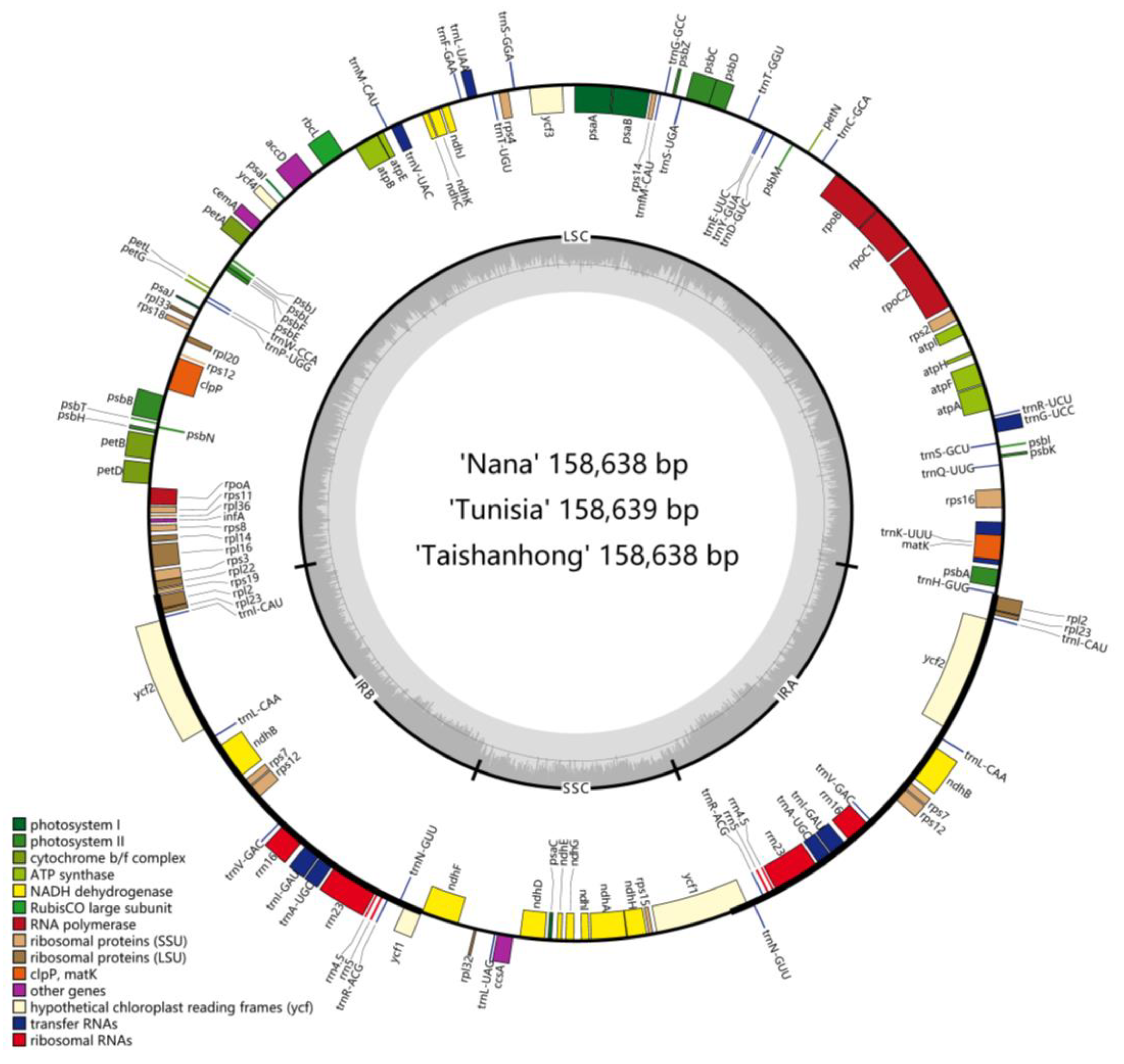
2.1. General Features of Pomegranate Chloroplast Genomes
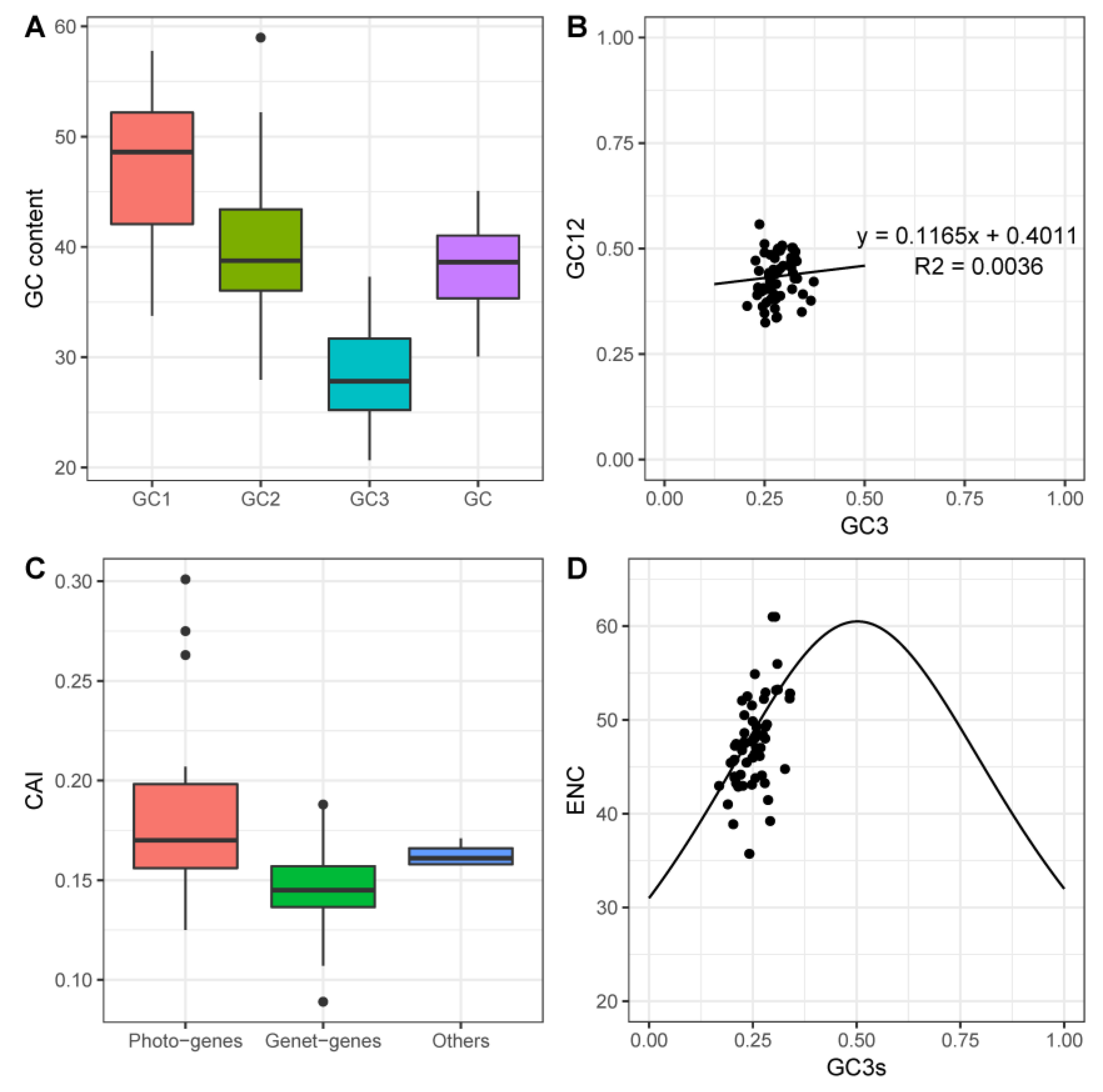
2.2. Codon Usage Bias
2.3. RNA Editing Sites
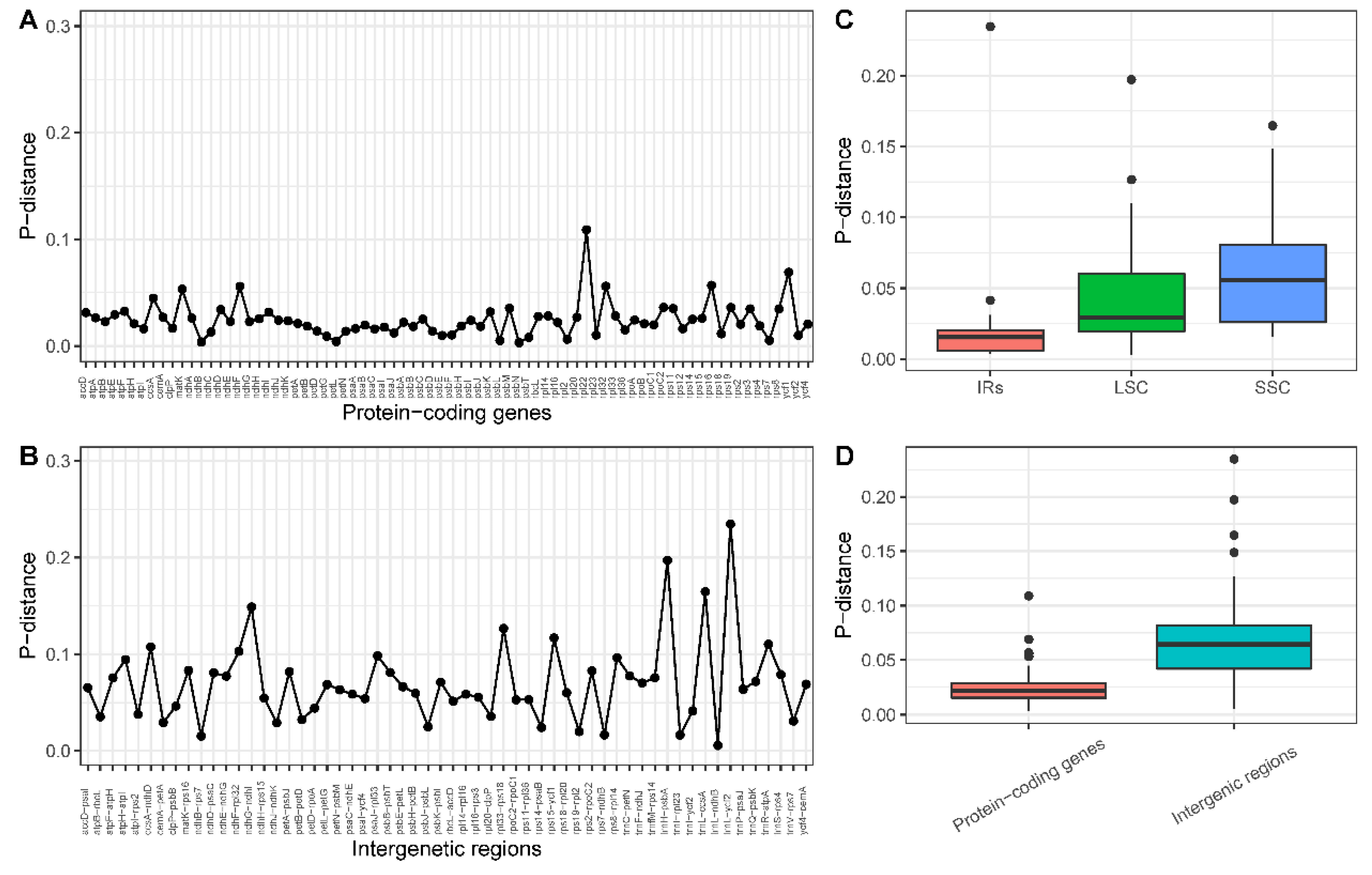
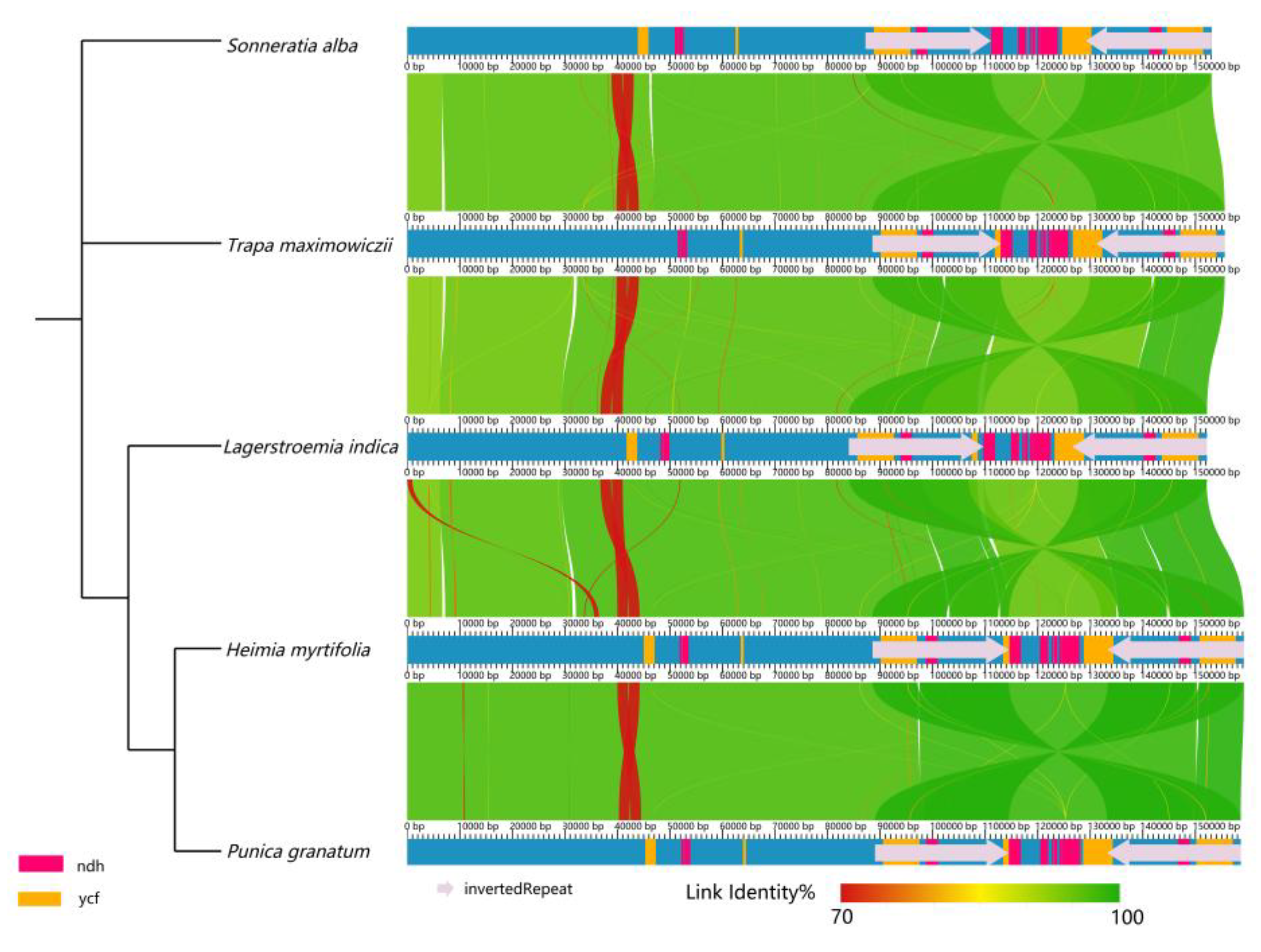
2.4. Sequence Diversity of the Chloroplast Genomes among Lythraceae Species
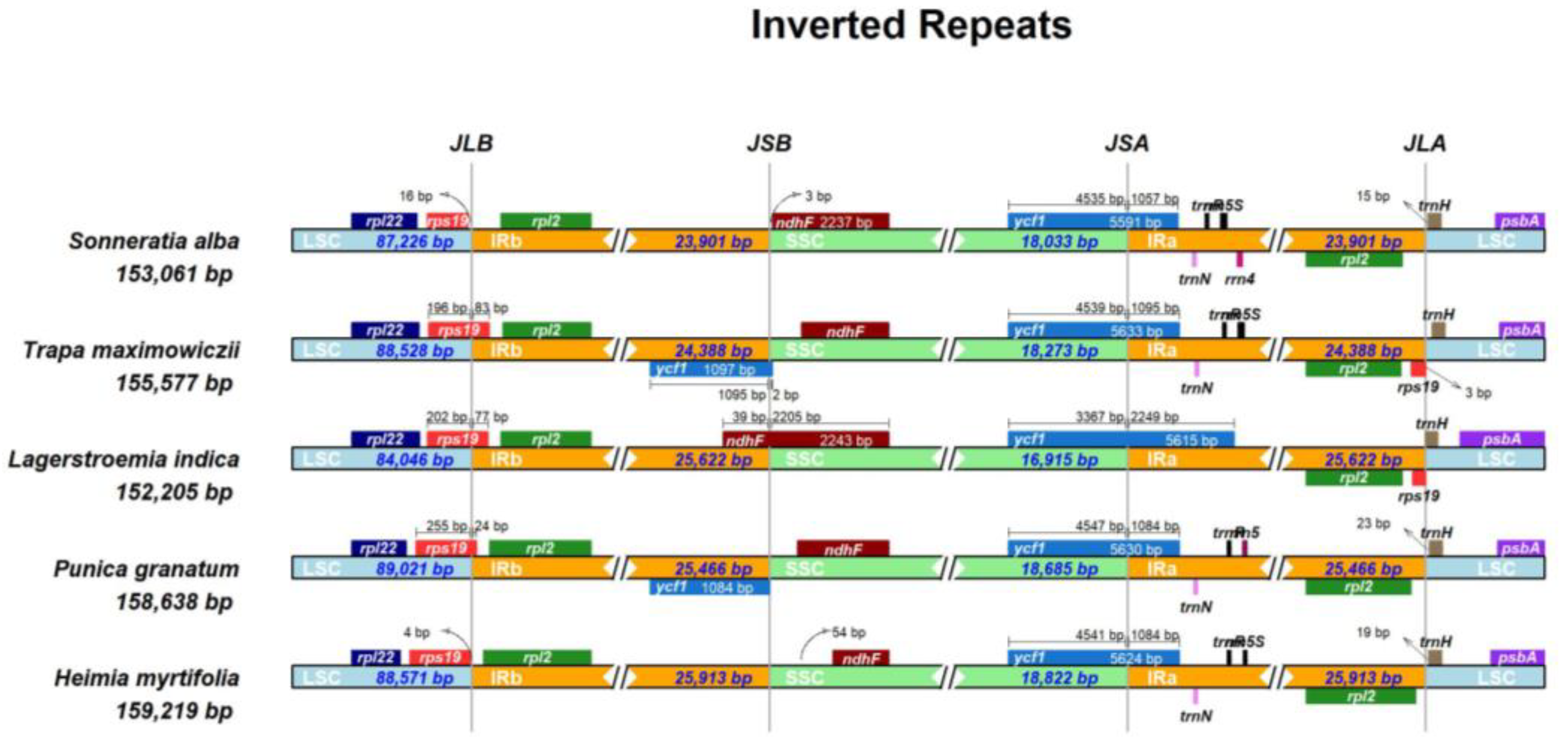
2.5. Structure Comparison among the Chloroplast Genomes of Lythraceae Species
2.6. Positive Selection Analysis
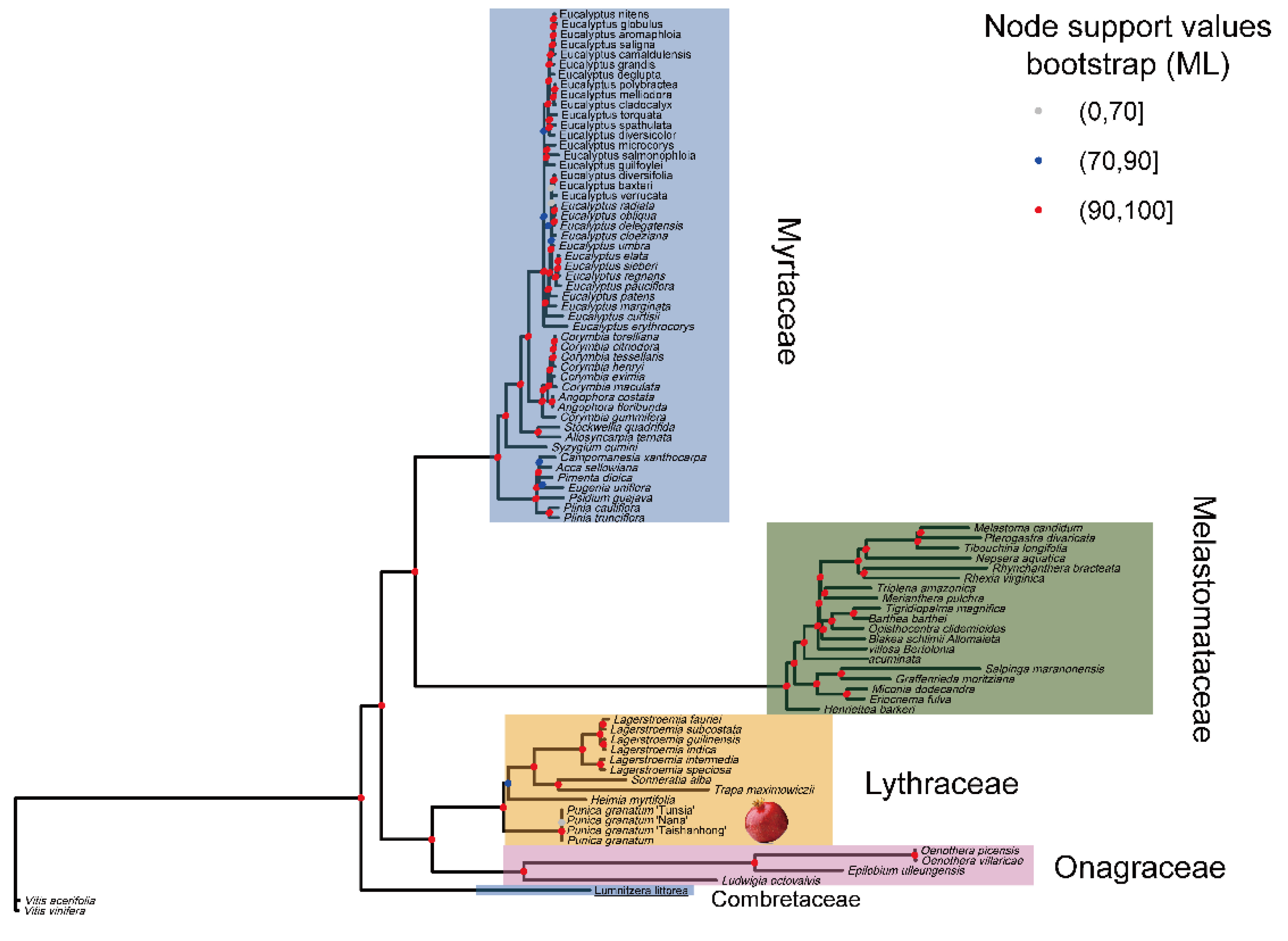
2.7. The Phylogenetic Position of P. granatum
3. Materials and Methods
3.1. Plant Material
3.2. DNA Sequencing, Genome Assembly, and Annotation
3.3. Codon Usage
3.4. RNA Editing Sites
3.5. Sequence Diversity
3.6. Structure Comparison
3.7. Positive Selection Analysis
3.8. Phylogenetic Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Al-Khayri, J.M.; Jain, S.M.; Johnson, D.V. Advances in Plant Breeding Strategies: Fruits; Springer: Berlin/Heidelberg, Germany, 2019; Volume 3. [Google Scholar]
- Shahindokht, B.J. Punica granatum (Pomegranate) activity in health promotion and cancer prevention. Oncol. Rev. 2018, 12, 345. [Google Scholar]
- Sreekumar, S.; Sithul, H.; Muraleedharan, P.; Azeez, J.-M.; Sreeharshan, S. Pomegranate Fruit as a Rich Source of Biologically Active Compounds. BioMed Res. Int. 2014, 2014, 686921. [Google Scholar] [CrossRef]
- Malviya, S.; Arvind; Jha, A.; Hettiarachchy, N. Antioxidant and antibacterial potential of pomegranate peel extracts. J. Food Sci. Technol. 2014, 51, 4132–4137. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Y.; Zhu, C.; Qian, T.-W.; Guo, H.; Wang, D.-D.; Zhang, F.; Yin, X.-X. Extracts of black bean peel and pomegranate peel ameliorate oxidative stress‑induced hyperglycemia in mice. Exp. Ther. Med. 2015, 9, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.A.T.D.; Rana, T.S.; Narzary, D.; Verma, N.; Meshram, D.T.; Ranade, S.A. Pomegranate biology and biotechnology: A review. Sci. Hortic. 2013, 160, 85–107. [Google Scholar] [CrossRef]
- Yuan, Z.-H.; Fang, Y.-M.; Zhang, T.-K.; Fei, Z.-J.; Han, F.-M.; Liu, C.-Y.; Liu, M.; Xiao, W.; Zhang, W.-J.; Wu, S. The pomegranate (Punica granatum L.) genome provides insights into fruit quality and ovule developmental biology. Plant Biotechnol. J. 2018, 16, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Sancho, R.; Cantalapiedra, C.-P.; Lopezalvarez, D.; Gordon, S.P.; Vogel, J.P.; Catalan, P.; Contrerasmoreira, B. Comparative plastome genomics and phylogenomics of Brachypodium: Flowering time signatures, introgression and recombination in recently diverged ecotypes. New Phytol. 2018, 218, 1631–1644. [Google Scholar] [CrossRef] [PubMed]
- Ruhfel, B.R.; Gitzendanner, M.A.; Soltis, P.S.; Soltis, D.E.; Burleigh, J.G. From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evolut. Biol. 2014, 14, 23. [Google Scholar] [CrossRef]
- Yang, J.-B.; Tang, M.; Li, H.-T.; Zhang, Z.-R.; Li, D.-Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evolut. Biol. 2013, 13, 84. [Google Scholar] [CrossRef]
- Chen, N.; Sha, L.-N.; Dong, Z.-Z.; Tang, C.; Wang, Y.; Kang, H.-Y.; Zhang, H.-Q.; Yan, X.-B.; Zhou, Y.-H.; Fan, X. Complete structure and variation of the chloroplast genome of Agropyron cristatum (L.) Gaertn. Gene 2018, 640, 86–96. [Google Scholar] [CrossRef]
- Mcfadden, G.I.; Van Dooren, G.G. Evolution: Red algal genome affirms a common origin of all plastids. Curr. Biol. 2004, 14, R514–R516. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genomo. Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, E.; Herz, M.A.G.; Fuchs, A.; Reifer, D.; Fuller, J.L.; Yanovsky, M.J.; Simpson, C.G.; Brown, J.W.S.; Barta, A.; Kalyna, M. A chloroplast retrograde signal regulates nuclear alternative splicing. Science 2014, 344, 427–430. [Google Scholar] [CrossRef]
- Godoy Herz, M.A.; Kubaczka, M.G.; Brzyzek, G.; Servi, L.; Krzyszton, M.; Simpson, C.; Brown, J.; Swiezewski, S.; Petrillo, E.; Kornblihtt, A.R. Light Regulates Plant Alternative Splicing through the Control of Transcriptional Elongation. Mol. Cell 2019, 73, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Alexia, T.; Cian, D.; Pooja, P.; Zachary, S.; María, E.S.; Lok, H.Y.; David, C.A.G.; Alexandre, Y.L.; Virendrasinh, K.; Andrew, D.W.; et al. Chloroplasts navigate towards the pathogen interface to counteract infection by the Irish potato famine pathogen. bioRxiv 2019, 516443. [Google Scholar] [CrossRef]
- Yang, J.; Feng, L.; Yue, M.; He, Y.L.; Zhao, G.F.; Li, Z.H. Species delimitation and interspecific relationships of the endangered herb genus Notopterygium inferred from multilocus variations. Mol. Phylogenet. Evolut. 2019, 133, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-L.; Zhou, J.-G.; Cui, Y.-X.; Wang, Y.; Duan, B.-Z.; Yao, H. Identification of Ligularia Herbs Using the Complete Chloroplast Genome as a Super-Barcode. Front. Pharmacol. 2018, 9, 695. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Li, J.-F.; Zhang, H.; Cai, B.-H.; Gao, Z.-H.; Qiao, Y.-S.; Mi, L. The complete chloroplast genome sequence of strawberry (Fragaria × ananassa Duch.) and comparison with related species of Rosaceae. PeerJ 2017, 5, e3919. [Google Scholar] [CrossRef] [PubMed]
- Redwan, R.-M.; Saidin, A.; Kumar, S.-V. Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass Commelinidae. BMC Plant Biol 2015, 15, 196. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.-H.; Tang, P.; Li, Z.-Z.; Li, D.-W.; Liu, Y.-F.; Huang, H.-W. The First Complete Chloroplast Genome Sequences in Actinidiaceae: Genome Structure and Comparative Analysis. PLoS ONE 2015, 10, e2109347. [Google Scholar] [CrossRef]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Depamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J. Methods for Obtaining and Analyzing Whole Chloroplast Genome Sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar] [PubMed]
- Khan, A.; Khan, I.-A.; Asif, H.M.; Azim, M. Current trends in chloroplast genome research. Afr. J. Biotechnol. 2010, 9, 3494–3500. [Google Scholar]
- Twyford, A.D.; Ness, R.W. Strategies for complete plastid genome sequencing. Mol. Ecol. Resour. 2017, 17, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, e18. [Google Scholar] [CrossRef]
- Ankenbrand, M.J.; Pfaff, S.; Terhoeven, N.; Qureischi, M.; Gundel, M.; Weis, C.L.; Hackl, T.; Forster, F. chloroExtractor: Extraction and assembly of the chloroplast genome from whole genome shotgun data. J. Open Sour. Softw. 2018, 3, 464. [Google Scholar] [CrossRef]
- Jin, J.; Yu, W.; Yang, J.; Song, Y.; Yi, T.; Li, D. GetOrganelle: A simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. bioRxiv 2018, 256479. [Google Scholar] [CrossRef]
- Ivanova, Z.; Sablok, G.; Daskalova, E.; Zahmanova, G.; Apostolova, E.; Yahubyan, G.; Baev, V. Chloroplast Genome Analysis of Resurrection Tertiary Relict Haberlea rhodopensis Highlights Genes Important for Desiccation Stress Response. Front. Plant Sci. 2017, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yao, X.; Tan, Y.-H.; Gan, Y.; Corlett, R.T. Complete chloroplast genome sequence of the avocado: Gene organization, comparative analysis, and phylogenetic relationships with other Lauraceae. Can. J. For. Res. 2016, 46, 1293–1301. [Google Scholar] [CrossRef]
- Yang, J.-B.; Yang, S.-X.; Li, H.-T.; Yang, J.; Li, D.-Z. Comparative Chloroplast Genomes of Camellia Species. PLoS ONE 2013, 8, e73053. [Google Scholar] [CrossRef]
- Wang, S.; Yang, C.; Zhao, X.; Chen, S.; Qu, G. Complete chloroplast genome sequence of Betula platyphylla: Gene organization, RNA editing, and comparative and phylogenetic analyses. BMC Genom. 2018, 19, 950. [Google Scholar] [CrossRef]
- Wickens, M.; Cox, M.M. Critical reviews in biochemistry and molecular biology. Introduction. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.-F.; Zang, M.-Y.; Li, M.-Z.; Fang, Y.-M. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Khan, A.L.; Khan, A.R.; Waqas, M.; Kang, S.; Khan, M.A.; Lee, S.; Lee, I. Complete Chloroplast Genome of Nicotiana otophora and its Comparison with Related Species. Front. Plant Sci. 2016, 7, 843. [Google Scholar] [CrossRef] [PubMed]
- Batnini, M.A.; Bourguiba, H.; Trififarah, N.; Krichen, L. Molecular diversity and phylogeny of Tunisian Prunus armeniaca L. by evaluating three candidate barcodes of the chloroplast genome. Sci. Hortic. 2019, 245, 99–106. [Google Scholar] [CrossRef]
- Norouzi, M.; Talebi, M.; Sayedtabatabaei, B. Chloroplast microsatellite diversity and population genetic structure of Iranian pomegranate (Punica granatum L.) genotypes. Sci. Hortic. 2012, 137, 114–120. [Google Scholar] [CrossRef]
- Feng, C.; Xu, C.-J.; Wang, Y.; Liu, W.-L.; Yin, X.-R.; Li, X.; Chen, M.; Chen, K.-S. Codon usage patterns in Chinese bayberry (Myrica rubra) based on RNA-Seq data. BMC Genom. 2013, 14, 732. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Cai, Z.-Q.; Xia, G.-M.; Wang, M.-C. Synonymous codon usage bias is correlative to intron number and shows disequilibrium among exons in plants. BMC Genom. 2013, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-Y.; Xing, H.-X.; Yuan, Y.-C.; Wang, X.-L.; Saeed, M.; Tao, J.-C.; Feng, W.; Zhang, G.-H.; Song, X.-L.; Sun, X.-Z. Genome-wide analysis of codon usage bias in four sequenced cotton species. PLoS ONE 2018, 13, e0194372. [Google Scholar] [CrossRef]
- Ning, L.I.; Sun, M.-H.; Jiang, Z.-S.; Shu, H.-R.; Zhang, S.-Z. Genome-wide analysis of the synonymous codon usage patterns in apple. J. Integr. Agric. 2016, 15, 983–991. [Google Scholar]
- Suzuki, H.; Morton, B.R. Codon Adaptation of Plastid Genes. PLoS ONE 2016, 11, e0154306. [Google Scholar] [CrossRef]
- Zhang, R.-Z.; Zhang, L.; Wang, W.; Zhang, Z.; Du, H.-H.; Qu, Z.; Li, X.-Q.; Xiang, H. Differences in Codon Usage Bias between Photosynthesis-Related Genes and Genetic System-Related Genes of Chloroplast Genomes in Cultivated and Wild Solanum Species. Int. J. Mol. Sci. 2018, 19, 3142. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.-J.; Deng, P.-C.; Feng, K.-W.; Liu, P.-X.; Du, X.-H.; You, F.M.; Song, W.-N. Comparative analysis of codon usage patterns in chloroplast genomes of the Asteraceae family. Plant Mol. Biol. Rep. 2014, 32, 828–840. [Google Scholar] [CrossRef]
- Wright, F. The effective number of codons used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Das, S.; Paul, S.; Dutta, C. Synonymous codon usage in adenoviruses: Influence of mutation, selection and protein hydropathy. Virus Res. 2006, 117, 227–236. [Google Scholar] [CrossRef]
- Harris, R.S.; Petersenmahrt, S.K.; Neuberger, M.S. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 2002, 10, 1247–1253. [Google Scholar] [CrossRef]
- Bentolila, S.; Oh, J.; Hanson, M.R.; Bukowski, R. Comprehensive High-Resolution Analysis of the Role of an Arabidopsis Gene Family in RNA Editing. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Saina, J.K.; Li, Z.; Gichira, A.W.; Liao, Y. The Complete Chloroplast Genome Sequence of Tree of Heaven (Ailanthus altissima (Mill.) (Sapindales: Simaroubaceae), an Important Pantropical Tree. Int. J. Mol. Sci. 2018, 19, 929. [Google Scholar] [CrossRef]
- Pinard, D.; Myburg, A.A.; Mizrachi, E. The plastid and mitochondrial genomes of Eucalyptus grandis. BMC Genom. 2019, 20, 132. [Google Scholar] [CrossRef]
- Rabah, S.O.; Lee, C.; Hajrah, N.H.; Makki, R.M.; Alharby, H.F.; Alhebshi, A.M.; Sabir, J.; Jansen, R.K.; Ruhlman, T.A. Plastome Sequencing of Ten Nonmodel Crop Species Uncovers a Large Insertion of Mitochondrial DNA in Cashew. Plant Genome 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, J.; Li, L.; Gao, L.; Xu, J.; Yang, M. Structural and Comparative Analysis of the Complete Chloroplast Genome of Pyrus hopeiensis—“Wild Plants with a Tiny Population”—and Three Other Pyrus Species. Int. J. Mol. Sci. 2018, 19, 3262. [Google Scholar] [CrossRef]
- Li, B.; Zheng, Y. Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci. Rep. 2018, 8, 9285. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Wang, R.; Zhang, N.; Fan, W.; Fang, M.; Li, Z. Molecular Evolution of Chloroplast Genomes of Orchid Species: Insights into Phylogenetic Relationship and Adaptive Evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.C.; Chen, S.L.; Xiao, P.G. Molecular evolution and positive Darwinian selection of the chloroplast maturase matK. J. Plant Res. 2010, 123, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Gitzendanner, M.; Soltis, P.; Wong, G.; Ruhfel, B.; Soltis, D. Plastid phylogenomic analysis of green plants: A billion years of evolutionary history. Am. J. Bot. 2018, 105, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Song, F.-H.; Zhu, J.-W.; Zhang, S.-S.; Yang, Y.-D.; Chen, T.-T.; Tang, B.; Dong, L.-L.; Ding, N.; Zhang, Q. GSA: Genome Sequence Archive. Genom. Proteom. Bioinform. 2017, 15, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhao, W.-M.; Xiao, J.-F.; Bao, Y.-M.; Wang, F.; Hao, L.-L.; Zhu, J.-W.; Chen, T.-T.; Zhang, S.-S.; Chen, X. Database Resources of the BIG Data Center in 2018. Nucleic Acids Res. 2018, 46, D14–D20. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbrichtjones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stoneshavas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Chen, X.; Jing, D.; Tong, C.-F.; Gong, X.-D.; Qiang, W.; Qiang, Z.-G. Analysis of Synonymous Codon Usage Patterns in Seven Different Citrus Species. Evol. Bioinform. Online 2013, 9, 215–228. [Google Scholar]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiryousefi, A.; Hyvonen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Ankenbrand, M.J.; Hohlfeld, S.; Hackl, T.; Forster, F. AliTV—Interactive visualization of whole genome comparisons. PeerJ 2017, 3. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evolut. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evolut. 2019, 9, 3891–3898. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Du, Z.; Shen, J.; Yang, H.; Liao, F. Genetic diversity and molecular evolution of Ornithogalum mosaic virus based on the coat protein gene sequence. PeerJ 2018, 6. [Google Scholar] [CrossRef]
- Bi, G.; Mao, Y.; Xing, Q.; Cao, M. HomBlocks: A multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics 2017, 110, 18–22. [Google Scholar] [CrossRef]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evolut. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evolut. 2017, 8, 28–36. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Genes | Gene Names |
|---|---|
| Photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| Cytochrome b/f complex | petA, petBa, petDa, petG, petL, petN |
| ATP synthase | atpA, atpB, atpE, atpFa, atpH, atpI |
| NADP dehydrogenase | ndhAa, ndhB*a, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| RubisCO large subunit | rbcL |
| RNA polymerase | rpoA, rpoB, rpoC1a, rpoC2 |
| Ribosomal proteins (SSU) | rps2, rps3, rps4, rps7*, rps8, rps11, rps12*b, rps14, rps15, rps16a, rps18, rps19 |
| Ribosomal proteins (LSU) | rpl2*, rpl14, rpl16a, rpl20, rpl22, rpl23*, rpl32, rpl33, rpl36 |
| Hypothetical chloroplast reading frames | ycf1, ycf2*, ycf3b, ycf4 |
| Translation initiation factor IF-1 | infA |
| Acetyl-CoA carboxylase | accD |
| Cytochrome c biogenesis Maturase | matK |
| ATP-dependent protease | clpPb |
| Inner membrane protein | cemA |
| Ribosomal RNAs | rna4.5S*, rna5S*, rna16S*, rna23S* |
| Transfer RNAs | trnA-UGC*a, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCCa, trnH-GUG, trnI-CAU*a, trnI-GAU*, trnK-UUUa, trnL-CAA*, trnL-UAAa, trnL-UAG, trnM-CAU, trnN-GUU*, trnP-UGG, trnQ-UUG, trnR-ACG*, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC*, trnV-UACa, trnW-CCA, trnY-GUA |
| Amino Acid | Codon | Codon Frequency | RSCU | AA | Codon | Codon Frequency | RSCU |
|---|---|---|---|---|---|---|---|
| Phe | UUU* | 4551 | 1.18 | Ser | UCU* | 2417 | 1.46 |
| UUC | 3143 | 0.82 | UCC | 1577 | 0.96 | ||
| Leu | UUA* | 3112 | 1.41 | UCA* | 2278 | 1.38 | |
| UUG* | 2920 | 1.32 | UCG | 1268 | 0.77 | ||
| CUU* | 2586 | 1.17 | Pro | CCU* | 1385 | 1.21 | |
| CUC | 1360 | 0.62 | CCC | 929 | 0.81 | ||
| CUA | 1958 | 0.89 | CCA* | 1387 | 1.21 | ||
| CUG | 1287 | 0.58 | CCG | 876 | 0.77 | ||
| Ile | AUU* | 4378 | 1.27 | Thr | ACU* | 1478 | 1.13 |
| AUC | 2723 | 0.79 | ACC | 1095 | 0.84 | ||
| AUA | 3246 | 0.94 | ACA* | 1759 | 1.35 | ||
| Met | AUG | 2760 | 1.00 | ACG | 886 | 0.68 | |
| Val | GUU* | 2045 | 1.34 | Ala | GCU* | 1389 | 1.45 |
| GUC | 1033 | 0.68 | GCC | 712 | 0.75 | ||
| GUA* | 1891 | 1.24 | GCA | 1145 | 1.20 | ||
| GUG | 1123 | 0.74 | GCG | 576 | 0.60 | ||
| Tyr | UAU* | 3606 | 1.37 | Cys | UGU* | 1410 | 1.17 |
| UAC | 1665 | 0.63 | UGC | 993 | 0.83 | ||
| TER | UAA* | 2029 | 1.03 | TER | UGA | 2003 | 1.01 |
| UAG | 1893 | 0.96 | Trp | UGG | 2392 | 1.00 | |
| His | CAU* | 1908 | 1.38 | Arg | CGU | 888 | 0.7 |
| CAC | 866 | 0.62 | CGC | 456 | 0.36 | ||
| Gln | CAA* | 2815 | 1.35 | CGA* | 1428 | 1.13 | |
| CAG | 1342 | 0.65 | CGG | 865 | 0.68 | ||
| Asn | AAU* | 3923 | 1.37 | Ser | AGU | 1441 | 0.87 |
| AAC | 1800 | 0.63 | AGC | 923 | 0.56 | ||
| Lys | AAA* | 4768 | 1.31 | Arg | AGA* | 2560 | 2.02 |
| AAG | 2538 | 0.69 | AGG | 1412 | 1.11 | ||
| Asp | GAU* | 2818 | 1.49 | Gly | GGU | 1642 | 1.01 |
| GAC | 962 | 0.51 | GGC | 886 | 0.54 | ||
| Glu | GAA* | 3632 | 1.37 | GGA* | 2409 | 1.48 | |
| GAG | 1689 | 0.63 | GGG | 1569 | 0.96 |
| Gene | Nucleotide Position | Amino Acid Position | Codon Conversion | Score |
|---|---|---|---|---|
| matK | 644 | 215 | GCA (A) => GTA (V) | 1 |
| 1177 | 393 | CGG (R) => TGG (W) | 1 | |
| 1187 | 396 | TCA (S) => TTA (L) | 0.86 | |
| 1246 | 416 | CAC (H) => TAC (Y) | 1 | |
| atpA | 791 | 264 | CCC (P) => CTC (L) | 1 |
| atpF | 92 | 31 | CCA (P) => CTA (L) | 0.86 |
| atpI | 23 | 8 | ACC (T) => ATC (I) | 1 |
| rps2 | 422 | 141 | TCG (S) => TTG (L) | 1 |
| rpoC2 | 3056 | 1019 | GCA (A) => GTA (V) | 0.86 |
| 3998 | 1333 | GCG (A) => GTG (V) | 0.86 | |
| rpoC1 | 41 | 14 | TCA (S) => TTA (L) | 1 |
| 1171 | 391 | CCA (P) => TCA (S) | 1 | |
| rpoB | 338 | 113 | TCT (S) => TTT (F) | 1 |
| 551 | 184 | TCA (S) => TTA (L) | 1 | |
| 566 | 189 | TCG (S) => TTG (L) | 1 | |
| 973 | 325 | CTC (L) => TTC (F) | 0.86 | |
| rps14 | 80 | 27 | TCA (S) => TTA (L) | 1 |
| 149 | 50 | TCA (S) => TTA (L) | 1 | |
| atpB | 1487 | 496 | TCG (S) => TTG (L) | 1 |
| accD | 794 | 265 | TCG (S) => TTG (L) | 0.8 |
| 1403 | 468 | CCT (P) => CTT (L) | 1 | |
| psbL | 2 | 1 | ACG (T) => ATG (M) | 1 |
| psbF | 77 | 26 | TCT (S) => TTT (F) | 1 |
| clpP | 559 | 187 | CAT (H) => TAT (Y) | 1 |
| ndhB | 28 | 10 | CTC (L) => TTC (F) | 1 |
| 149 | 50 | TCA (S) => TTA (L) | 1 | |
| 467 | 156 | CCA (P) => CTA (L) | 0.8 | |
| 586 | 196 | CAT (H) => TAT (Y) | 1 | |
| 611 | 204 | TCA (S) => TTA (L) | 1 | |
| 737 | 246 | CCA (P) => CTA (L) | 1 | |
| 746 | 249 | TCT (S) => TTT (F) | 1 | |
| 830 | 277 | TCA (S) => TTA (L) | 1 | |
| 836 | 279 | TCA (S) => TTA (L) | 1 | |
| 1255 | 419 | CAT (H) => TAT (Y) | 1 | |
| 1481 | 494 | CCA (P) => CTA (L) | 1 | |
| ndhF | 160 | 54 | CTT (L) => TTT (F) | 1 |
| 586 | 196 | CTT (L) => TTT (F) | 0.8 | |
| ccsA | 89 | 30 | TCG (S) => TTG (L) | 1 |
| ndhD | 2 | 1 | ACG (T) => ATG (M) | 1 |
| 185 | 62 | ACC (T) => ATC (I) | 1 | |
| 313 | 105 | CGG (R) => TGG (W) | 0.8 | |
| 383 | 128 | TCA (S) => TTA (L) | 1 | |
| 674 | 225 | TCG (S) => TTG (L) | 1 | |
| 845 | 282 | ACA (T) => ATA (I) | 0.8 | |
| 878 | 239 | TCA (S) => TTA (L) | 1 | |
| 887 | 296 | CCA (P) => CTA (L) | 1 | |
| 1405 | 469 | CTT (L) => TTT (F) | 0.8 | |
| ndhG | 155 | 52 | CCA (P) => CTA (L) | 1 |
| 166 | 56 | CAT (H) => TAT (Y) | 0.8 | |
| 314 | 105 | ACA (T) => ATA (I) | 0.8 | |
| ndhA | 341 | 114 | TCA (S) => TTA (L) | 1 |
| 566 | 189 | TCA (S) => TTA (L) | 1 | |
| 1073 | 358 | TCC (S) => TTC (F) | 1 |
| Species | Punica granatum | Lagerstromeia indica | Sonneratia alba | Trapa maximowicizz | Heimia myrtifolia |
|---|---|---|---|---|---|
| Genome size | 158,638 | 152,025 | 153,061 | 155,577 | 159,219 |
| LSC size | 89,021 | 84,046 | 87,226 | 88,528 | 88,571 |
| SSC size | 18,684 | 16,914 | 18,032 | 18,272 | 18,821 |
| IR size | 25,467 | 25,623 | 23,902 | 24,389 | 25,914 |
| Number of genes | 113 | 113 | 107 | 110 | 112 |
| Protein-coding genes | 79 (6) | 79 (7) | 79 (6) | 77 (5) | 78 (7) |
| tRNA genes | 30 (7) | 30 (7) | 24 (5) | 29 (9) | 30 (6) |
| rRNA genes | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) |
| Number of genes duplicated in IR | 17 | 18 | 15 | 18 | 17 |
| GC content | 36.92 | 37.59 | 37.29 | 36.4 | 36.95 |
| GenBank accession | MK603511 | NC_030484 | NC_039975 | NC_037023 | MG921615 |
| Gene Name | Models (Number of Parameters) | lnL | Likelihood Ratio Test p-Value | Positively Selected Sites |
|---|---|---|---|---|
| accD | M8 (12) | −2534.400824 | 0.0962956 | 125 G 0.955 * |
| M7 (10) | −2536.741156 | |||
| ndhF | M8(12) | −4446.871610 | 0.000000002 | 292 N 0.961 *; 486 R 0.999 **; 487 I 0.975 *; 490 K 0.985 *; 518 N 0.969 *; 648 S 0.983 *; 738 F 0.995 ** |
| M7(10) | −4466.787914 | |||
| ndhJ | M8(12) | −806.644367 | 0.003291615 | 121 R 0.970 * |
| M7(10) | −812.360744 | |||
| psaI | M8(12) | −139.985819 | 0.031547151 | 26 H 0.959* |
| M7(10) | −143.4420 | |||
| rpl22 | M8(12) | −942.833567 | 0.000497160 | 4 L 0.972 *; 5 Y 0.961 *; 73 P 0.962 *; 125 A 0.993 **; 126 R 0.994 ** |
| M7(10) | −950.440165 | |||
| rps12 | M8(12) | −529.317591 | 0.002536102 | 117 K 0.974 * |
| M7(10) | −535.294718 | |||
| rps2 | M8(12) | −1171.788526 | 0.008240568 | 173 E 0.982 * |
| M7(10) | −1176.587212 | |||
| rps4 | M8(12) | −994.666749 | 0.007084882 | 28 P 0.959 * |
| M7(10) | −999.616541 | |||
| rps7 | M8(12) | −672.815064 | 0.000000001 | 84 T 1.000 ** |
| M7(10) | −693.433775 | |||
| rps8 | M8(12) | −718.655799 | 0.001840922 | 59 L 0.989 * |
| M7(10) | −724.953288 | |||
| ycf1 | M8(12) | −11,993.590817 | 0.000001936 | 205 V 0.977 *; 206 F 0.975 *; 341 S 0.974 *; 495 S 0.952 *; 534 A 0.951 *; 1073 A 0.963 *; 1290 R 0.978 *; 1446 E 0.963 *; 1701 K 0.976 *; 1728 T 0.950 * |
| M7(10) | −12,006.745724 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, M.; Zhao, X.; Zhou, J.; Huo, Y.; Ding, Y.; Yuan, Z. The Complete Chloroplast Genomes of Punica granatum and a Comparison with Other Species in Lythraceae. Int. J. Mol. Sci. 2019, 20, 2886. https://doi.org/10.3390/ijms20122886
Yan M, Zhao X, Zhou J, Huo Y, Ding Y, Yuan Z. The Complete Chloroplast Genomes of Punica granatum and a Comparison with Other Species in Lythraceae. International Journal of Molecular Sciences. 2019; 20(12):2886. https://doi.org/10.3390/ijms20122886
Chicago/Turabian StyleYan, Ming, Xueqing Zhao, Jianqing Zhou, Yan Huo, Yu Ding, and Zhaohe Yuan. 2019. "The Complete Chloroplast Genomes of Punica granatum and a Comparison with Other Species in Lythraceae" International Journal of Molecular Sciences 20, no. 12: 2886. https://doi.org/10.3390/ijms20122886