Kinase-Inactivated EGFR Is Required for the Survival of Wild-Type EGFR-Expressing Cancer Cells Treated with Tyrosine Kinase Inhibitors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
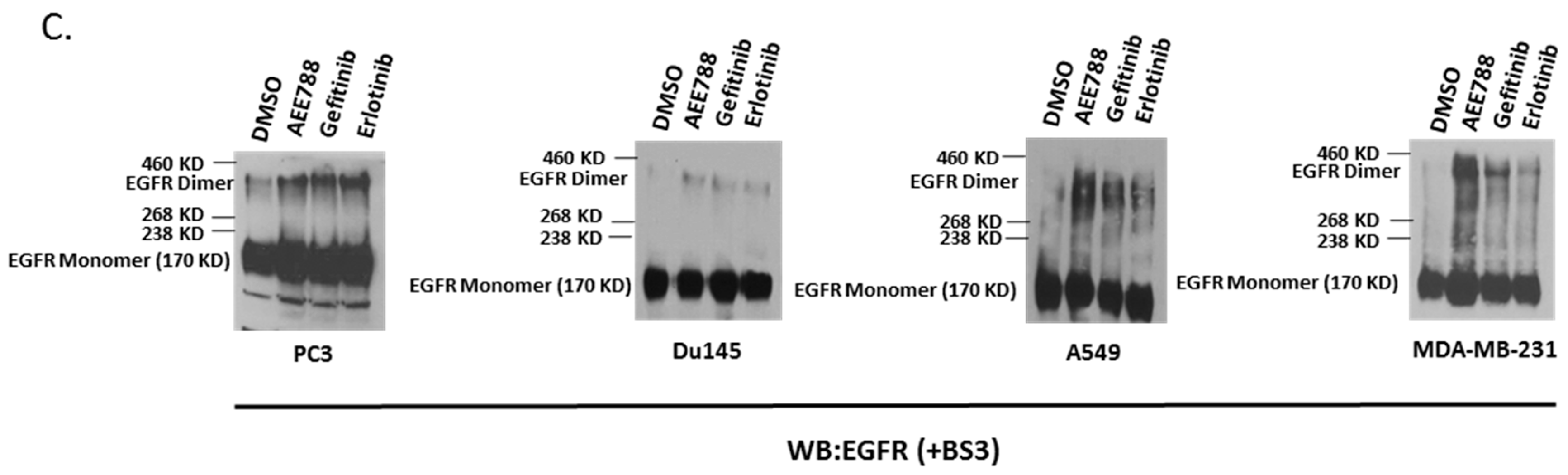
2.1. Epidermal Growth Factor Receptor (EGFR) Dimerization in Response to Acute Treatment of Tyrosine Kinase Inhibitors (TKIs)
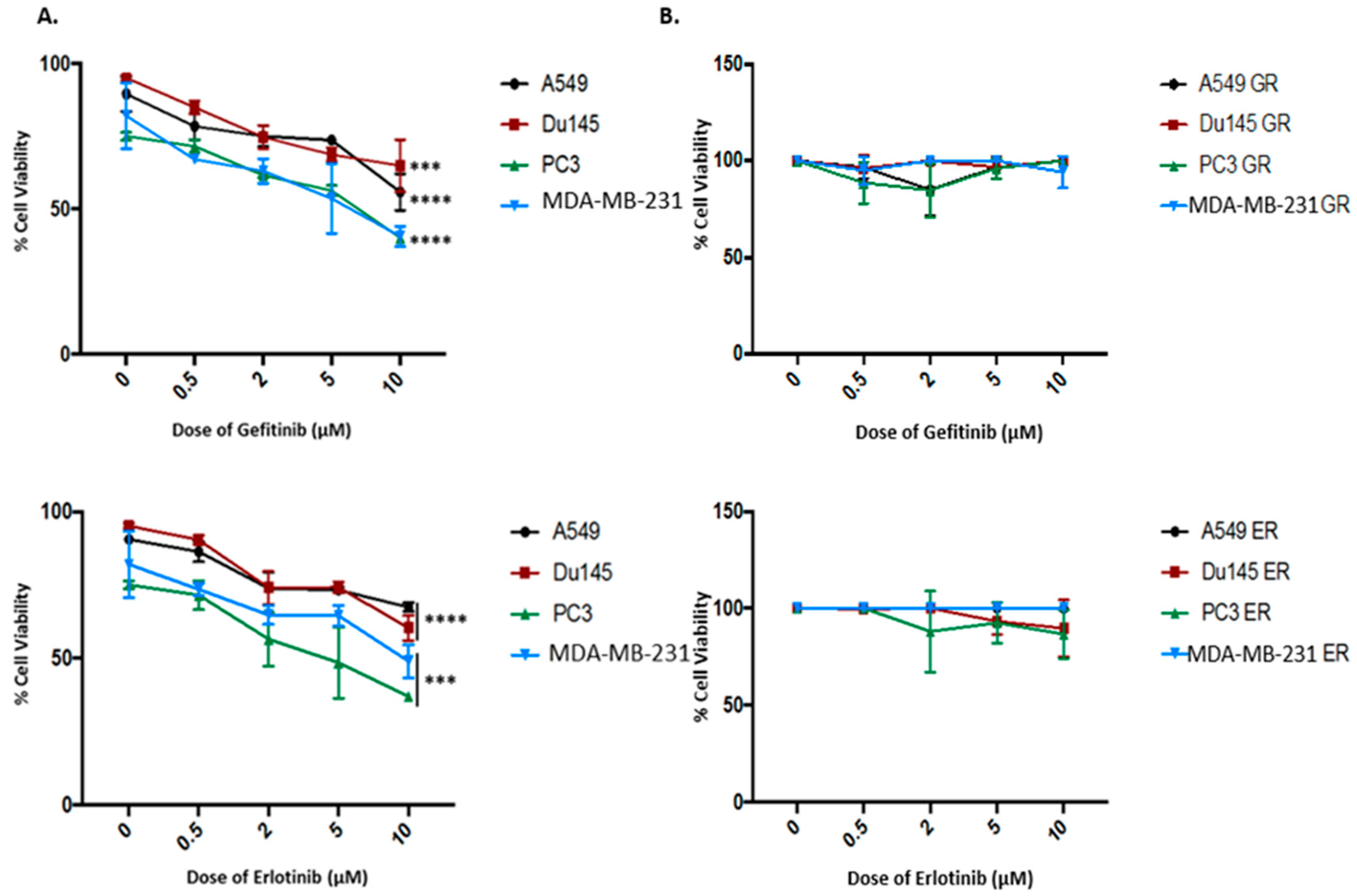
2.2. Kinase-Inactivated EGFR Dimers Increased in TKI Resistant Cells
2.3. Inhibition of Palmitoylation Abolishes TKI-Induced EGFR Dimer Formation
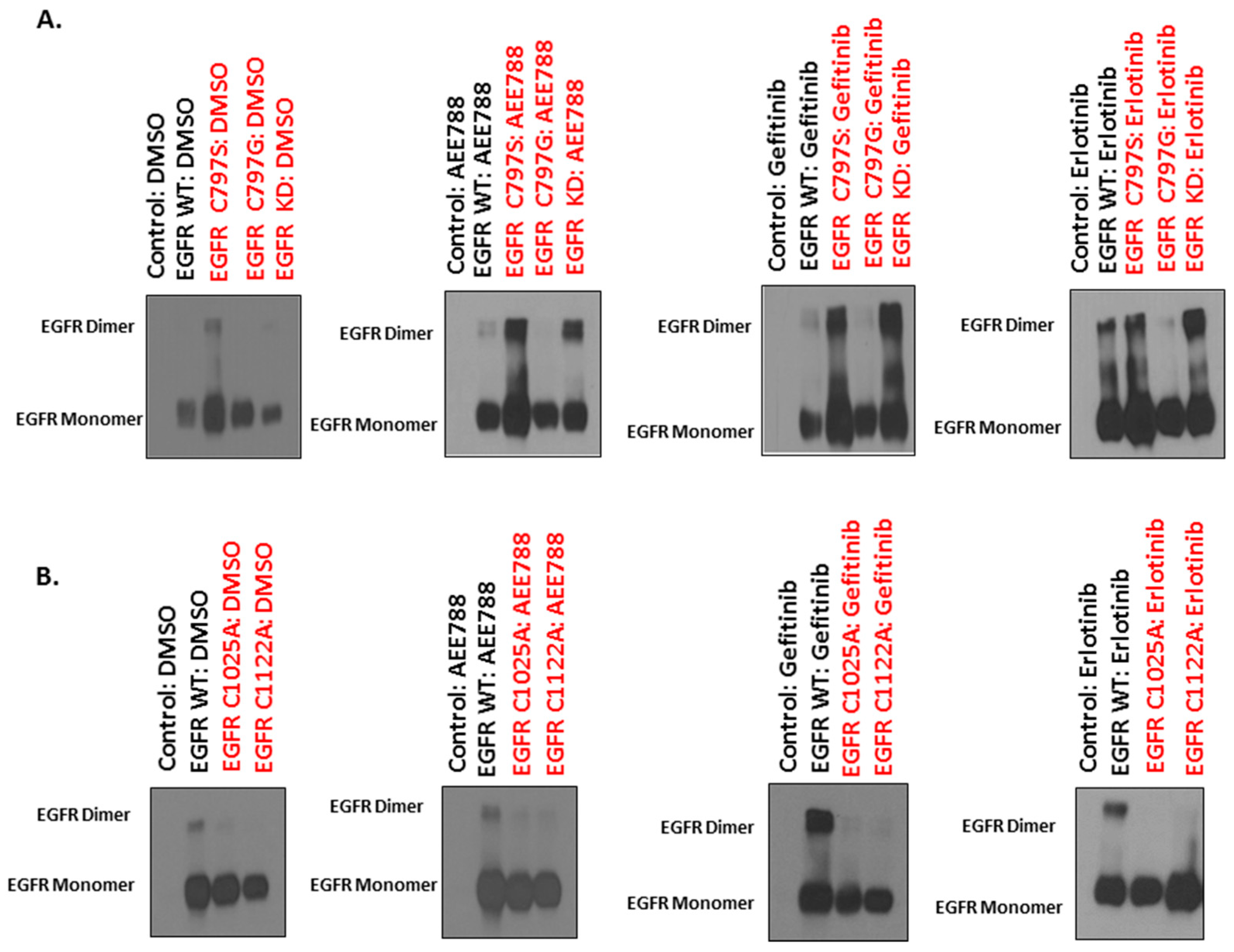
2.4. Mutations of Cysteine Residues Critical for EGFR Palmitoylation Abolished TKI-Induced Dimerization, and the Kinase Activity of EGFR Is Not Required for TKI-Induced Dimerization of EGFR
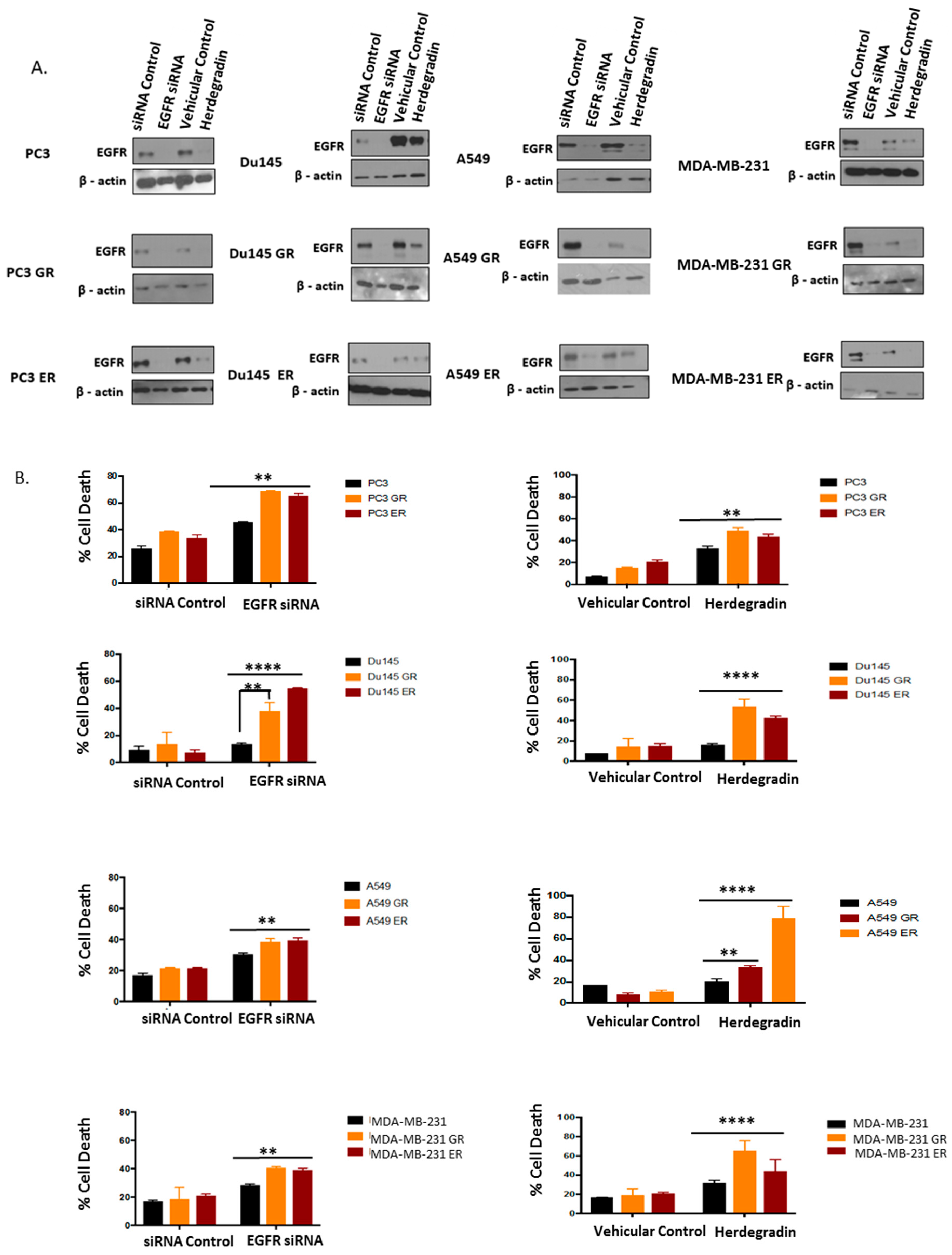
2.5. Both Inhibition of Palmitoylation and Targeted Reduction of EGFR Are Lethal to TKI-Resistant Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Reagents
4.2. Establishment of Gefitinib- and Erlotinib-Resistant Cells
4.3. Plasmids, siRNAs, and Peptide
4.4. Transfections and Western Blotting
4.5. EGFR Dimerization Assay
4.6. MTT Cell Proliferation/Cell Viability Assay
4.7. Trypan Blue Exclusion Assay
4.8. Statistics
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Roskoski, R.J. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharm. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in Cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, X.; Jin, H. EGFR-TKI resistance in NSCLC patients, mechanisms and strategies. Am. J. Cancer Res. 2014, 4, 411–435. [Google Scholar]
- Weihua, Z.; Tsan, R.; Huang, W.C.; Wu, Q.; Chiu, C.H.; Fidler, I.J.; Hung, M.C. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008, 13, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J. The epidermal growth factor receptor as a target for cancer therapy. Endocr. Relat. Cancer 2001, 8, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.R.; Cepero, V.; Giordano, S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol. Cancer 2010, 9, 1–13. [Google Scholar] [CrossRef]
- Nan, X.; Xie, C.; Yu, X.; Liu, J. EGFR TKI as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer. Oncotarget 2017, 8, 75712–75726. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors, from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.E.; Mueller, K.L.; Bohin, N.; Ge, Y.; Boerner, J.L. Lipid raft localization of EGFR alters the response of cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J. Cell Physiol. 2011, 226, 2316–2328. [Google Scholar] [CrossRef] [PubMed]
- Cossu-Rocca, P.; Muroni, M.R.; Sanges, F.; Sotgiu, G.; Asunis, A.; Tanca, L.; Onnis, D.; Pira, G.; Manca, A.; Dore, S.; et al. EGFR kinase-dependent and kinase-independent roles in clear cell renal cell carcinoma. Am. J. Cancer Res. 2016, 6, 71–83. [Google Scholar]
- Riely, G.J.; Yu, H.A. EGFR: The paradigm of an oncogene-driven lung cancer. Clin. Cancer Res. 2015, 21, 2221–2226. [Google Scholar] [CrossRef]
- Ricordel, C.; Friboulet, L.; Facchinetti, F.; Soria, J.C. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann. Oncol. 2018, 29, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer, mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumors: learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef]
- Husain, H.; Scur, M.; Murtuza, A.; Bui, N.; Woodward, B.; Kurzrock, R. Strategies to overcome bypass mediating clinical resistance to EGFR tyrosine kinase inhibition in lung cancer. Mol. Cancer Ther. 2017, 16, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Cortot, A.B.; Janne, P.A. Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung cancer adenocarcinoma. Eur. Respir. Rev. 2014, 23, 356–366. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimen at the time of acquired resistance to EGFR-TKI therapy in 155 Patients with EGFR mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Canil, C.M.; Moore, M.J.; Winquist, E.; Baetz, T.; Pollack, M.; Chi, K.N.; Berry, S.; Ernst, D.S.; Douglas, L.; Brundage, M.; et al. Randomized phase II study of two doses of gefitinib in hormone-refractory prostate cancer, a trial of the national cancer institute of canada-clinical trials group. J. Clin. Oncol. 2005, 23, 455–460. [Google Scholar] [CrossRef]
- Liu, Q.; Yu, S.; Zhao, W.; Qin, S.; Chu, Q.; Wu, K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol. Cancer 2018, 17, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lisberg, A.; Garon, E.B. Epidermal growth factor tyrosine kinase inhibitor therapy inferior to second-line chemotherapy in EGFR wild-type non-small cell lung cancer patients: results of French nationwide observational study. Transl. Lung Cancer Res. 2017, 6, S39–S40. [Google Scholar] [CrossRef] [Green Version]
- Ojemuyiwa, M.A.; Madan, R.A.; Dahut, W.L. Tyrosine kinase inhibitors in the treatment of prostate cancer: taking the next step in clinical development. Expert Opin. Emerg. Drugs 2014, 19, 459–470. [Google Scholar] [CrossRef]
- Mahipal, A.; Kothan, N.; Gupta, S. Epidermal growth factor receptor inhibitors: coming of age. Cancer Control 2014, 21, 74–79. [Google Scholar] [CrossRef]
- Matsuda, N.; Lim, B.; Wang, X.; Ueno, N.T. Early clinical development of epidermal growth factor receptor targeted therapy in breast cancer. Expert Opin. Investig. Drugs 2017, 26, 463–479. [Google Scholar] [CrossRef]
- Ilaria, B.; Bracarda, S.; Caserta, C.; Minelli, A. Targeting of EGFR tyrosine kinase by ZD1839 (“Iressa”) in androgen-responsive prostate cancer in vitro. Mol. Genet. Metab. 2006, 88, 114–122. [Google Scholar] [CrossRef]
- Festuccia, C.; Gravia, G.L.; Biordi, L.; D’Ascenzo, S.; Dolo, V.; Corrado, F.; Ricevuto, E.; Tombolini, V. Effects of EGFR tyrosine kinase inhibitor erlotinib in prostate cancer cells in vitro. Prostate 2009, 69, 1529–1537. [Google Scholar] [CrossRef]
- Anderson, N.G.; Ahmad, T.; Chan, K.; Dobson, R.; Bundred, N.J. ZD1839 (Iressa), a novel epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, potently inhibits the growth of EGFR-positive cancer cell lines with or without erbB2 overexpression. Int. J. Cancer 2001, 94, 774–782. [Google Scholar] [CrossRef]
- Zhu, H.; Cao, X.; Ali-Osman, F.; Keir, S.; Lo, H.W. EGFR and EGFRvIII interact with PUMA to inhibit mitochondrial translocation of PUMA and PUMA-mediated apoptosis independent of EGFR kinase. Cancer Lett. 2015, 294, 101–110. [Google Scholar] [CrossRef]
- Tan, X.; Thapa, N.; Sun, Y.; Anderson, R.A. A kinase-independent role for EGF receptor in autophagy initiation. Cell 2015, 160, 145–160. [Google Scholar] [CrossRef]
- Bollu, L.R.; Ren, J.; Blessing, A.M.; Katreddy, R.R.; Gao, G.; Xu, L.; Wang, J.; Su, F.; Weihua, Z. Involvement of de novo synthesized palmitate and mitochondrial EGFR in EGF induced mitochondrial fusion of cancer cells. Cell Cycle 2014, 13, 2415–2430. [Google Scholar] [CrossRef] [Green Version]
- Tsuchihashi, K.; Okazaki, S.; Ohmura, M.; Ishikawa, M.; Sampetrean, O.; Onishi, H.; Wakimoto, H.; Yoshikawa, M.; Seishima, R.; Iwasaki, Y.; et al. The Egf receptor promotes the malignant potential of glioma by regulating amino acid transport system Xc(-). Cancer Res. 2016, 76, 2954–2963. [Google Scholar] [CrossRef]
- Katreddy, R.R.; Bollu, L.R.; Su, F.; Xian, N.; Srivastava, S.; Thomas, R.; Dai, Y.; Wu, B.; Xu, Y.; Rea, M.A.; et al. Targeted reduction of the EGFR protein, but not inhibition of its kinase activity, induces mitophagy and death of cancer cells through activation of mTORC2 and Akt. Oncogenesis 2018, 7, 1–13. [Google Scholar] [CrossRef]
- Cho, J.; Kim, S.; Du, J.; Meyerson, M. Autophosphorylation of the carboxy-terminal domain is not required for oncogenic transformation by lung cancer derived mutants. Int. J. Cancer 2018, 143, 679–685. [Google Scholar] [CrossRef]
- Munoz, L. Non-kinase targets of protein kinase inhibitors. Nat. Rev. 2017, 16, 424–440. [Google Scholar] [CrossRef]
- Bjorkelund, H.; Gedda, L.; Barta, P.; Malmqvist, M.; Anderson, K. Gefitinib induces epidermal growth factor receptor dimers which alters the interaction characteristics with 25I-EGF. PLoS ONE 2011, 6, 1–11. [Google Scholar] [CrossRef]
- Lu, C.; Mi, L.Z.; Schurpf, T.; Walz, T.; Springer, T.A. Mechanisms for kinase-mediated dimerization of the epidermal growth factor receptor. J. Biol. Chem. 2012, 45, 38244–38253. [Google Scholar] [CrossRef]
- Bublil, E.M.; Pines, G.; Patel, G.; Fruhwirth, G.; Ng, T.; Yarden, Y. Kinase-mediated quasi-dimers of EGFR. FASEB J. 2010, 24, 4744–4755. [Google Scholar] [CrossRef]
- Traxler, P.; Allegrini, P.B.; Brandt, R.; Brueggen, J.; Cozens, R.; Fabbro, D.; Grosios, K.; Lane, H.A.; McSheehy, P.; Mestan, J.; et al. AEE788: A dual family epidermal growth factor receptor/ErbB2 and vascular endothelial growth factor receptor tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2004, 64, 4931–4941. [Google Scholar] [CrossRef]
- Wong, S.S. Chemistry of Protein Conjugation and Crosslinking; CRC Press: Boca Raton, FL, USA, 1993; pp. 75–103. [Google Scholar]
- Fukata, Y.; Murakami, T.; Yokoi, N.; Fukata, M. Local palmitoylation cycles and specialized membrane domain organization. Curr. Top. Membr. 2016, 77, 97–141. [Google Scholar] [CrossRef]
- Anderson, A.M.; Ragan, M.A. Palmitoylation, a protein S-acylation with implications for breast cancer. NPJ Breast Cancer 2016, 2, 1–10. [Google Scholar] [CrossRef]
- Bollu, L.R.; Katreddy, R.R.; Blessing, A.M.; Nguyen, P.; Zheng, B.; Wu, X.; Weihua, Z. Intracellular activation of EGFR by fatty acid synthase dependent palmitoylation. Oncotarget 2015, 6, 34992–35003. [Google Scholar] [CrossRef] [Green Version]
- Runkle, K.B.; Kharbanda, A.; Stypulkowski, E.; Cao, X.J.; Wang, W.; Garcia, B.A.; Witze, E.S. Inhibition of DHHC20mediated EGFR palmitoylation creates a dependence on EGFR signaling. Mol. Cell 2016, 62, 385–396. [Google Scholar] [CrossRef]
- Jennings, B.C.; Nadolski, M.J.; Ling, Y.; Baker, M.B.; Harrison, M.L.; Deschenes, R.J.; Linder, M.E. 2-Bromopalmitate and 2-(2-hydroxy-5-nitro-benzylidene)-benzo[b]thiophen-3-one inhibit DHHC-mediated palmitoylation in vitro. J. Lipid Res. 2009, 50, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Lado, M. Fatty acid synthase as a potential therapeutic target in cancer 2010. Future Oncol. 2010, 6, 551–562. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Chu, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 560–562. [Google Scholar] [CrossRef]
- Chan, C.; Gill, G.N. Mutational analysis of the nucleotide binding site of the epidermal growth factor receptor and v-Src protein-tyrosine kinases. J. Biol. Chem. 1996, 271, 22619–22623. [Google Scholar] [CrossRef]
- Morgillo, F.; Corte, C.M.D.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR targeted therapies: lung cancer. ESMO Open 2016, 1, 1–9. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel) 2017, 9, 52. [Google Scholar]
- Macdonald-Obermann, J.L.; Piwnica-Worms, D.; Pike, L.J. Mechanics of EGFR receptors/ErbB2 kinase activation revealed by luciferase fragment complementation imaging. Proc. Natl. Acad. Sci. USA 2012, 109, 137–142. [Google Scholar] [CrossRef]
- Coban, O.; Zanetti-Dominguez, L.C.; Matthews, D.R.; Rolfe, D.J.; Weitsman, G.; Barber, P.R.; Barbeau, J.; Devauges, V.; Kampmeier, F.; Winn, M.; et al. Effect of phosphorylation on EGFR dimer stability probed by single molecule dynamics and FRET/FLIM. Biophys. J. 2016, 108, 1013–1026. [Google Scholar] [CrossRef]
- Wang, S.; Tsui, S.T.; Liu, C.; Song, Y.; Liu, D. EGFR C797S mutation mediates resistance to third- generation inhibitors in T790M-positive non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 1–5. [Google Scholar] [CrossRef]
- Wang, J.; Wang, B.; Chu, H.; Yao, Y. Intrinsic resistance to EGFR tyrosine kinase inhibitors in advanced non-small cell lung cancer with EGFR activating mutations. OncoTargets Ther. 2016, 9, 3711–3726. [Google Scholar] [CrossRef]
- Di Lorenzo, G.; Tortora, G.; D’Armiento, F.P.; de Rossa, G.; Staibano, S.; Autorino, R.; D’Armiento, M.; de Placido, S.; Catalano, G.; Bianco, A.R.; et al. Expression of epidermal growth factor receptor correlates with disease relaspse and progression to androgen-independence in human prostate cancer. Clin. Cancer Res. 2002, 8, 3438–3444. [Google Scholar]
- Scher, H.I.; Sarkis, A.; Reuters, V.; Cohen, D.; Netto, G.; Petrylak, D.; Lianes, P.; Fuks, Z.; Mendelsohn, J.; Cordon-Cardo, C. Changing pattern of expression of the epidermal growth factor receptor and transforming growth factor alpha in the progression of prostatic neoplasms. Clin. Cancer Res. 1995, 1, 545–550. [Google Scholar]
- Gross, M.; Higano, C.; Pantuck, A.; Castellanos, O.; Green, E.; Nguyen, K.; Agus, D.B. A phase II trial of docetaxel and erlotinib as first line therapy for elderly patients with androgen-independent prostate cancer. BMC Cancer 2007, 7, 142. [Google Scholar] [CrossRef]
- Li, S.; Chen, S.; Jiang, Y.; Liu, J.; Yang, X.; Quan, S. Synergistic interaction between MEK inhibitor and gefitinib in EGFR-TKI resistant human lung cancer cells. Oncol. Lett. 2015, 10, 2652–2656. [Google Scholar] [CrossRef]
- Lee, P.C.; Fang, Y.F.; Yamaguchi, H.; Wang, W.J.; Chen, T.S.; Hong, X.; Ke, B.; Xia, W.; Wei, Y.; Zha, Z.; et al. Targeting PKCδ as a therapeutic strategy against heterogeneous mechanisms of EGFR inhibitor resistance in EGFR-mutant lung cancer. Cancer Cell 2018, 34, 954–969. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Gee, H.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Chong, C.R.; Janne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2014, 19, 1389–1400. [Google Scholar] [CrossRef]
- Li, H.; You, L.; Xie, J.; Pan, H.; Han, W. The roles of subcellularly located EGFR in Autophagy. Cell Signal. Technol. 2017, 35, 223–230. [Google Scholar] [CrossRef]
- Ren, J.; Bollu, L.R.; Su, F.; Gao, G.; Xu, L.; Huang, W.C.; Hung, M.C.; Weihua, Z. EGFR-SGLT1 interaction does not respond to EGFR modulators, but inhibition of SGLT1 sensitizes prostate cancer cells to EGFR tyrosine kinase inhibitors. Prostate 2013, 73, 1453–1461. [Google Scholar] [CrossRef]
- Fung, C.; Chen, X.; Grandis, J.R.; Duvvui, U. EGFR Tyrosine Kinase Inhibition Induces Autophagy in Cancer Cells. Cancer Biol. Ther. 2012, 13, 1417–1424. [Google Scholar] [CrossRef]
- Lambert, S.; Kezunovic, D.V.; Karvinen, S.; Gniadecki, R. Ligand independent activation of EGFR by lipid raft disruption. J. Investig. Dermatol. 2006, 954–962. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Menendez, J.A.; Vellon, L.; Lupu, R. Targeting fatty acid synthase-driven lipid rafts, a novel strategy to overcome trastuzumab resistance in breast cancer cells. Med. Hypotheses 2005, 64, 997–1001. [Google Scholar] [CrossRef]
- Scaltriti, M.; Baselga, J. The epidermal growth factor receptor pathway, A Model for Targeted Therapy. Clin. Cancer Res. 2006, 12, 5268–5272. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, R.; Srivastava, S.; Katreddy, R.R.; Sobieski, J.; Weihua, Z. Kinase-Inactivated EGFR Is Required for the Survival of Wild-Type EGFR-Expressing Cancer Cells Treated with Tyrosine Kinase Inhibitors. Int. J. Mol. Sci. 2019, 20, 2515. https://doi.org/10.3390/ijms20102515
Thomas R, Srivastava S, Katreddy RR, Sobieski J, Weihua Z. Kinase-Inactivated EGFR Is Required for the Survival of Wild-Type EGFR-Expressing Cancer Cells Treated with Tyrosine Kinase Inhibitors. International Journal of Molecular Sciences. 2019; 20(10):2515. https://doi.org/10.3390/ijms20102515
Chicago/Turabian StyleThomas, Rintu, Shivangi Srivastava, Rajasekhara Reddy Katreddy, Jason Sobieski, and Zhang Weihua. 2019. "Kinase-Inactivated EGFR Is Required for the Survival of Wild-Type EGFR-Expressing Cancer Cells Treated with Tyrosine Kinase Inhibitors" International Journal of Molecular Sciences 20, no. 10: 2515. https://doi.org/10.3390/ijms20102515