Atorvastatin and Fenofibrate Increase the Content of Unsaturated Acyl Chains in HDL and Modify In Vivo Kinetics of HDL-Cholesteryl Esters in New Zealand White Rabbits

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Biochemical Analyses
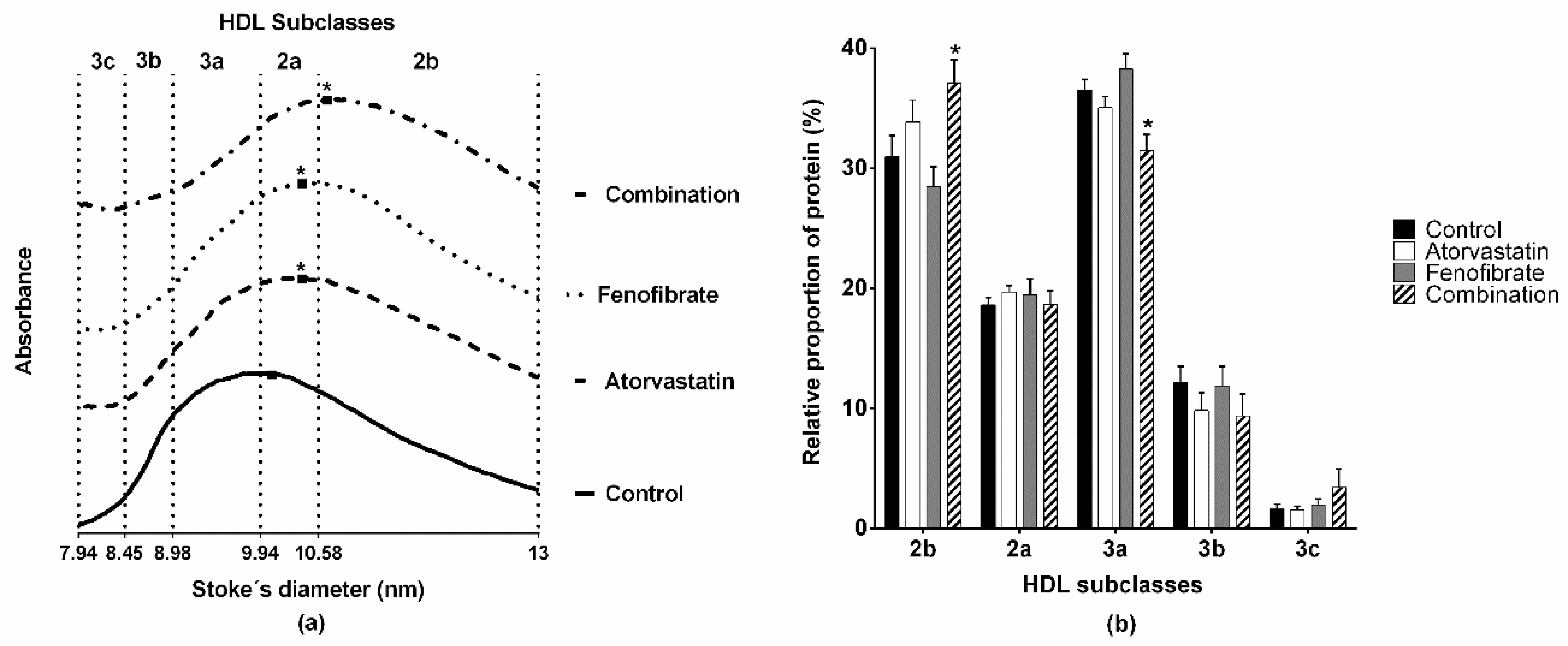
2.2. Structure and Lipid Composition of HDL
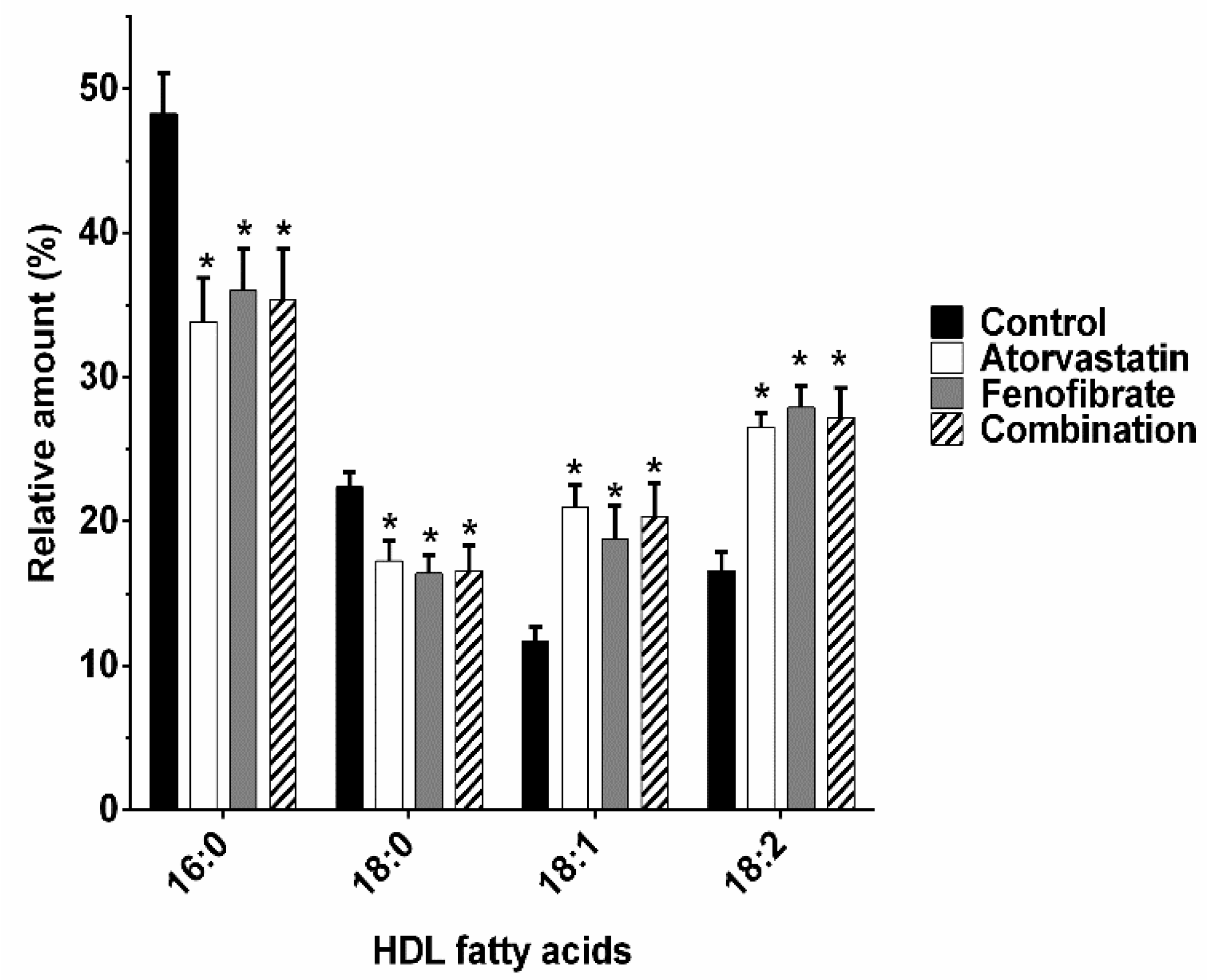
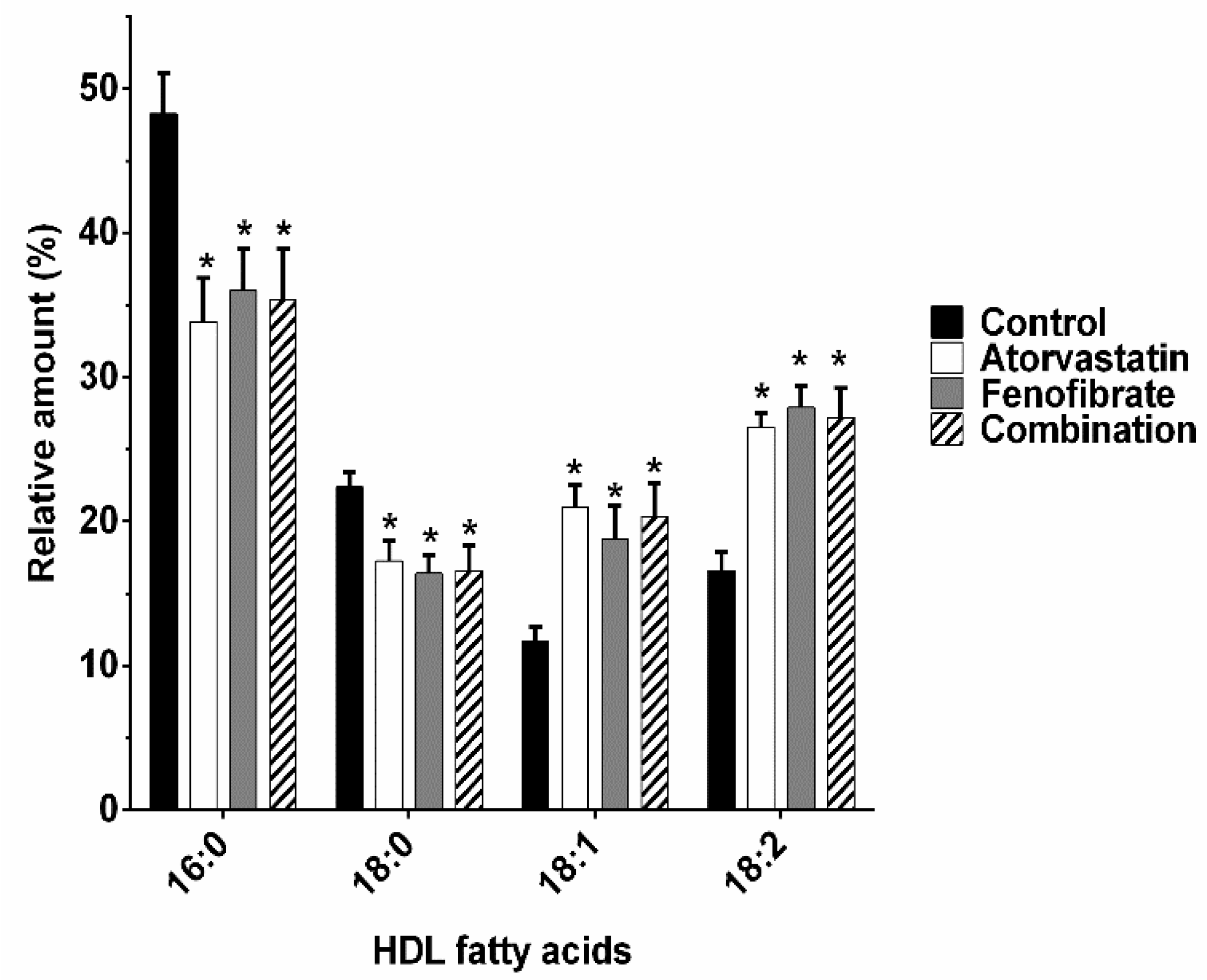
2.3. Fatty Acids Composition of HDL
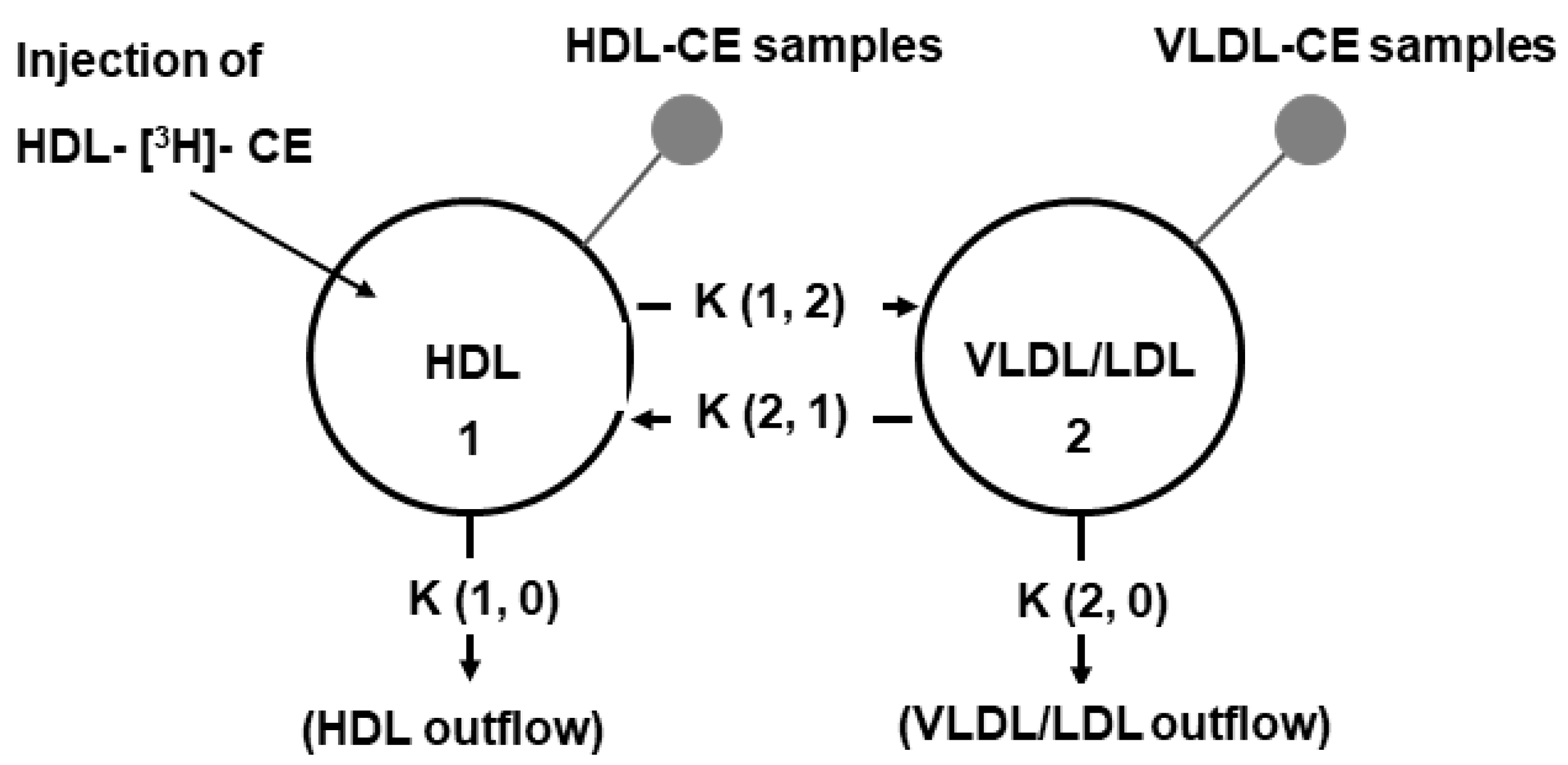
2.4. In Vivo Kinetic Studies of HDL-CE
2.5. Activity of the Cholesteryl Ester Transfer Protein (CETP)
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Blood Samples
4.3. Biochemical Analyses
4.4. Isolation of HDL
4.5. Determination of HDL Lipid Composition
4.6. Determination of Fatty Acids Composition of HDL
4.7. Preparation of Labeled HDL with [3H]-Cholesterol
4.8. In Vivo Kinetic Studies of HDL-[3H]-Cholesteryl Esters
4.9. Compartmental Analysis
4.10. Cholesteryl Ester Transfer Protein Activity Assay
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HDL-C | High-density lipoproteins-cholesterol |
| HDL | High-density lipoproteins |
| Apo A-I | Apolipoprotein A-I |
| CE | Cholesteryl esters |
| RCT | Reverse cholesterol transport |
| HDL-CE | High-density lipoproteins-cholesteryl esters |
| VLDL/LDL | Very low-density lipoproteins/low-density lipoproteins |
| HDL-Tg | High-density lipoproteins-triglycerides |
| HDL-Ph | High-density lipoproteins-phospholipids |
| GC-MS | Gas chromatography–mass spectrometry |
| CETP | Cholesteryl ester transfer protein |
References
- Assmann, G.; Schulte, H.; von Eckardstein, A.; Huang, Y. High-density lipoprotein cholesterol as a predictor of coronary heart disease risk. The procam experience and pathophysiological implications for reverse cholesterol transport. Atherosclerosis 1996, 124, S11–S20. [Google Scholar] [CrossRef]
- Navab, M.; Reddy, S.T.; Van Lenten, B.J.; Fogelman, A.M. Hdl and cardiovascular disease: Atherogenic and atheroprotective mechanisms. Nat. Rev. Cardiol. 2011, 8, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low hdl cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [PubMed]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the field study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef]
- Rached, F.H.; Chapman, M.J.; Kontush, A. Hdl particle subpopulations: Focus on biological function. Biofactors 2015, 41, 67–77. [Google Scholar] [CrossRef]
- Pérez-Méndez, O.; Pacheco, H.G.; Martinez-Sanchez, C.; Franco, M. Hdl-cholesterol in coronary artery disease risk: Function or structure? Clin. Chim. Acta 2014, 429, 111–122. [Google Scholar] [CrossRef]
- Williams, P.T.; Krauss, R.M.; Nichols, A.V.; Vranizan, K.M.; Wood, P.D. Identifying the predominant peak diameter of high-density and low-density lipoproteins by electrophoresis. J. Lipid Res. 1990, 31, 1131–1139. [Google Scholar] [PubMed]
- Florentin, M.; Liberopoulos, E.N.; Wierzbicki, A.S.; Mikhailidis, D.P. Multiple actions of high-density lipoprotein. Curr. Opin. Cardiol. 2008, 23, 370–378. [Google Scholar] [CrossRef]
- Bhalodkar, N.C.; Blum, S.; Rana, T.; Kitchappa, R.; Bhalodkar, A.N.; Enas, E.A. Comparison of high-density and low-density lipoprotein cholesterol subclasses and sizes in asian indian women with caucasian women from the framingham offspring study. Clin. Cardiol. 2005, 28, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yan, B.; Fu, M.; Xu, Y.; Tian, Y. Relationship between plasma lipid concentrations and hdl subclasses. Clin. Chim. Acta 2005, 354, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Carreón-Torres, E.; Juárez-Meaveapeña, M.; Cardoso-Saldaña, G.; Gómez, C.H.; Franco, M.; Fievet, C.; Luc, G.; Juárez-Oropeza, M.A.; Pérez-Méndez, O. Pioglitazone increases the fractional catabolic and production rates of high-density lipoproteins apo ai in the new zealand white rabbit. Atherosclerosis 2005, 181, 233–240. [Google Scholar] [CrossRef]
- Huesca-Gómez, C.; Luc, G.; Duhal, N.; Lacroix, B.; Fruchart, J.; Pérez-Méndez, O. Ciprofibrate increases synthesis and catabolism of hdl apo ai and aii in patients with hypertriglyceridemia (abstract). Atherosclerosis 2004, 5, 64. [Google Scholar]
- Garvey, W.T.; Kwon, S.; Zheng, D.; Shaughnessy, S.; Wallace, P.; Hutto, A.; Pugh, K.; Jenkins, A.J.; Klein, R.L.; Liao, Y. Effects of insulin resistance and type 2 diabetes on lipoprotein subclass particle size and concentration determined by nuclear magnetic resonance. Diabetes 2003, 52, 453–642. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Méndez, O.; Torres-Tamayo, M.; Posada-Romero, C.; Vidaure-Garcés, V.; Carreón-Torres, E.; Mendoza-Pérez, E.; Medina-Urrutia, A.; Huesca-Gómez, C.; Zamora González, J.; Aguilar-Herrera, B. Abnormal hdl subclasses distribution in overweight children with insulin resistance or type 2 diabetes mellitus. Clin. Chim. Acta 2007, 376, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; John, C.; Schaltenberg, N.; Berbee, J.F.P.; Worthmann, A.; Cherradi, M.L.; Schlein, C.; Piepenburg, J.; Boon, M.R.; Rinninger, F.; et al. Thermogenic adipocytes promote hdl turnover and reverse cholesterol transport. Nat. Commun. 2017, 8, 15010. [Google Scholar] [CrossRef]
- Luna-Luna, M.; Cruz-Robles, D.; Avila-Vanzzini, N.; Herrera-Alarcon, V.; Martinez-Reding, J.; Criales-Vera, S.; Sandoval-Zarate, J.; Vargas-Barron, J.; Martinez-Sanchez, C.; Tovar-Palacio, A.R.; et al. Differential expression of osteopontin, and osteoprotegerin mrna in epicardial adipose tissue between patients with severe coronary artery disease and aortic valvular stenosis: Association with hdl subclasses. Lipids Health Dis. 2017, 16, 156. [Google Scholar] [CrossRef]
- Fielding, C.J.; Fielding, P.E. Molecular physiology of reverse cholesterol transport. J. Lipid Res. 1995, 36, 211–228. [Google Scholar] [PubMed]
- Van der Velde, A.E. Reverse cholesterol transport: From classical view to new insights. World J. Gastroenterol. 2010, 16, 5908–5915. [Google Scholar]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of hdl cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef]
- Rye, K.A.; Bursill, C.A.; Lambert, G.; Tabet, F.; Barter, P.J. The metabolism and anti-atherogenic properties of hdl. J. Lipid Res. 2009, 50, S195–S200. [Google Scholar] [CrossRef]
- Mutharasan, R.K.; Thaxton, C.S.; Berry, J.; Daviglus, M.L.; Yuan, C.; Sun, J.; Ayers, C.; Lloyd-Jones, D.M.; Wilkins, J.T. Hdl efflux capacity, hdl particle size, and high-risk carotid atherosclerosis in a cohort of asymptomatic older adults: The chicago healthy aging study. J. Lipid Res. 2017, 58, 600–606. [Google Scholar] [CrossRef]
- Sola, R.; Baudet, M.F.; Motta, C.; Maille, M.; Boisnier, C.; Jacotot, B. Effects of dietary fats on the fluidity of human high-density lipoprotein: Influence of the overall composition and phospholipid fatty acids. Biochim. Biophys. Acta 1990, 1043, 43–51. [Google Scholar] [CrossRef]
- Davidson, W.S.; Gillotte, K.L.; Lund-Katz, S.; Johnson, W.J.; Rothblat, G.H.; Phillips, M.C. The effect of high density lipoprotein phospholipid acyl chain composition on the efflux of cellular free cholesterol. J. Biol. Chem. 1995, 270, 5882–5890. [Google Scholar] [CrossRef]
- Jove, M.; Naudi, A.; Portero-Otin, M.; Cabre, R.; Rovira-Llopis, S.; Banuls, C.; Rocha, M.; Hernandez-Mijares, A.; Victor, V.M.; Pamplona, R. Plasma lipidomics discloses metabolic syndrome with a specific hdl phenotype. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 5163–5171. [Google Scholar] [CrossRef]
- Skeggs, J.W.; Morton, R.E. Ldl and hdl enriched in triglyceride promote abnormal cholesterol transport. J. Lipid Res. 2002, 43, 1264–1274. [Google Scholar]
- Flores-Castillo, C.; Zamora-Perez, J.A.; Carreon-Torres, E.; Arzola-Paniagua, A.; Aguilar-Salinas, C.; Lopez-Olmos, V.; Fragoso, J.M.; Luna-Luna, M.; Rodriguez-Perez, J.M.; Franco, M.; et al. Atorvastatin and fenofibrate combination induces the predominance of the large hdl subclasses and increased apo ai fractional catabolic rates in new zealand white rabbits with exogenous hypercholesterolemia. Fundam Clin. Pharm. 2015, 29, 362–370. [Google Scholar] [CrossRef]
- Kee, P.; Caiazza, D.; Rye, K.A.; Barret, P.H.; Morehouse, L.A.; Barter, P.J. Effect of inhibiting cholesteryl ester transfer protein on the kinetics of high-density lipoprotein cholesteryl ester transport in plasma: In vivo studies in rabbits. Arter. Thromb. Vasc. Biol. 2006, 26, 884–890. [Google Scholar] [CrossRef]
- Carreón-Torres, E.; Rendón-Sauer, K.; Monter-Garrido, M.; Toledo-Ibelles, P.; Gamboa, R.; Menjivar, M.; Lopéz Marure, R.; Luc, G.; Fievet, C.; Cruz, D.; et al. Rosiglitazone modifies hdl structure and increases hdl-apo ai synthesis and catabolism. Clin. Chim. Acta 2009, 401, 37–41. [Google Scholar]
- Martinez-Ramirez, M.; Flores-Castillo, C.; Sanchez-Lozada, L.G.; Bautista-Perez, R.; Carreon-Torres, E.; Fragoso, J.M.; Rodriguez-Perez, J.M.; Garcia-Arroyo, F.E.; Lopez-Olmos, V.; Luna-Luna, M.; et al. Hyperuricemia is associated with increased apo ai fractional catabolic rates and dysfunctional hdl in new zealand rabbits. Lipids 2017, 52, 999–1006. [Google Scholar] [CrossRef]
- López-Olmos, V.; Carreón-Torres, E.; Luna-Luna, M.; Flores-Castillo, C.; Martínez-Ramírez, M.; Bautista-Pérez, R.; Franco, M.; Sandoval-Zárate, J.; Roldán, F.J.; Aranda-Fraustro, A.; et al. Increased hdl size and enhanced apo a-i catabolic rates are associated with doxorubicin-induced proteinuria in new zealand white rabbits. Lipids 2016, 51, 311–320. [Google Scholar] [CrossRef]
- Fournier, N.; Tuloup-Minguez, V.; Pourci, M.L.; Therond, P.; Jullian, J.C.; Wien, F.; Leroy, M.; Dallongeville, J.; Paul, J.L.; Leroy, A. Fibrate treatment induced quantitative and qualitative hdl changes associated with an increase of sr-bi cholesterol efflux capacities in rabbits. Biochimie 2013, 95, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Priyadharsini, R.P. Animal models to evaluate anti-atherosclerotic drugs. Fundam Clin. Pharm. 2015, 29, 329–340. [Google Scholar] [CrossRef]
- Zhao, S.P.; Wu, Z.H.; Hong, S.C.; Ye, H.J.; Wu, J. Effect of atorvastatin on sr-bi expression and hdl-induced cholesterol efflux in adipocytes of hypercholesterolemic rabbits. Clin. Chim. Acta 2006, 365, 119–124. [Google Scholar] [CrossRef]
- Hennuyer, N.; Poulain, P.; Madsen, L.; Berge, R.K.; Houdebine, L.M.; Branellec, D.; Fruchart, J.C.; Fievet, C.; Duverger, N.; Staels, B. Beneficial effects of fibrates on apolipoprotein a-i metabolism occur independently of any peroxisome proliferative response. Circulation 1999, 99, 2445–2451. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Uffelman, K.D.; Barrett, P.H.; Lewis, G.F. Effect of atorvastatin on high-density lipoprotein apolipoprotein a-i production and clearance in the new zealand white rabbit. Circulation 2002, 106, 2955–2960. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Khanna, D.; Kalra, S.; Balakumar, P. Chronic oral administration of low-dose combination of fenofibrate and rosuvastatin protects the rat heart against experimentally induced acute myocardial infarction. Fundam Clin. Pharm. 2016, 30, 394–405. [Google Scholar] [CrossRef]
- Chapman, M.J.; Le Goff, W.; Guerin, M.; Kontush, A. Cholesteryl ester transfer protein: At the heart of the action of lipid-modulating therapy with statins, fibrates, niacin, and cholesteryl ester transfer protein inhibitors. Eur. Heart J. 2010, 31, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D.; McLean, M.P. Activation of the rat scavenger receptor class b type i gene by pparalpha. Mol. Cell. Endocrinol. 2006, 251, 67–77. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Q.; Zhou, J.; Xu, Q.; Chu, X.; Sun, T.; Liu, X.; Cai, S. Rosuvastatin attenuates atherosclerosis in rats via activation of scavenger receptor class b type i. Eur. J. Pharm. 2014, 723, 23–28. [Google Scholar] [CrossRef]
- Shen, W.J.; Azhar, S.; Kraemer, F.B. Sr-b1: A unique multifunctional receptor for cholesterol influx and efflux. Annu. Rev. Physiol. 2018, 80, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.C.; Nanjee, M.N.; Olszewski, W.L.; Miller, N.E. Ldl and hdl transfer rates across peripheral microvascular endothelium agree with those predicted for passive ultrafiltration in humans. J. Lipid Res. 2015, 56, 122–128. [Google Scholar] [CrossRef]
- Muñoz-Vega, M.; Masso, F.; Paez, A.; Vargas-Alarcon, G.; Coral-Vazquez, R.; Mas-Oliva, J.; Carreon-Torres, E.; Perez-Mendez, O. Hdl-mediated lipid influx to endothelial cells contributes to regulating intercellular adhesion molecule (icam)-1 expression and enos phosphorylation. Int. J. Mol. Sci. 2018, 19, pii: E3394. [Google Scholar]
- Huang, Z.; Zhou, X.; Nicholson, A.C.; Gotto, A.M., Jr.; Hajjar, D.P.; Han, J. Activation of peroxisome proliferator-activated receptor-alpha in mice induces expression of the hepatic low-density lipoprotein receptor. Br. J. Pharmacol. 2008, 155, 596–605. [Google Scholar] [CrossRef]
- Gao, Y.; Shen, W.; Lu, B.; Zhang, Q.; Hu, Y.; Chen, Y. Upregulation of hepatic vldlr via pparalpha is required for the triglyceride-lowering effect of fenofibrate. J. Lipid Res. 2014, 55, 1622–1633. [Google Scholar] [CrossRef]
- Chapman, M.J.; Orsoni, A.; Robillard, P.; Therond, P.; Giral, P. Duality of statin action on lipoprotein subpopulations in the mixed dyslipidemia of metabolic syndrome: Quantity vs quality over time and implication of cetp. J. Clin. Lipidol. 2018, 12, 784–800. [Google Scholar] [CrossRef]
- Beyer, T.P.; Chen, Y.; Porter, R.K.; Lu, D.; Schmidt, R.J.; Mantlo, N.B.; Konrad, R.J.; Cao, G. Peroxisome proliferator-activated receptor alpha agonists regulate cholesterol ester transfer protein. Lipids 2008, 43, 611–618. [Google Scholar] [CrossRef]
- Lund-Katz, S.; Hammerschlag, B.; Phillips, M.C. Kinetics and mechanism of free cholesterol exchange between human serum high- and low-density lipoproteins. Biochemistry 1982, 21, 2964–2969. [Google Scholar] [CrossRef] [PubMed]
- Gillard, B.K.; Rosales, C.; Xu, B.; Gotto, A.M., Jr.; Pownall, H.J. Rethinking reverse cholesterol transport and dysfunctional high-density lipoproteins. J. Clin. Lipidol. 2018, 12, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Padro, T.; Cubedo, J.; Camino, S.; Bejar, M.T.; Ben-Aicha, S.; Mendieta, G.; Escola-Gil, J.C.; Escate, R.; Gutierrez, M.; Casani, L.; et al. Detrimental effect of hypercholesterolemia on high-density lipoprotein particle remodeling in pigs. J. Am. Coll. Cardiol. 2017, 70, 165–178. [Google Scholar] [CrossRef]
- Cazzola, R.; Cassani, E.; Barichella, M.; Cestaro, B. Impaired fluidity and oxidizability of hdl hydrophobic core and amphipathic surface in dyslipidemic men. Metabolism 2013, 62, 986–991. [Google Scholar] [CrossRef]
- Massey, J.B.; Pownall, H.J. Surface properties of native human plasma lipoproteins and lipoprotein models. Biophys. J. 1998, 74, 869–878. [Google Scholar] [CrossRef]
- Martin-Fuentes, P.; Garcia-Otin, A.L.; Calvo, L.; Gomez-Coronado, D.; Civeira, F.; Cenarro, A. Atorvastatin decreases stearoyl-coa desaturase gene expression in thp-1 macrophages incubated with oxidized ldl. Lipids 2009, 44, 115–123. [Google Scholar] [CrossRef]
- Yamazaki, T.; Okada, H.; Sakamoto, T.; Sunaga, K.; Tsuda, T.; Mitsumoto, A.; Kudo, N.; Kawashima, Y. Differential induction of stearoyl-coa desaturase 1 and 2 genes by fibrates in the liver of rats. Biol. Pharm. Bull. 2012, 35, 116–120. [Google Scholar] [CrossRef]
- Montanaro, M.A.; Bernasconi, A.M.; Gonzalez, M.S.; Rimoldi, O.J.; Brenner, R.R. Effects of fenofibrate and insulin on the biosynthesis of unsaturated fatty acids in streptozotocin diabetic rats. ProstaglandinsLeukot. Essent. Fat. Acids 2005, 73, 369–378. [Google Scholar] [CrossRef]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the hdl lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef]
- Schonewille, M.; de Boer, J.F.; Mele, L.; Wolters, H.; Bloks, V.W.; Wolters, J.C.; Kuivenhoven, J.A.; Tietge, U.J.; Brufau, G.; Groen, A.K. Statins increase hepatic cholesterol synthesis and stimulate fecal cholesterol elimination in mice. J. Lipid Res. 2016, 57, 1455–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Post, S.M.; Duez, H.; Gervois, P.P.; Staels, B.; Kuipers, F.; Princen, H.M. Fibrates suppress bile acid synthesis via peroxisome proliferator-activated receptor-alpha-mediated downregulation of cholesterol 7alpha-hydroxylase and sterol 27-hydroxylase expression. Arter. Thromb. Vasc. Biol. 2001, 21, 1840–1845. [Google Scholar] [CrossRef]
- National Research Council (US) Institute for Laboratory Animal Research. Guide for the Care and Use of Laboratory Animals; National Academic Press (USA): Washington, DC, USA, 1996.
- Huesca-Gómez, C.; Franco, M.; Luc, G.; Montaño, L.F.; Massó, F.; Posada-Romero, C.; Pérez-Méndez, O. Chronic hypothyroidism induces abnormal structure of high-density lipoproteins and impaired kinetics of apolipoprotein a-i in the rat. Metabolism 2002, 51, 443–450. [Google Scholar] [CrossRef]
- Gárcia-Sánchez, C.; Torres-Tamayo, M.; Juárez-Meaveapeña, M.; López-Osorio, C.; Toledo-Ibelles, P.; Monter-Garrido, M.; Cruz-Robles, D.; Carreón-Torres, E.; Vargas-Alarcón, G.; Pérez-Méndez, O. Lipid plasma concentrations of hdl subclasses determined by enzymatic staining on polyacrylamide electrophoresis gels in children with metabolic syndrome. Clin. Chim. Acta 2011, 412, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Ibelles, P.; Gárcia-Sánchez, C.; Ávila-Vazzini, N.; Carreon-Torres, E.; Posada-Romero, C.; Vargas-Alarcón, G.; Pérez-Méndez, O. Enzymatic assesment of cholesterol on electrophoresis gels for estimating hdl size distribution and plasma concentrations of hdl subclasses. J. Lipid Res. 2010, 51, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Romero-Aguilar, L.; Pardo, J.P.; Lomeli, M.M.; Bocardo, O.I.L.; Juarez Oropeza, M.A.; Guerra Sanchez, G. Lipid droplets accumulation and other biochemical changes induced in the fungal pathogen ustilago maydis under nitrogen-starvation. Arch. Microbiol. 2017, 199, 1195–1209. [Google Scholar] [CrossRef]
- Huesca-Gómez, C.; Carreón-Torres, E.; Nepomuceno-Mejía, T.; Sánchez-Solorio, M.; Galicia-Hidalgo, M.; Mejía, A.M. Contribution of cholesteryl ester transfer protein and lecithin: Cholesterol acyl tranferase to hdl size distribution. Endocr. Res. 2004, 30, 403–415. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Biochemical Parameters | Control n = 6 | Atorvastatin n = 6 | Fenofibrate n = 6 | Combination n = 6 |
|---|---|---|---|---|
| Cholesterol (mmol/L) | 1.41 ± 0.06 | 1.19 ± 0.11 | 1.26 ± 0.17 | 1.30 ± 0.07 |
| Triglycerides (mmol/L) | 0.75 ± 0.05 | 0.93 ± 0.23 | 0.66 ± 0.08 | 0.75 ± 0.11 |
| Glucose (mmol/L) | 6.25 ± 0.32 | 6.28 ± 0.14 | 6.39 ± 0.28 | 6.10 ± 0.10 |
| HDL-C (mmol/L) | 0.79 ± 0.03 | 0.79 ± 0.05 | 0.89 ± 0.10 | 1.14 ± 0.12 *,**,*** |
| HDL-Tg (mmol/L) | 0.42 ± 0.06 | 0.44 ± 0.06 | 0.37 ± 0.02 | 0.46 ± 0.10 |
| HDL-Ph (mmol/L) | 2.47 ± 0.22 | 2.39 ± 0.10 | 2.32 ± 0.23 | 3.07 ± 0.27 **,*** |
| Apo A-I (mg/L) | 398.6 ± 32.6 | 408.1 ± 20.8 | 617.6 ± 10.8* | 560.2 ± 77.0 * |
| HDL Subclasses | Control n = 6 | Atorvastatin n = 6 | Fenofibrate n = 6 | Combination n = 6 |
|---|---|---|---|---|
| C (µmol/L) | ||||
| HDL2b | 411.1 ± 36.9 | 416.2 ± 38.3 | 438.2 ± 61.3 | 586.1 ± 90.8 * |
| HDL2a | 141.4 ± 3.5 | 144.3 ± 8.9 | 154.8 ± 19.4 | 199.2 ± 29.3 *,** |
| HDL3a | 181.5 ± 12.4 | 178.0 ± 19.8 | 201.3 ± 28.4 | 245.9 ± 28.3** |
| HDL3b | 44.9 ± 8.6 | 38.8 ± 7.4 | 67.4 ± 8.2 | 64.4 ± 13.6 |
| HDL3c | 12.4 ± 3.3 | 11.3 ± 2.3 | 29.9 ± 3.8 | 41.3 ± 24.7 |
| Tg (µmol/L) | ||||
| HDL2b | 206.3 ± 32.4 | 212.9 ± 38.9 | 172.1 ± 15.9 | 208.5 ± 35.9 |
| HDL2a | 71.9 ± 10.8 | 74.8 ± 9.7 | 64.1 ± 5.8 | 75.6 ± 15.8 |
| HDL3a | 101.4 ± 16.1 | 106.1 ± 13.9 | 93.2 ± 5.3 | 116.2 ± 28.1 |
| HDL3b | 30.2 ± 6.6 | 33.6 ± 11.9 | 29.7 ± 6.4 | 43.0 ± 14.1 |
| HDL3c | 8.1 ± 3.6 | 14.3 ± 6.8 | 12.2 ± 4.2 | 21.7 ± 12.1 |
| Ph (µmol/L) | ||||
| HDL2b | 1347.8 ± 145.4 | 1308.6 ± 78.8 | 1273 ± 147.2 | 1826.2 ± 164.8 *,**,*** |
| HDL2a | 411.2 ± 29.4 | 414.9 ± 15.9 | 403.7 ± 31.8 | 473.5 ± 33.9 *,**,*** |
| HDL3a | 527.4 ± 44.5 | 521.0 ± 35.2 | 513.7 ± 37.4 | 572.7 ± 60.5 |
| HDL3b | 147.6 ± 19.5 | 108.6 ± 16.8 | 103.5 ± 17.1 | 148.2 ± 34.2 |
| HDL3c | 38.8 ± 6.0 | 35.6 ± 6.5 | 25.2 ± 7.2 | 50.9 ± 14.9 |
| Constants | Transfer of CE | Control n = 6 | Atorvastatin n = 6 | Fenofibrate n = 6 | Combination n = 6 |
|---|---|---|---|---|---|
| K (1, 2) | HDL to VLDL/LDL | 10.60 (8.09–12.29) | 71.30 * (43.45–84.11) | 32.74 (9.23–64.87) | 38.92 * (21.65–51.77) |
| K (2,1) | VLDL/LDL to HDL | 11.46 (5.19–22.16) | 80.63* (52.42–101.25) | 23.37 (12.62–68.91) | 82.81 * (10.55–190.81) |
| K (1, 0) | HDL outflow | 3.28 (3.06–5.14) | 4.69 (3.22–6.70) | 2.34 (1.66–5.56) | 5.69 * (5.20–6.31) |
| K (2, 0) | VLDL/LDL outflow | 8.08 (5.07–9.55) | 7.26 (6.67–9.19) | 3.94 *, ** (2.22–4.98) | 5.83 (3.89–18.91) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Castillo, C.; Luna-Luna, M.; Carreón-Torres, E.; López-Olmos, V.; Frías, S.; Juárez-Oropeza, M.A.; Franco, M.; Fragoso, J.M.; Vargas-Alarcón, G.; Pérez-Méndez, Ó. Atorvastatin and Fenofibrate Increase the Content of Unsaturated Acyl Chains in HDL and Modify In Vivo Kinetics of HDL-Cholesteryl Esters in New Zealand White Rabbits. Int. J. Mol. Sci. 2019, 20, 2521. https://doi.org/10.3390/ijms20102521
Flores-Castillo C, Luna-Luna M, Carreón-Torres E, López-Olmos V, Frías S, Juárez-Oropeza MA, Franco M, Fragoso JM, Vargas-Alarcón G, Pérez-Méndez Ó. Atorvastatin and Fenofibrate Increase the Content of Unsaturated Acyl Chains in HDL and Modify In Vivo Kinetics of HDL-Cholesteryl Esters in New Zealand White Rabbits. International Journal of Molecular Sciences. 2019; 20(10):2521. https://doi.org/10.3390/ijms20102521
Chicago/Turabian StyleFlores-Castillo, Cristóbal, María Luna-Luna, Elizabeth Carreón-Torres, Victoria López-Olmos, Sara Frías, Marco Antonio Juárez-Oropeza, Martha Franco, José Manuel Fragoso, Gilberto Vargas-Alarcón, and Óscar Pérez-Méndez. 2019. "Atorvastatin and Fenofibrate Increase the Content of Unsaturated Acyl Chains in HDL and Modify In Vivo Kinetics of HDL-Cholesteryl Esters in New Zealand White Rabbits" International Journal of Molecular Sciences 20, no. 10: 2521. https://doi.org/10.3390/ijms20102521