Gastric Leptin and Tumorigenesis: Beyond Obesity

Division of Host Defense, Department of Life Sciences, Faculty of Life and Environmental Sciences, Prefectural University of Hiroshima, 5562 Nanatsuka, Shobara, Hiroshima 727-0023, Japan
Int. J. Mol. Sci. 2019, 20(11), 2622; https://doi.org/10.3390/ijms20112622
Submission received: 1 May 2019
/
Revised: 24 May 2019
/
Accepted: 24 May 2019
/
Published: 28 May 2019
(This article belongs to the Special Issue Leptin and Relatives: Molecules at the Crossroad of Inflammation and Immunometabolism)
Abstract
:Leptin, an adipocyte-derived hormone and its receptor (ObR) expressed in the hypothalamus are well known as an essential regulator of appetite and energy expenditure. Obesity induces abundant leptin production, however, reduced sensitivity to leptin leads to the development of metabolic disorders, so called leptin resistance. The stomach has been identified as an organ that simultaneously expresses leptin and ObR. Accumulating evidence has shown gastric leptin to perform diverse functions, such as those in nutrient absorption and carcinogenesis in the gastrointestinal system, independent of its well-known role in appetite regulation and obesity. Overexpression of leptin and phosphorylated ObR is implicated in gastric cancer in humans and in murine model, and diet-induced obesity causes precancerous lesions in the stomach in mice. While the underlying pathomechanisms remain unclear, leptin signaling can affect gastric mucosal milieu. In this review, we focus on the significant role of the gastric leptin signaling in neoplasia and tumorigenesis in stomach in the context of hereditary and diet-induced obesity.
1. Introduction
Humans have been exposed to malnutrition due to starvation. However, recently, malnutrition in the form of over-nutrition has become a serious social problem, because diet-induced obesity due to an increase in nutrient intake and insufficient energetic expenditure, has dramatically increased worldwide and is implicated in numerous metabolic disorders, including cardiovascular diseases and type 2 diabetes (T2D). Furthermore, obesity is a critical risk factor for cancers in various tissues. Prevalence of obesity has been steadily increasing over the past a few decades in not only developed, but also in developing countries, shaping up as a worldwide health problem for children to adult.
Adipocyte-derived leptin, discovered in 1994, modulates appetite and body weight. However, in the case of leptin resistance in obese patients, despite the high concentrations of leptin in blood, appetite is not suppressed, due to decreased sensitivity of its receptor signaling [1,2]. Gastric leptin was discovered in 1998, has been shown to accelerate the absorption of nutrients, although its significance is still being investigated. Leptin exerts pleiotropic effects on immunity and cell differentiation/proliferation in the physiological state [3]. Leptin is mainly produced by adipocytes, besides other tissues, including the stomach [4], and placenta [5]. Leptin secreted by such tissues seems to be independent of the satiety regulation. However, overexpression of leptin and activation of ObR has been reported in various cancers, such as those of the stomach and mammary gland, and is mediated by inflammation, angiogenesis, stemness [6], and epithelial–mesenchymal transition (EMT) and progression [7] (Figure 1).
Gastric cancer (GC) is the fourth most common malignancy and the second leading cause of cancer-related deaths [8,9]. There are critical geographical differences regarding its incidence, mortality, social, culture, and economic entities. More than 100,000 new cases were diagnosed in the world in the last 10 years [10,11]. In Europe, 140,000 individuals were diagnosed in 2014 and 107,000 patients died that same year [12]. One of the reasons for the high mortality in GC patients is the lack of a biomarker for diagnosis at the early stage of GC, besides the high relapse rate after tumor resection. Helicobacter pylori (H. pylori) infection is well known as a causative factor of GC. through an initiation of epigenetics reprogramming of host cells [13] and dysregulation of β-defensins, antimicrobial peptides [14]. However, H. pylori-infected individuals constitute approximately half of the world population, with only a small portion of them eventually developing GC. Recently development of gastric cardia adenocarcinoma has been reported to be strongly linked to obesity [15,16], suggesting the possibility of causes other than H. pylori infection. In this review, we focus on the role of leptin as an inducer of carcinogenesis and its implications in GC.
2. Background of Leptin and Its Receptor Signaling
Leptin is a 16-kD non-glycosylated protein, consisting of a 167-amino acid polypeptide with 21-amino acids signal sequence at the amino-terminus, which is cleaved following the translocation of leptin into microsomes and is then secreted into the blood stream. The significance of leptin was first discovered by parabiosis experiments. Mutant mice (ob/ob) were found to exhibit severe obesity with decreased basic metabolism and physical activity [17], whereas crossing them with wild-type mice led to restoration of normal body weight [18]. Later, ob gene was identified to be response for obesity in ob/ob mice and subsequently named ‘leptin’, which is derived from a Greek word, ‘leptos’ (‘thin’) [1]. Leptin receptor (ObR) encoded by the db gene in mice, is a member of class I cytokine receptor family, similar to gp130 [3,19,20]. ObR consists of six isoforms (ObRa, ObRb, ObRc, ObRd, ObRe and ObRf), formed from alternative RNA splicing of the db gene. These isoforms have a common leptin-binding domain, and differ in their intracellular domains [21,22]. Among them, ObRb, named as the long form, is the only isoform that contains the intracellular motif necessary for the leptin-mediated JAK-STAT pathway, whereas four short forms (ObRa, c, d, and f), differ in their cytosolic carboxy terminals. ObRe is a soluble form that lacks the trans-membrane domain and is involved in blood to brain leptin transport by antagonizing leptin endocytosis [23]. The extracellular domain of the ObRb is constituted of homology region 2 (CHR2), which contains flexible hinge regions [24]. Upon leptin binding, ObRb dimerizes, resulting in the activation and tyrosine (Y) phosphorylation of JAK2, subsequently forming PY-985 and PY-1138 in ObRb. PY-1138 of ObRb recruits the SH2 domain-containing transcription factor STAT3, resulting in tyrosine phosphorylation of STAT3 and its translocation to the nucleus [25]. In addition, PY-1077 promotes the recruitment and activation of STAT5 [26]. The lack of STAT5 in the central nervous system causes to obesity with hyperphagia, whereas STAT5 activation in hypothalamic neurons suppresses food intake [27], suggesting that not only STAT3, but the JAK2-STAT5 pathway also contributes to the prevention of obesity. PY-985, to which SOCS3 binds to recruit the SH2 domain-containing protein-tyrosine phosphatase SHP-2, functions upstream of extracellular signal regulated kinase (ERK) activation and c-fos transcription [28]. Once inside the nucleus, STAT3 mediates gene transcription, including transcription of the suppressor of cytokine signaling, SOCS3, which in turn binds to PY-985 and JAK2 to attenuate leptin receptor signaling (Figure 2).
3. Adipocyte-Derived Leptin in the Central Nervous System
Leptin secreted by adipose tissue reaches the hypothalamus by receptor-mediated transcytosis rather than by the blood-brain barrier system [29]. ObRb is located in the arcuate nucleus of the hypothalamus, which is responsible for the secretion of neuropeptides and neurotransmitters that suppress appetite and body weight [2,21]. Further studies on gene-targeted mice have revealed that leptin’s action in the central nervous system only is sufficient to regulate body weight, feeding, energy expenditure, glucose metabolism, and behavior [30]. However, leptin’s anorexigenic effects are suppressed in obese individuals and high-fat diet-induced obese animals, despite the elevated serum leptin levels is; when leptin signaling does not function, a condition termed “leptin resistance” [31,32,33,34,35]. In humans, congenital mutation of the ob gene is rare, although patients with early-onset extreme obesity have been reported; cousins from a consanguineous Pakistani family, involved the absence of leptin due to homozygous frame shift mutation in leptin gene [36], and a boy of Turkish consanguinity, whose parents were of normal weight was reported to be obese due to homozygous transversion (c.298G -> T), resulting in mutated leptin that failed to bind ObR [37].
SOCS3 has been proposed as a potential mediator of leptin resistance. Peripheral administration of leptin to ob/ob mice was found to specifically induce SOCS3 mRNA in regions of the hypothalamus that are known to be important for feeding behavior [38]. Mice with SOCS3 deletion either in the whole brain or in proopiomelanocortin (POMC) neurons (the key leptin target neurons in the arcuate nucleus of the hypothalamus) are resistant to high-fat diet-induced obesity [39,40]. Furthermore, studies using either POMC or ObRb-expressed neuron-specific SOCS3 transgenic mice indicated that SOCS3 upregulation alone in proopiomelanocortin (POMC) neurons, but not in ObRb neurons, is sufficient to cause leptin resistance and obesity, mediated by the antagonization of phosphorylated STAT3 and mTOR-S6K signaling due to SOCS3 upregulation [41].
4. Gastric Leptin as a Causative Factor of Carcinogenesis in Stomach
In the gastrointestinal system, leptin is constitutively produced in the stomach, by parietal cells that secrete gastric acid, and chief cells that produce pepsinogen and gastric lipase, but not in bowels, whereas ObR is expressed in both the stomach and bowels [4,42,43]. Thus, the stomach is a unique organ, which constitutively expresses both leptin and ObR, and can transduce autocrine leptin signaling. Two main effects of gastric leptin may be considered: First, its effect on intestinal function. Leptin is secreted in the gastric lumen and enters the intestine to directly act on the intestinal epithelium to enhance absorption of peptides by Pept-1, glucose and galactose by Glut-5 in the physiological state [44,45], promotes cell invasive capacity via the PI3K, Rho- and Rac-dependent pathway in colon cells [46], along with cell proliferation [47], which is beneficial effect for both normal and repaired mucosa such as that after inflammation. Leptin-deficient ob/ob mice, which underwent resection of the small intestine, showed reduced cell proliferation and enhanced apoptosis compared to lean mice that received the same treatment [48]; however, contradictory reports showed increased apoptosis and accelerated intestinal adaptation [49]. Unlike leptin signaling in the hypothalamus, that in the gut is independent of the obesity development. Intestinal epithelium specific-ObR-deficient mice using villin-promoter are not spontaneously obese [50], implying that intestinal leptin signaling is independent of the metabolism and appetite. Rather, villin-ObRb conditional KO (cKO) mice showed a reduced number of aberrant crypt foci and colon tumors induced by azoxymethane, mediated through activation of ObRb-STAT3 signaling [51], thus indicating that intestinal leptin signaling controls the development and cell fate of epithelial cell carcinogenesis. In terms of intestinal infection, Entamoeba histolytica, which is an anaerobic parasitic amoebozoan, induces severe diarrheal diseases. Both db/db mice and villin-ObR cKO mice are susceptible to E. histolytica infection to exhibit severe mucosal destruction of the intestine, indicating leptin signal to be an essential factor for host resistance against amebiasis [52]. Leptin acts as a pro-inflammatory cytokine that promotes the Th1 immune response, and Th1-type cytokines, in particular IFN-γ are critical for the protection against E. histolytica. Both db/db mice and villin-ObR cKO mice were unable to transduce leptin signaling to facilitate higher susceptibility to E. histolytica. Second, the effect of gastric leptin on gastric function is still an enigma, although both leptin and ObR are expressed in parietal cells and chief cells [4,53,54]. First report of enhanced gastric leptin was in GC caused by Helicobacter pylori infection [55]. H. pylori infection significantly increased in gastric leptin levels while its cure decreased the levels, whereas serum leptin remained unaffected by H. pylori. Leptin stimulates cell proliferation, and its effect on the development and progression of GC is seeded in GC cells, including advanced GC cells [56,57,58]; expression of gastric leptin might be a prognostic marker in poorly differentiated GC [59]. Lee et al. had demonstrated that both leptin and ObR are overexpressed in gastric adenoma, and in early or advanced GC, and that leptin activates STAT3-ERK1/2 pathway leading to VEGF expression in GC cell line [60]. In vitro, leptin induces the migration of AGS cell (adenocarninoma) and MKN-45 cell (poorly differentiated adenocarcinoma) by upregulating ICAM-1 expression [61], hence suggesting that leptin can activate or modulate a variety of signaling cascades. We demonstrated that overexpression of leptin and its signaling was stimulated by SOCS3 deletion in gastrointestinal epithelium-specific using T3b-promoter [62]. T3b-SOCS3 cKO mice showed spontaneous gastric tumor by enhancing the ObRb-STAT3 pathway. Although T3b-SOCS3 cKO mice underwent SOCS3 deletion in both small and large intestines as well as in stomach, the tumor was restricted in the stomach. All T3b-SOCS3 cKO mice died within half year. The cKO mice exhibited hyperplasia at 3 weeks of age and developed tumors by 8 weeks, indicating that leptin can stimulate rapid tumorigenesis of epithelial cells in the stomach. This was supported by excess of leptin and ObR being associated with tumorigenesis in patients with early stage of GC [58]. As another unique point of this model, gastric tumor formation occurs prior to the inflammatory lesions. Chronic inflammation is considered to raise the incidence of cancers in various tissues. Gp130 is ubiquitously expressed and its mutant gp130757F mice developed inflammation-associated gastric adenoma because mutated gp130 cannot bind to SOCS3 with decreased in leptin expression in the gastric mucosa [63]. In contrast, in T3b-SOCS3 cKO mice, CD45+ infiltrated cells clearly appeared at 15 weeks of age, when gastric tumor formation was already completed. The evidence demonstrated that leptin is a trigger that stimulates malignancy and carcinogenesis; in particular, the stomach possibly prompts autocrine leptin signaling. Tumor-initiating stem cells have been reported to potently express ObR, resulting in tumor progression mediated by the activation of STAT3 and induction of pluripotency-associated transcription factors, such as Oct4 and Sox2 [64]. This evidence implied that leptin actively affects tumor formation and supports cell proliferation and pluripotency for tumorigenesis.
Murine models of GC, which closely mimic human GC pathology, have enriched our understanding of the disease (Table 1), either chemical carcinogen-induced or by bacterial infection, or using gene-targeting model focused on gastric function. N-methyl-N-nitrosourea (MNU) is an effective chemical for carcinogenesis, although limited to the antral region, and rarely in fundus [65,66]. H. pylori is considered to account for major of gastric cancer, however, human H. pylori infection is difficult to induce in murine models, with the exception of Mongolian gerbils. While CagA is a virulence factor of H. pylori, only a small portion of H/K-ATPase promoter-driven cagA Tg mice develop adenocarinoma after 72 weeks [67]. Thus, H. felis is frequently used as an alternative for murine model of gastric Helicobacter infection. Moreover, molecular-based approach using gene-targeting models has revealed a variety of processed of tumorigenesis [68,69]. TFF1 is a mucin-associated peptide secreted by gastric pit and neck cells, and found to be frequently downregulation in gastric carcinoma [70,71]. Gastrin is released by G cells of the stomach, and stimulates the secretion of gastric acids by the parietal cells. ATP4a and ATP4β genes encode α and β subunits of H+K+ATPase, which are responsible for the secretion of gastric acid. Despite these molecules being critical for the maintenance of normal functioning of gastric mucosa, mice with TFF1, gastrin or ATP4a deleted developed precancerous lesions or incomplete cancer [72,73]. K-ras has been shown to be strongly linked to the development of human cancers. Ubiquitous K-ras activation rapidly induces hyperplasia, metaplasia and adenomas in the stomach, while carcinogenesis has been observed in other tissues such as the oral cavity, colon, liver and lung [74]. In terms of cytokine signaling, deletion of TGF-β1 caused 40% incidence of adenocarcinoma [75], and lack of its downstream molecules Smad3 and Smad4 caused carcinoma, based on E-cadherin downregulation and invasive neoplasia [76,77]. Tgfβ1-C33S crossed with Rag2−/− chimeric mice exhibited reduced inflammation and tumors, implying that TGF-β1-mediated immune responses can suppress tumor development. IL-1β is strong association with increased risk of GC, whereas ATP4b prompter-driven IL-1β Tg mice exhibited only 30% incidence of adenocarcinoma [78]. These studies using gene-targeted mice, collectively indicate that mice can rarely develop perfectly spontaneous GC at young age, and that leptin signaling in the stomach can potently and rapidly induce GC.
5. Gastric Neoplasia Triggered by Diet-Induced Obesity
Obesity is one of the causative factors of GC. Excessive leptin secretion/augmentation of leptin signaling occurs in HFD-feeding. We demonstrated that high-fat diet (HFD) induces the loss of parietal cells and glandular cells, and promotes intestinal metaplasia, which transforms the gastric epithelium to an intestine-like epithelium, or precancerous lesions [85]. The stomach is so sensitive to HFD that 1week of HFD feeding resulted in hyperplasia. At 3 months of feeding, complete loss of zymogenic and glandular metaplasia occurred, and remarkable nuclear atypia such as nuclear elongation and pseudostratification was seen at 12w and no normal gastric gland and at 20 weeks. HFD induces ectopic expression of the following in the stomach: Muc2, an intestinal type mucus protein; Cdx2, a transcription factor that directs development of the intestinal epithelium; PLA2, a Paneth cell marker; in contrast, it causes the loss of H+K+ATPase, a parietal cell marker and integral membrane protein responsible for gastric acid secretion. Interestingly, these changes similar to those in carcinogenesis, were suppressed in db/db mice. The db/db mice are extraordinarily obese, yet showed suppressed pathogenesis.
HFD feeding also induces ectopic expression of fat in the stomach [86]. Accumulating ectopic fat can cause cell injury and functional impairment in a variety of tissues, also known as ‘lypotoxicity’ [87]. In terms of localization, fat accumulation was observed in the cytosol, not in the nucleus. Such a state of lipid stress in non-adipose tissues affects organelle homeostasis. Examining lysosome, mitochondria, Golgi apparatus, and endoplasmic reticulum, we found LAMP2A, a lysosomal marker protein, to be elevated in the early period after HFD feeding [86]. LAMP2A is a lysosome-associated membrane protein, which acts as a receptor for the substrates of chaperon-mediated autophagy, and is highly expressed in gastric cancer [88]. In addition, acidic lipase, which is produced by the chief cells in the gastric fundus and functions in the lysosome to maintain low pH of the gastric mucosal milieu [89], is ectopically expressed in the duodenum. In contrast, lipoprotein lipase, which is a classical lipase produced and secreted by pancreas into the duodenum, is expressed and in the gastric mucosa. HFD might induce not only intestinal metaplasia, but also gastric metaplasia. Recently, accumulating evidence has revealed the link between lysosomal dysfunction and obesity. HFD downregulates autophagy by reducing autophagosomes and lowering acidity of lysosomes both in vitro and in vivo [90,91]. Although HFD-related lipid changes have been reported to reduce LAMP2A expression in neurodegenerative diseases [92], earliest changes of LAMP2A may be considered as one of the host defense mechanism occurring in the stomach. Following LAMP2A change, COX IV-2 expression was increased, which is the hypoxia marker molecule in the mitochondria of the stomach by HFD feeding. Hypoxia is a common condition in most of tumors, referring to a non-physiological oxygen level, leading to the acquisition of EMT [93]. Hypoxia-inducible factor 1α (HIF-1α) is a master regulator of adaptive responses to hypoxia and promotes critical steps in tumor progression [94] as well as obesity [95]. HIF-1α occurs downstream of STAT3 and regulates Hes-1 expression [96]. Hes-1 belongs to helix-loop-helix family of the transcription factors and regulates the differentiation of stem/progenitor cells to absorptive cells in intestinal tract or in tumor cells, in addition to the normal cells of gastrointestine [97]. Hes-1 is highly expressed in the gastric mucosa, showing intestinal metaplasia induced by HFD feeding [86]. Thus, excessive leptin signaling might be involved in neoplasia and metaplasia in the stomach, mediated by the STAT3-HIF-1-Hes-1 axis. In triple-negative breast cancer, leptin-dependent mechanisms, which lead to cancer, mediated by the upregulation of stem cell (CSC)- and EMT-related gene expression [98], induces estrogen receptor-α (ER-α) expression via JAK-STAT pathway [99]. Leptin-induced ZEB-1, an EMT-induced transcription factor via activation of ERK signaling, was seen to be promoted in lung cancer [100].
β-Catenin is a multi-functional molecule with a dual role. First, it is a cytoplasmic anchoring protein of E-cadherin, required for cell-cell junction as adherens junctions [101]. Secondly, it acts as an intracellular signal transducer in Wnt pathway [102,103]. Target molecules of β-catenin include stem cell and CSC markers, namely Lgr5, CD44, EpCAM [104], Nanog, Oct4, and c-Myc [105]. Suppression of E-cadherin expression and nuclear accumulation of β-catenin are correlated with tumor progression in many cancers including GC [103]. In murine models with upregulation of leptin signaling in the stomach, β-catenin localization was clearly shown; the gastric mucosa due to SOCS3 deletion showed completed nuclear accumulation of β-catenin [62], and HFD-fed gastric mucosa exhibited accumulated intracellular or perinuclear localization [86]. The alteration was observed in an early stage of the pathogenesis (Figure 3).
6. Therapeutic Implications
The rising incidence of GC and its poor clinical outcome using current therapeutic approaches urge the discovery of new medicinal approaches, since the survival rate is 40-50% after 1 year, and less than 15% over a 5-year duration after initial therapy treatment, due to relapse [106]. Cisplatin and 5-fluorouracil (5-FU), in addition to trastuzumab, have been used as effective treatments for patients with gastro-esophageal adenocarcinoma, with human epidermal growth factor 2 (HER2) [107], a proto-oncogene, being expressed frequently in the proximal stomach compared to that in the distal [108]. In addition, in HER2-negative GC, oxaliplatin-based regimens could improve tolerance effectively. Another drug, ramucirumab, a monoclonal antibody against vascular endothelial growth factor receptor 2, was shown to be effective in patients with GC that had failed or progressed after the first line therapy [109]. In relation to angiogenesis, although these approaches demonstrate success to some extent, treatment-associated toxicity is a pronounced side-effect. Recently, leptin has been reported as a candidate target molecule for GC therapy since it is considered as a tumorigenesis-associated molecule. Thus, the use of leptin antagonist could be a useful therapeutic strategy against cancer. Leptin-expressing GC cells also exhibited chemoresistance [110,111]. Leptin attenuated the effect of 5-FU on apoptosis via downregulation of pro-apoptotic molecules (BAX, Caspase-3) and upregulation of anti-apoptotic molecules (BcL-xL, RIP) in pancreatic cancer [112]. Moreover, inhibition of leptin-induced NLRP3 inflammasome prevented cancer cell proliferation of MCF-7 breast cancer cells [113]. Recently, leptin has been reported to induce antibodies to H. pylori in mice vaccination, suggesting the possibility of adjuvant tool in the development of effective vaccine [114]. All these studies encourage the clinical inhibition of leptin and its signaling as a novel therapeutic approach against GC.
7. Conclusions
Gastric cancer and obesity are currently considered as major public health concerns worldwide. Leptin potently affects metaplasia and cancer cell proliferation. In most cases, augmentation of leptin and its signaling promotes carcinogenesis at an early stage. Gastric leptin signaling is a critical checkpoint for the onset and development of gastric neoplasia. Thus, silencing of leptin or leptin-associated upregulated molecules may be considered as a therapeutic treatment.
Funding
K.I.O. is supported by Grants-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (17K08790).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef] [PubMed]
- La Cava, A.; Matarese, G. The weight of leptin in immunity. Nat. Rev. Immunol. 2004, 4, 371–379. [Google Scholar] [CrossRef]
- Bado, A.; Levasseur, S.; Attoub, S.; Kermorgant, S.; Laigneau, J.P.; Bortoluzzi, M.N.; Moizo, L.; Lehy, T.; Guerre-Millo, M.; Le Marchand-Brustel, Y.; et al. The stomach is a source of leptin. Nature 1998, 394, 790–793. [Google Scholar] [CrossRef]
- Masuzaki, H.; Ogawa, Y.; Sagawa, N.; Hosoda, K.; Matsumoto, T.; Mise, H.; Nishimura, H.; Yoshimasa, Y.; Tanaka, I.; Mori, T.; et al. Nonadipose tissue production of leptin: Leptin as a novel placenta-derived hormone in humans. Nat. Med. 1997, 3, 1029–1033. [Google Scholar] [CrossRef]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Ando, S.; Catalano, S. Leptin, obesity and breast cancer: Progress to understanding the molecular connections. Curr. Opin. Pharmacol. 2016, 31, 83–89. [Google Scholar] [CrossRef]
- Olea-Flores, M.; Juarez-Cruz, J.C.; Mendoza-Catalan, M.A.; Padilla-Benavides, T.; Navarro-Tito, N. Signaling pathways induced by leptin during epithelial(-)mesenchymal transition in breast cancer. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Vannella, L.; Lahner, E.; Annibale, B. Risk for gastric neoplasias in patients with chronic atrophic gastritis: A critical reappraisal. World J. Gastroenterol. 2012, 18, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Asombang, A.W.; Ibdah, J.A. Characteristics of gastric cancer in asia. World J. Gastroenterol. 2014, 20, 4483–4490. [Google Scholar] [CrossRef]
- Rivera, F.; Romero, C.; Jimenez-Fonseca, P.; Izquierdo-Manuel, M.; Salud, A.; Martinez, E.; Jorge, M.; Arrazubi, V.; Mendez, J.C.; Garcia-Alfonso, P.; et al. Phase ii study to evaluate the efficacy of trastuzumab in combination with capecitabine and oxaliplatin in first-line treatment of her2-positive advanced gastric cancer: Herxo trial. Cancer Chemother. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed]
- Chiariotti, L.; Angrisano, T.; Keller, S.; Florio, E.; Affinito, O.; Pallante, P.; Perrino, C.; Pero, R.; Lembo, F. Epigenetic modifications induced by helicobacter pylori infection through a direct microbe-gastric epithelial cells cross-talk. Med. Microbiol. Immunol. 2013, 202, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Pero, R.; Coretti, L.; Nigro, E.; Lembo, F.; Laneri, S.; Lombardo, B.; Daniele, A.; Scudiero, O. Beta-defensins in the fight against helicobacter pylori. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Lee, D.H.; Oh, H.S.; Seo, J.Y.; Lee, D.H.; Kim, N.; Jeong, S.H.; Kim, J.W.; Hwang, J.H.; Park, Y.S.; et al. Higher prevalence of obesity in gastric cardia adenocarcinoma compared to gastric non-cardia adenocarcinoma. Dig. Dis. Sci. 2012, 57, 2687–2692. [Google Scholar] [CrossRef] [PubMed]
- O’Doherty, M.G.; Freedman, N.D.; Hollenbeck, A.R.; Schatzkin, A.; Abnet, C.C. A prospective cohort study of obesity and risk of oesophageal and gastric adenocarcinoma in the nih-aarp diet and health study. Gut 2012, 61, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J. Hered. 1950, 41, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Coleman, D.L. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia 1973, 9, 294–298. [Google Scholar] [CrossRef]
- Niswender, K.D.; Morton, G.J.; Stearns, W.H.; Rhodes, C.J.; Myers, M.G., Jr.; Schwartz, M.W. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature 2001, 413, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Al-Qassab, H.; Smith, M.A.; Irvine, E.E.; Guillermet-Guibert, J.; Claret, M.; Choudhury, A.I.; Selman, C.; Piipari, K.; Clements, M.; Lingard, S.; et al. Dominant role of the p110beta isoform of pi3k over p110alpha in energy homeostasis regulation by pomc and agrp neurons. Cell. Metab. 2009, 10, 343–354. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, ob-r. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef]
- Bjorbaek, C.; Uotani, S.; da Silva, B.; Flier, J.S. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J. Biol. Chem. 1997, 272, 32686–32695. [Google Scholar] [CrossRef]
- Pan, W.; Hsuchou, H.; Tu, H.; Kastin, A.J. Developmental changes of leptin receptors in cerebral microvessels: Unexpected relation to leptin transport. Endocrinology 2008, 149, 877–885. [Google Scholar] [CrossRef]
- Mancour, L.V.; Daghestani, H.N.; Dutta, S.; Westfield, G.H.; Schilling, J.; Oleskie, A.N.; Herbstman, J.F.; Chou, S.Z.; Skiniotis, G. Ligand-induced architecture of the leptin receptor signaling complex. Mol. Cell. 2012, 48, 655–661. [Google Scholar] [CrossRef]
- Howard, J.K.; Flier, J.S. Attenuation of leptin and insulin signaling by socs proteins. Trends Endocrinol. Metab. 2006, 17, 365–371. [Google Scholar] [CrossRef]
- Gong, Y.; Ishida-Takahashi, R.; Villanueva, E.C.; Fingar, D.C.; Munzberg, H.; Myers, M.G., Jr. The long form of the leptin receptor regulates stat5 and ribosomal protein s6 via alternate mechanisms. J. Biol. Chem. 2007, 282, 31019–31027. [Google Scholar] [CrossRef]
- Lee, J.Y.; Muenzberg, H.; Gavrilova, O.; Reed, J.A.; Berryman, D.; Villanueva, E.C.; Louis, G.W.; Leinninger, G.M.; Bertuzzi, S.; Seeley, R.J.; et al. Loss of cytokine-stat5 signaling in the cns and pituitary gland alters energy balance and leads to obesity. PLoS ONE 2008, 3, e1639. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G., Jr. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef]
- Di Spiezio, A.; Sandin, E.S.; Dore, R.; Muller-Fielitz, H.; Storck, S.E.; Bernau, M.; Mier, W.; Oster, H.; Johren, O.; Pietrzik, C.U.; et al. The lepr-mediated leptin transport across brain barriers controls food reward. Mol. Metab. 2018, 8, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Gautron, L.; Elmquist, J.K. Sixteen years and counting: An update on leptin in energy balance. J. Clin. Invest. 2011, 121, 2087–2093. [Google Scholar] [CrossRef]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob rna in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Townsend, K.L.; Lorenzi, M.M.; Widmaier, E.P. High-fat diet-induced changes in body mass and hypothalamic gene expression in wild-type and leptin-deficient mice. Endocrine 2008, 33, 176–188. [Google Scholar] [CrossRef] [PubMed]
- White, C.L.; Whittington, A.; Barnes, M.J.; Wang, Z.; Bray, G.A.; Morrison, C.D. Hf diets increase hypothalamic ptp1b and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E291–E299. [Google Scholar] [CrossRef]
- Ogus, S.; Ke, Y.; Qiu, J.; Wang, B.; Chehab, F.F. Hyperleptinemia precipitates diet-induced obesity in transgenic mice overexpressing leptin. Endocrinology 2003, 144, 2865–2869. [Google Scholar] [CrossRef]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J.; Sewter, C.P.; Digby, J.E.; Mohammed, S.N.; Hurst, J.A.; et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997, 387, 903–908. [Google Scholar] [CrossRef]
- Wabitsch, M.; Funcke, J.B.; Lennerz, B.; Kuhnle-Krahl, U.; Lahr, G.; Debatin, K.M.; Vatter, P.; Gierschik, P.; Moepps, B.; Fischer-Posovszky, P. Biologically inactive leptin and early-onset extreme obesity. N Engl. J. Med. 2015, 372, 48–54. [Google Scholar] [CrossRef]
- Bjorbaek, C.; Elmquist, J.K.; Frantz, J.D.; Shoelson, S.E.; Flier, J.S. Identification of socs-3 as a potential mediator of central leptin resistance. Mol. Cell. 1998, 1, 619–625. [Google Scholar] [CrossRef]
- Mori, H.; Hanada, R.; Hanada, T.; Aki, D.; Mashima, R.; Nishinakamura, H.; Torisu, T.; Chien, K.R.; Yasukawa, H.; Yoshimura, A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat. Med. 2004, 10, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Kievit, P.; Howard, J.K.; Badman, M.K.; Balthasar, N.; Coppari, R.; Mori, H.; Lee, C.E.; Elmquist, J.K.; Yoshimura, A.; Flier, J.S. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in pomc-expressing cells. Cell. Metab. 2006, 4, 123–132. [Google Scholar] [CrossRef]
- Reed, A.S.; Unger, E.K.; Olofsson, L.E.; Piper, M.L.; Myers, M.G., Jr.; Xu, A.W. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 2010, 59, 894–906. [Google Scholar] [CrossRef]
- Mix, H.; Widjaja, A.; Jandl, O.; Cornberg, M.; Kaul, A.; Goke, M.; Beil, W.; Kuske, M.; Brabant, G.; Manns, M.P.; et al. Expression of leptin and leptin receptor isoforms in the human stomach. Gut 2000, 47, 481–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilmeau, S.; Buyse, M.; Bado, A. Gastric leptin: A new manager of gastrointestinal function. Curr. Opin. Pharmacol. 2004, 4, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Morton, N.M.; Emilsson, V.; Liu, Y.L.; Cawthorne, M.A. Leptin action in intestinal cells. J. Biol. Chem. 1998, 273, 26194–26201. [Google Scholar] [CrossRef]
- Buyse, M.; Berlioz, F.; Guilmeau, S.; Tsocas, A.; Voisin, T.; Peranzi, G.; Merlin, D.; Laburthe, M.; Lewin, M.J.; Roze, C.; et al. Pept1-mediated epithelial transport of dipeptides and cephalexin is enhanced by luminal leptin in the small intestine. J. Clin. Invest. 2001, 108, 1483–1494. [Google Scholar] [CrossRef]
- Attoub, S.; Noe, V.; Pirola, L.; Bruyneel, E.; Chastre, E.; Mareel, M.; Wymann, M.P.; Gespach, C. Leptin promotes invasiveness of kidney and colonic epithelial cells via phosphoinositide 3-kinase-, rho-, and rac-dependent signaling pathways. FASEB J. 2000, 14, 2329–2338. [Google Scholar] [CrossRef]
- Hardwick, J.C.; Van Den Brink, G.R.; Offerhaus, G.J.; Van Deventer, S.J.; Peppelenbosch, M.P. Leptin is a growth factor for colonic epithelial cells. Gastroenterology 2001, 121, 79–90. [Google Scholar] [CrossRef]
- Kiely, J.M.; Noh, J.H.; Pitt, H.A.; Swartz-Basile, D.A. Impaired intestinal cell proliferation and cell death in leptin-deficient obese mice. JPEN J. Parenter. Enteral. Nutr. 2005, 29, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Sukhotnik, I.; Coran, A.G.; Mogilner, J.G.; Shamian, B.; Karry, R.; Lieber, M.; Shaoul, R. Leptin affects intestinal epithelial cell turnover in correlation with leptin receptor expression along the villus-crypt axis after massive small bowel resection in a rat. Pediatr. Res. 2009, 66, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Rajala, M.W.; Patterson, C.M.; Opp, J.S.; Foltin, S.K.; Young, V.B.; Myers, M.G., Jr. Leptin acts independently of food intake to modulate gut microbial composition in male mice. Endocrinology 2014, 155, 748–757. [Google Scholar] [CrossRef]
- Higurashi, T.; Endo, H.; Uchiyama, T.; Uchiyama, S.; Yamada, E.; Ohkubo, H.; Sakai, E.; Takahashi, H.; Maeda, S.; Wada, K.; et al. Conditional knockout of the leptin receptor in the colonic epithelium revealed the local effects of leptin receptor signaling in the progression of colonic tumors in mice. Carcinogenesis 2014, 35, 2134–2141. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Roberts, M.R.; Becker, S.M.; Podd, B.; Zhang, Y.; Chua, S.C., Jr.; Myers, M.G., Jr.; Duggal, P.; Houpt, E.R.; Petri, W.A., Jr. Leptin signaling in intestinal epithelium mediates resistance to enteric infection by entamoeba histolytica. Mucosal. Immunol. 2011, 4, 294–303. [Google Scholar] [CrossRef]
- Sobhani, I.; Bado, A.; Vissuzaine, C.; Buyse, M.; Kermorgant, S.; Laigneau, J.P.; Attoub, S.; Lehy, T.; Henin, D.; Mignon, M.; et al. Leptin secretion and leptin receptor in the human stomach. Gut 2000, 47, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Cinti, S.; Matteis, R.D.; Pico, C.; Ceresi, E.; Obrador, A.; Maffeis, C.; Oliver, J.; Palou, A. Secretory granules of endocrine and chief cells of human stomach mucosa contain leptin. Int. J. Obes. Relat. Metab. Disord 2000, 24, 789–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, T.; Suto, H.; Ito, Y.; Ohtani, M.; Dojo, M.; Kuriyama, M.; Kato, T. Gastric leptin and helicobacter pylori infection. Gut 2001, 49, 324–329. [Google Scholar] [CrossRef]
- Pai, R.; Lin, C.; Tran, T.; Tarnawski, A. Leptin activates stat and erk2 pathways and induces gastric cancer cell proliferation. BioChem. Biophys Res. Commun. 2005, 331, 984–992. [Google Scholar] [CrossRef]
- Zhao, L.; Shen, Z.X.; Luo, H.S.; Shen, L. Possible involvement of leptin and leptin receptor in developing gastric adenocarcinoma. World J. Gastroenterol. 2005, 11, 7666–7670. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Kitayama, J.; Nagawa, H. Expression pattern of leptin and leptin receptor (ob-r) in human gastric cancer. World J. Gastroenterol. 2006, 12, 5517–5522. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, K.; Zhu, Z.; Chen, S.; Hu, R. Correlation between expression of leptin and clinicopathological features and prognosis in patients with gastric cancer. J. Gastroenterol. Hepatol. 2007, 22, 1317–1321. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.N.; Choi, H.S.; Yang, S.Y.; Park, H.K.; Lee, Y.Y.; Lee, O.Y.; Yoon, B.C.; Hahm, J.S.; Paik, S.S. The role of leptin in gastric cancer: Clinicopathologic features and molecular mechanisms. BioChem. Biophys Res. Commun. 2014, 446, 822–829. [Google Scholar] [CrossRef]
- Dong, Z.; Fu, S.; Xu, X.; Yang, Y.; Du, L.; Li, W.; Kan, S.; Li, Z.; Zhang, X.; Wang, L.; et al. Leptin-mediated regulation of icam-1 is rho/rock dependent and enhances gastric cancer cell migration. Br. J. Cancer 2014, 110, 1801–1810. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Mayuzumi, H.; Kato, S.; Minokoshi, Y.; Otsubo, T.; Kawamura, Y.I.; Dohi, T.; Matsuzaki, G.; Yoshimura, A. Enhancement of leptin receptor signaling by socs3 deficiency induces development of gastric tumors in mice. Oncogene 2014, 33, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Tebbutt, N.C.; Giraud, A.S.; Inglese, M.; Jenkins, B.; Waring, P.; Clay, F.J.; Malki, S.; Alderman, B.M.; Grail, D.; Hollande, F.; et al. Reciprocal regulation of gastrointestinal homeostasis by shp2 and stat-mediated trefoil gene activation in gp130 mutant mice. Nat. Med. 2002, 8, 1089–1097. [Google Scholar] [CrossRef]
- Feldman, D.E.; Chen, C.; Punj, V.; Tsukamoto, H.; Machida, K. Pluripotency factor-mediated expression of the leptin receptor (ob-r) links obesity to oncogenesis through tumor-initiating stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Yamachika, T.; Nakanishi, H.; Inada, K.; Tsukamoto, T.; Shimizu, N.; Kobayashi, K.; Fukushima, S.; Tatematsu, M. N-methyl-n-nitrosourea concentration-dependent, rather than total intake-dependent, induction of adenocarcinomas in the glandular stomach of balb/c mice. Jpn. J. Cancer Res. 1998, 89, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Furihata, C.; Ogiu, T.; Tsukamoto, T.; Inada, K.; Hirano, K.; Tatematsu, M. Independent variation in susceptibilities of six different mouse strains to induction of pepsinogen-altered pyloric glands and gastric tumor intestinalization by n-methyl-n-nitrosourea. Cancer Lett. 2002, 179, 121–132. [Google Scholar] [CrossRef]
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of helicobacter pylori caga induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008. [Google Scholar] [CrossRef]
- Sakagami, T.; Dixon, M.; O’Rourke, J.; Howlett, R.; Alderuccio, F.; Vella, J.; Shimoyama, T.; Lee, A. Atrophic gastric changes in both helicobacter felis and helicobacter pylori infected mice are host dependent and separate from antral gastritis. Gut 1996, 39, 639–648. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, N.; Choi, Y.J.; Nam, R.H.; Choi, Y.J.; Lee, S.; Choi, D.; Lee, H.S.; Kim, J.W.; Lee, D.H. Effect of n-methyl-n-nitrosourea on helicobacter-induced gastric carcinogenesis in c57bl/6 mice. J. Cancer Prev. 2016, 21, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Zhang, Y.; Yuan, H.; Bai, R.; Zheng, J.; Zhang, J.; Song, M. DNA methylation of trefoil factor 1 (tff1) is associated with the tumorigenesis of gastric carcinoma. Mol. Med. Rep. 2014, 9, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, O.; Chenard, M.P.; Masson, R.; Linares, J.; Dierich, A.; LeMeur, M.; Wendling, C.; Tomasetto, C.; Chambon, P.; Rio, M.C. Gastric mucosa abnormalities and tumorigenesis in mice lacking the ps2 trefoil protein. Science 1996, 274, 259–262. [Google Scholar] [CrossRef]
- Zavros, Y.; Eaton, K.A.; Kang, W.; Rathinavelu, S.; Katukuri, V.; Kao, J.Y.; Samuelson, L.C.; Merchant, J.L. Chronic gastritis in the hypochlorhydric gastrin-deficient mouse progresses to adenocarcinoma. Oncogene 2005, 24, 2354–2366. [Google Scholar] [CrossRef] [PubMed]
- Judd, L.M.; Andringa, A.; Rubio, C.A.; Spicer, Z.; Shull, G.E.; Miller, M.L. Gastric achlorhydria in h/k-atpase-deficient (atp4a(−/−)) mice causes severe hyperplasia, mucocystic metaplasia and upregulation of growth factors. J. Gastroenterol. Hepatol. 2005, 20, 1266–1278. [Google Scholar] [CrossRef]
- Ray, K.C.; Bell, K.M.; Yan, J.; Gu, G.; Chung, C.H.; Washington, M.K.; Means, A.L. Epithelial tissues have varying degrees of susceptibility to kras(g12d)-initiated tumorigenesis in a mouse model. PLoS ONE 2011, 6, e16786. [Google Scholar] [CrossRef]
- Ota, M.; Horiguchi, M.; Fang, V.; Shibahara, K.; Kadota, K.; Loomis, C.; Cammer, M.; Rifkin, D.B. Genetic suppression of inflammation blocks the tumor-promoting effects of tgf-beta in gastric tissue. Cancer Res. 2014, 74, 2642–2651. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.T.; O’Neal, R.; Lee, Y.S.; Lee, Y.C.; Coffey, R.J.; Goldenring, J.R. Gastric tumor development in smad3-deficient mice initiates from forestomach/glandular transition zone along the lesser curvature. Lab. Invest. 2012, 92, 883–895. [Google Scholar] [CrossRef]
- Xu, X.; Brodie, S.G.; Yang, X.; Im, Y.H.; Parks, W.T.; Chen, L.; Zhou, Y.X.; Weinstein, M.; Kim, S.J.; Deng, C.X. Haploid loss of the tumor suppressor smad4/dpc4 initiates gastric polyposis and cancer in mice. Oncogene 2000, 19, 1868–1874. [Google Scholar] [CrossRef]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008, 14, 408–419. [Google Scholar] [CrossRef]
- Oshima, H.; Matsunaga, A.; Fujimura, T.; Tsukamoto, T.; Taketo, M.M.; Oshima, M. Carcinogenesis in mouse stomach by simultaneous activation of the wnt signaling and prostaglandin e2 pathway. Gastroenterology 2006, 131, 1086–1095. [Google Scholar] [CrossRef]
- Wang, T.C.; Dangler, C.A.; Chen, D.; Goldenring, J.R.; Koh, T.; Raychowdhury, R.; Coffey, R.J.; Ito, S.; Varro, A.; Dockray, G.J.; et al. Synergistic interaction between hypergastrinemia and helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000, 118, 36–47. [Google Scholar] [CrossRef]
- Syder, A.J.; Karam, S.M.; Mills, J.C.; Ippolito, J.E.; Ansari, H.R.; Farook, V.; Gordon, J.I. A transgenic mouse model of metastatic carcinoma involving transdifferentiation of a gastric epithelial lineage progenitor to a neuroendocrine phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 4471–4476. [Google Scholar] [CrossRef] [Green Version]
- Shimada, S.; Mimata, A.; Sekine, M.; Mogushi, K.; Akiyama, Y.; Fukamachi, H.; Jonkers, J.; Tanaka, H.; Eishi, Y.; Yuasa, Y. Synergistic tumour suppressor activity of e-cadherin and p53 in a conditional mouse model for metastatic diffuse-type gastric cancer. Gut 2012, 61, 344–353. [Google Scholar] [CrossRef]
- Lee, M.P.; Ravenel, J.D.; Hu, R.J.; Lustig, L.R.; Tomaselli, G.; Berger, R.D.; Brandenburg, S.A.; Litzi, T.J.; Bunton, T.E.; Limb, C.; et al. Targeted disruption of the kvlqt1 gene causes deafness and gastric hyperplasia in mice. J. Clin. Invest. 2000, 106, 1447–1455. [Google Scholar] [CrossRef]
- Ito, K.; Chuang, L.S.; Ito, T.; Chang, T.L.; Fukamachi, H.; Salto-Tellez, M.; Ito, Y. Loss of runx3 is a key event in inducing precancerous state of the stomach. Gastroenterology 2011, 140, 1536–1546.e8. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Okamoto, S.; Takagi, K.; Saito, K.; Arita, S.; Tang, L.; Hori, T.; Kataoka, H.; Matsumoto, S.; Minokoshi, Y. Leptin receptor signaling is required for high-fat diet-induced atrophic gastritis in mice. Nutr. Metab. 2016, 13. [Google Scholar] [CrossRef]
- Arita, S.; Kinoshita, Y.; Ushida, K.; Enomoto, A.; Inagaki-Ohara, K. High-fat diet feeding promotes stemness and precancerous changes in murine gastric mucosa mediated by leptin receptor signaling pathway. Arch. BioChem. Biophys 2016, 610, 16–24. [Google Scholar] [CrossRef]
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys Acta 2010, 1801, 209–214. [Google Scholar] [CrossRef]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The low-density lipoprotein pathway and its relation to atherosclerosis. Annu. Rev. BioChem. 1977, 46, 897–930. [Google Scholar] [CrossRef]
- Las, G.; Serada, S.B.; Wikstrom, J.D.; Twig, G.; Shirihai, O.S. Fatty acids suppress autophagic turnover in beta-cells. J. Biol. Chem. 2011, 286, 42534–42544. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Kimura, T.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Matsusaka, T.; et al. High-fat diet-induced lysosomal dysfunction and impaired autophagic flux contribute to lipotoxicity in the kidney. J. Am. Soc. Nephrol. 2017, 28, 1534–1551. [Google Scholar] [CrossRef]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone mediated autophagy in the crosstalk of neurodegenerative diseases and metabolic disorders. Front. Endocrinol. (Lausanne) 2018, 9. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Targeting hif-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M.; Velloso, L.A. Hypoxia inducible factor as a central regulator of metabolism—Implications for the development of obesity. Front. NeuroSci. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh, S.; Yazdanparast, R. Activation of stat3/hif-1alpha/hes-1 axis promotes trastuzumab resistance in her2-overexpressing breast cancer cells via down-regulation of pten. Biochim. Biophys Acta Gen. Subj. 2017, 1861, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Goto, N.; Ueo, T.; Fukuda, A.; Kawada, K.; Sakai, Y.; Miyoshi, H.; Taketo, M.M.; Chiba, T.; Seno, H. Distinct roles of hes1 in normal stem cells and tumor stem-like cells of the intestine. Cancer Res. 2017, 77, 3442–3454. [Google Scholar] [CrossRef]
- Bowers, L.W.; Rossi, E.L.; McDonell, S.B.; Doerstling, S.S.; Khatib, S.A.; Lineberger, C.G.; Albright, J.E.; Tang, X.; deGraffenried, L.A.; Hursting, S.D. Leptin signaling mediates obesity-associated csc enrichment and emt in preclinical tnbc models. Mol. Cancer Res. 2018, 16, 869–879. [Google Scholar] [CrossRef]
- Haque, I.; Ghosh, A.; Acup, S.; Banerjee, S.; Dhar, K.; Ray, A.; Sarkar, S.; Kambhampati, S.; Banerjee, S.K. Leptin-induced er-alpha-positive breast cancer cell viability and migration is mediated by suppressing ccn5-signaling via activating jak/akt/stat-pathway. BMC Cancer 2018, 18. [Google Scholar] [CrossRef]
- Xu, M.; Cao, F.L.; Li, N.; Gao, X.; Su, X.; Jiang, X. Leptin induces epithelial-to-mesenchymal transition via activation of the erk signaling pathway in lung cancer cells. Oncol. Lett. 2018, 16, 4782–4788. [Google Scholar] [CrossRef]
- Kimelman, D.; Xu, W. Beta-catenin destruction complex: Insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, R.L.; Xu, A.M. Epithelial-mesenchymal transition in gastric cancer. Am. J. Transl. Res. 2015, 7, 2141–2158. [Google Scholar]
- Rassouli, F.B.; Matin, M.M.; Saeinasab, M. Cancer stem cells in human digestive tract malignancies. Tumour Biol. 2016, 37, 7–21. [Google Scholar] [CrossRef]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef]
- Kunz, P.L.; Gubens, M.; Fisher, G.A.; Ford, J.M.; Lichtensztajn, D.Y.; Clarke, C.A. Long-term survivors of gastric cancer: A california population-based study. J. Clin. Oncol. 2012, 30, 3507–3515. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of her2-positive advanced gastric or gastro-oesophageal junction cancer (toga): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Abrahao-Machado, L.F.; Scapulatempo-Neto, C. Her2 testing in gastric cancer: An update. World J. Gastroenterol. 2016, 22, 4619–4625. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (regard): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Bain, G.H.; Collie-Duguid, E.; Murray, G.I.; Gilbert, F.J.; Denison, A.; McKiddie, F.; Ahearn, T.; Fleming, I.; Leeds, J.; Phull, P.; et al. Tumour expression of leptin is associated with chemotherapy resistance and therapy-independent prognosis in gastro-oesophageal adenocarcinomas. Br. J. Cancer 2014, 110, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Blank, S.; Deck, C.; Dreikhausen, L.; Weichert, W.; Giese, N.; Falk, C.; Schmidt, T.; Ott, K. Angiogenic and growth factors in gastric cancer. J. Surg. Res. 2015, 194, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Harbuzariu, A.; Gonzalez-Perez, R.R. Leptin-notch axis impairs 5-fluorouracil effects on pancreatic cancer. Oncotarget 2018, 9, 18239–18253. [Google Scholar] [CrossRef] [PubMed]
- Raut, P.K.; Kim, S.H.; Choi, D.Y.; Jeong, G.S.; Park, P.H. Growth of breast cancer cells by leptin is mediated via activation of the inflammasome: Critical roles of estrogen receptor signaling and reactive oxygen species production. BioChem. Pharmacol. 2019, 161, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Alti, D.; Sambamurthy, C.; Kalangi, S.K. Emergence of leptin in infection and immunity: Scope and challenges in vaccines formulation. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
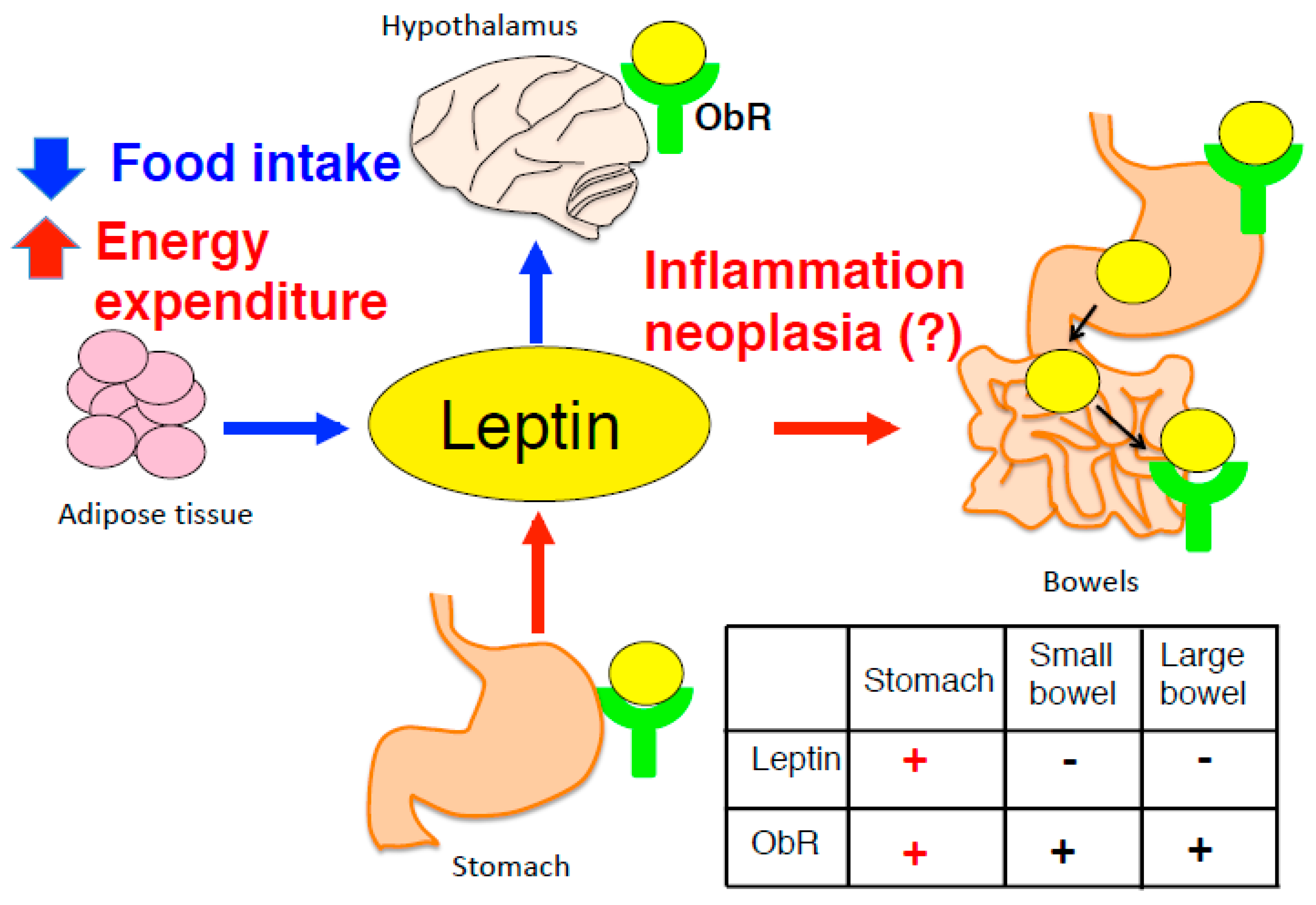
Figure 1.
Adipocyte-derived leptin modulates the suppression of appetite and increased energy expenditure mediated by leptin signaling in hypothalamus. By contrast, stomach expresses both leptin and ObR, however, physiological significance of gastric leptin remains unclear. Table inside figure shows expression of leptin and ObR in the gastrointestinal tract.
Figure 1.
Adipocyte-derived leptin modulates the suppression of appetite and increased energy expenditure mediated by leptin signaling in hypothalamus. By contrast, stomach expresses both leptin and ObR, however, physiological significance of gastric leptin remains unclear. Table inside figure shows expression of leptin and ObR in the gastrointestinal tract.

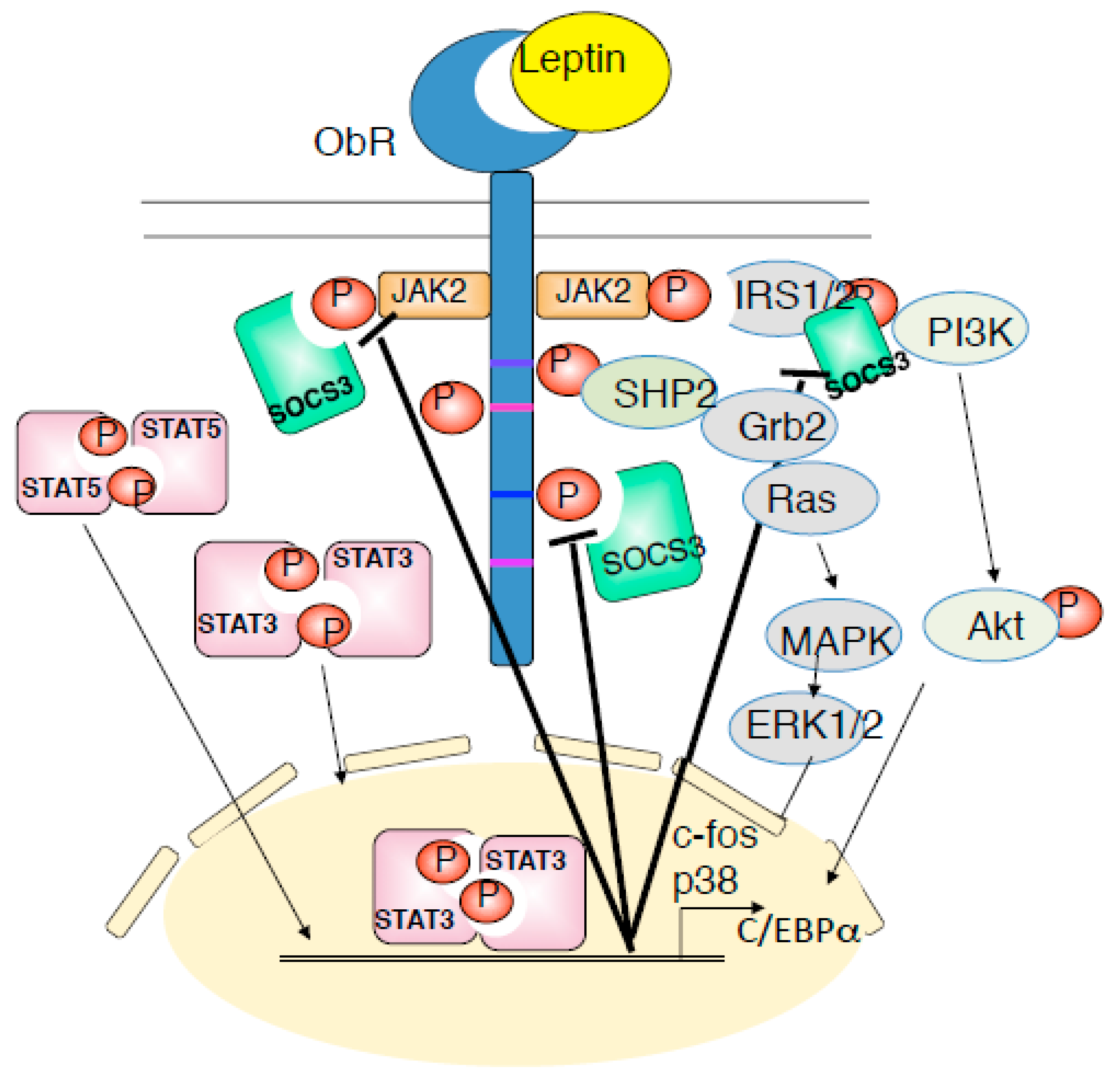
Figure 2.
Leptin receptor signaling is mediated by JAK-STAT, PI3K-Akt, and SHP2-ERK pathway. Leptin binds to ObRb and activates JAK2, and induced to the phosphorylation of Tyr985, Tyr1077, and Tyr1138 of ObRb. PY-985, PY-1077, and PY-1138 bind to their downstream molecules and proceeds to phosphorylation of JAK2-STAT3, JAk2-STAT5, PI3K-IRS-Akt, SHP2-ERK pathways. These signaling are negatively regulated by SOCS3. Dysregulation of the leptin receptor signaling is involved in the onset of leptin resistance, inflammation and cancer.
Figure 2.
Leptin receptor signaling is mediated by JAK-STAT, PI3K-Akt, and SHP2-ERK pathway. Leptin binds to ObRb and activates JAK2, and induced to the phosphorylation of Tyr985, Tyr1077, and Tyr1138 of ObRb. PY-985, PY-1077, and PY-1138 bind to their downstream molecules and proceeds to phosphorylation of JAK2-STAT3, JAk2-STAT5, PI3K-IRS-Akt, SHP2-ERK pathways. These signaling are negatively regulated by SOCS3. Dysregulation of the leptin receptor signaling is involved in the onset of leptin resistance, inflammation and cancer.

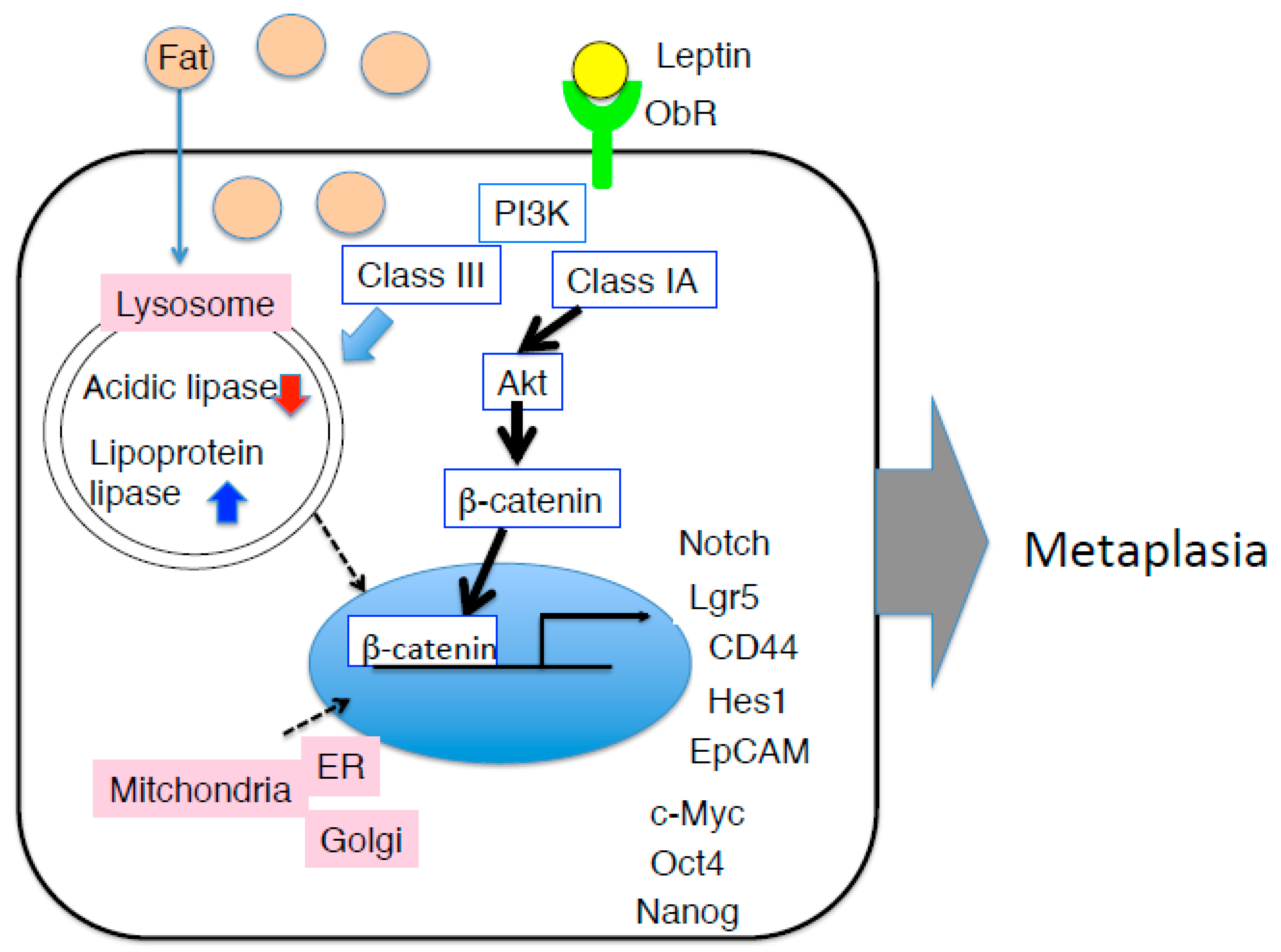
Figure 3.
Hypothesis mechanism of intestinal metaplasia of the gastric mucosa in HFD-induced obese mice. In addition to JAK-STAT pathway, by PI3K-Akt-β-catenin pathway may be strongly induce pluripotent gene, leading to the onset of intestinal metaplasia. Amount of HFD causes lipotoxic milieu in the gastric epithelium, in particular, function of lysosome and mitochondria is impaired, although it remains unclear why gastric leptin is increased due to HFD feeding.
Figure 3.
Hypothesis mechanism of intestinal metaplasia of the gastric mucosa in HFD-induced obese mice. In addition to JAK-STAT pathway, by PI3K-Akt-β-catenin pathway may be strongly induce pluripotent gene, leading to the onset of intestinal metaplasia. Amount of HFD causes lipotoxic milieu in the gastric epithelium, in particular, function of lysosome and mitochondria is impaired, although it remains unclear why gastric leptin is increased due to HFD feeding.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Murine model of gastric cancer.
| Model | Incidence (%) | Periods or Age Onset | Phenotype | Causative Mechanism | References No. |
|---|---|---|---|---|---|
| Chemical carcinogen/Bacterial infection induced | |||||
| MNU | (1) 60% (2) 19% | > 50 weeks | Adenocarcinoma | An alkylating reagent in experimental gastric carcinogenesis | (1) [65] (2) [66] |
| H. felis | 80% | < 6months | Severe gastritis | More susceptible Helicobacter spp for C57BL/6 murine model | [68] |
| MNU + H. felis | 40% | 9 months | Adenoma | [69] | |
| Gene-targeting | |||||
| pS2 (TFF1)−/− | 30% | 5 months | Dysplasia | Lacking normal gastric mucus | [71] |
| Gan (K19-Wnt1/C2mETg) | 100% | < 10 months | Carcinoma | Excess of COX-2 and microsomal prostaglandin E synthase-1 | [79] |
| INS-GAS | 85% | > 20 months | Intramucosal carcinoma | Hyperexpression of gastrin | [80] |
| INS-GAS + H. felis | < 8 months | ||||
| GAS−/− | 60% | 1 year | Displasia | Lacking gastrin | [72] |
| Atp4a−/− | 100% | 1 year | Incomplete intestinal metaplasia | Lacking H+K+ ATPase | [73] |
| Atp4b-SV40 | 60% | 1 year | Carcinoma, invasion (Lymphatic–vascular), metastasis (liver) | Expression of SV40 in parietal cells | [81] |
| Atp4b- (CDH1xTrp53)−/− | 100% | 1 year | Metastasized to lymph nodes | Lacking E-cadherin and p53 in parietal cells | [82] |
| ATP4b-hIL-1b | 30% | 1 year | Dysplasia, Adenocarcinoma | MDSCs recruitment via IL-1RI/NF-κB pathway | [78] |
| Kvlqt1−/− | 100% | 3 months | Hyperplasia in gastric neck cells | Lacking potassium channel | [83] |
| K-ras G12D (systemic) | 100% | < 18 days | Metaplasia | Hyperactivation of MAPK by K-ras mutation | [74] |
| Tgfβ1-C33S | 40% | 4-5 months | Well-differentiated adenocarcinomas | Unable forming latent TGF-β binding protein-1 | [75] |
| Smad3−/− | 100% | 10 months | Tumors, invasive neoplasia | Excess of cytosolic E-cadherin | [76] |
| Smad4−/− | 100% | > 1 year | Invasive carcinoma | Increased cyclin1 and upregulation of TGF-β1 | [77] |
| RUNX3−/− | 70% | > 1 year | Hyperplasia, Chief cells loss and increased cdx2 | Enhanced Wnt-β catenin signaling by RUNX3 loss | [84] |
| gp130757F | 100% | 3 months | Adenoma | Abrogating SHP2-Ras-ERK signaling | [63] |
| T3b-SOCS3−/− | 100% | 2 months | Carcinoma | Augment of leptin expression and ObR-STAT3 signaling by gastrointestinal cell- specific SOCS3 loss | [62] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Inagaki-Ohara, K. Gastric Leptin and Tumorigenesis: Beyond Obesity. Int. J. Mol. Sci. 2019, 20, 2622. https://doi.org/10.3390/ijms20112622
AMA Style
Inagaki-Ohara K. Gastric Leptin and Tumorigenesis: Beyond Obesity. International Journal of Molecular Sciences. 2019; 20(11):2622. https://doi.org/10.3390/ijms20112622
Chicago/Turabian StyleInagaki-Ohara, Kyoko. 2019. "Gastric Leptin and Tumorigenesis: Beyond Obesity" International Journal of Molecular Sciences 20, no. 11: 2622. https://doi.org/10.3390/ijms20112622
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.