Integration of mRNA and miRNA Analysis Reveals the Molecular Mechanism Underlying Salt and Alkali Stress Tolerance in Tobacco

Abstract
:1. Introduction
2. Results
2.1. mRNA Sequencing Data Analysis
2.2. Differentially Expressed Genes under Salt-Alkali Stress
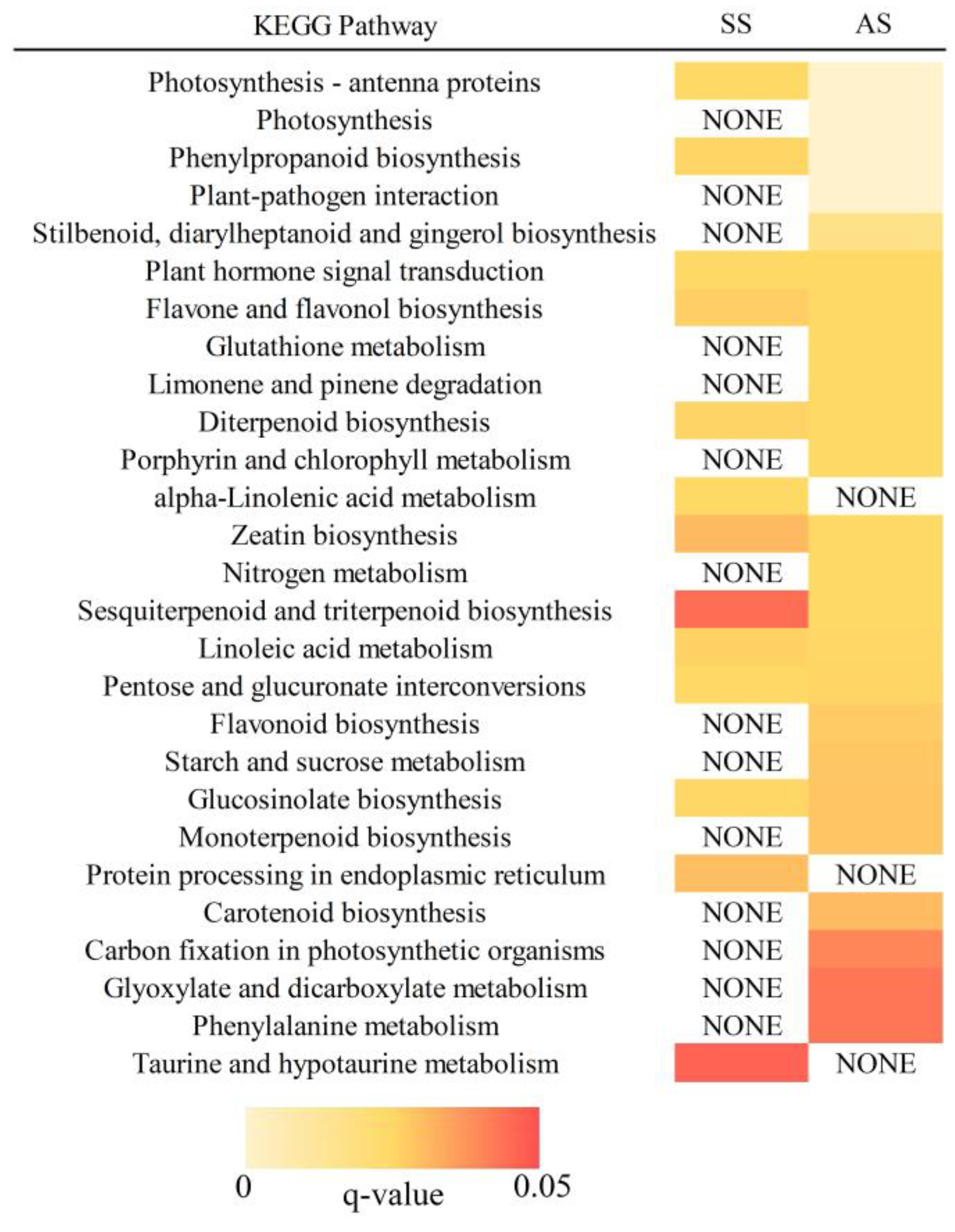
2.3. Functional Classification of Salt-Alkali Responsive Genes
2.4. miRNA Sequencing Data Analysis
2.5. Differential Expression of miRNAs in Response to Salt-Alkali Stress
2.6. Integration Analysis of the miRNAs and mRNAs
2.7. RT-qPCR Validation of DE mRNAs and miRNAs
3. Discussion
3.1. mRNA Profiling in Leaves of Tobacco under Salt-Alkali Stress
3.2. miRNA Profiling of the Salt-Alkali Response in Tobacco
4. Materials and Methods
4.1. Plant Materials and Salt-Alkali Treatment
4.2. Construction and Sequencing of mRNA-seq and sRNA Libraries
4.3. Mapping of mRNA-seq Reads
4.4. Analysis of Differential Gene Expression in Response to Salt-Alkali Stress
4.5. GO and KEGG Pathway Enrichment Analysis
4.6. Identification of Known and Novel miRNAs and Prediction of miRNA Targets
4.7. Detection of the Differential Expression of miRNAs in Response to Salt-Alkali Stress
4.8. Validation of mRNA and miRNA Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar]
- Zhang, B.; Chen, X.; Lu, X.; Shu, N.; Wang, X.; Yang, X.; Wang, S.; Wang, J.; Guo, L.; Wang, D.; et al. Transcriptome Analysis of Gossypium hirsutum L. Reveals Different Mechanisms among NaCl, NaOH and Na2CO3 Stress Tolerance. Sci. Rep. 2018, 8, 13527. [Google Scholar] [CrossRef]
- Razzaghi, F.; Jacobsen, S.-E.; Jensen, C.R.; Andersen, M.N. Ionic and photosynthetic homeostasis in quinoa challenged by salinity and drought—Mechanisms of tolerance. Functional Plant Biol. 2015, 42, 136–148. [Google Scholar] [CrossRef]
- Gupta, B.; Huang, B. Mechanism of Salinity Tolerance in Plants: Physiological, Biochemical, and Molecular Characterization. Int. J. Genom. 2014. [Google Scholar] [CrossRef]
- Hagemann, M. Molecular biology of cyanobacterial salt acclimation. Fems Microbiol. Rev. 2011, 35, 87–123. [Google Scholar] [CrossRef] [Green Version]
- Aghaei, K.; Ehsanpour, A.A.; Komatsu, S. Potato Responds to Salt Stress by Increased Activity of Antioxidant Enzymes. J. Integrative Plant Biol. 2009, 51, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Jagendorf, A.; Zhu, J.K. Understanding and improving salt tolerance in plants. Crop Sci. 2005, 45, 437–448. [Google Scholar] [CrossRef]
- Tang, J.; Liu, Q.Q.; Yuan, H.Y.; Zhang, Y.X.; Huang, S.Z. Molecular analysis of a novel alkaline metal salt (NaCl)-responsive WRKY transcription factor gene IlWRKY1 from the halophyte Iris lactea var. chinensis. Int. Biodeterior. Biodegrad. 2018, 127, 139–145. [Google Scholar] [CrossRef]
- Budak, H.; Akpinar, B.A. Plant miRNAs: Biogenesis, organization and origins. Funct. Integrative Genom. 2015, 15, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Li, Y.-F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Zhang, J.; Ma, Y.; Pan, X.; Dong, C.; Pang, S.; He, S.; Deng, L.; Yi, S.; Zheng, Y.; et al. Combined analysis of mRNA and miRNA identifies dehydration and salinity responsive key molecular players in citrus roots. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Shenoy, A.; Blelloch, R.H. Regulation of microRNA function in somatic stem cell proliferation and differentiation. Nat. Rev. Mol. Cell Biol. 2014, 15, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Wang, Y.; Gruber, M.Y.; Hannoufa, A. miR 156/SPL10 Modulates Lateral Root Development, Branching and Leaf Morphology in Arabidopsisby Silencing AGAMOUS-LIKE 79. Front. Plant Sci. 2018, 8. [Google Scholar] [CrossRef]
- Cagirici, H.B.; Biyiklioglu, S.; Budak, H. Assembly and Annotation of Transcriptome Provided Evidence of miRNA Mobility between Wheat and Wheat Stem Sawfly. Front. Plant Sci. 2017, 8, 14. [Google Scholar] [CrossRef]
- Poethig, R.S. Small RNAs and developmental timing in plants. Curr. Opin. Genet. Dev. 2009, 19, 374–378. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.-Y.; Zhou, Y.; He, F.; Dong, X.; Liu, L.-Y.; Coupland, G.; Turck, F.; de Meaux, J. miR824-Regulated AGAMOUS-LIKE16 Contributes to Flowering Time Repression in Arabidopsis. Plant Cell 2014, 26, 2024–2037. [Google Scholar] [CrossRef] [PubMed]
- Hajyzadeh, M.; Turktas, M.; Khawar, K.M.; Unver, T. miR408 overexpression causes increased drought tolerance in chickpea. Gene 2015, 555, 186–193. [Google Scholar] [CrossRef]
- Giusti, L.; Mica, E.; Bertolini, E.; De Leonardis, A.M.; Faccioli, P.; Cattivelli, L.; Crosatti, C. microRNAs differentially modulated in response to heat and drought stress in durum wheat cultivars with contrasting water use efficiency. Funct. Integr. Genom. 2017, 17, 293–309. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, G.; Sutoh, K.; Zhu, J.-K.; Zhang, W. Identification of cold-inducible microRNAs in plants by transcriptome analysis. Biochimica Et Biophysica Acta-Gene Regulatory Mechanisms 2008, 1779, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Stewart, C.N., Jr.; Taki, F.A.; He, Q.; Liu, H.; Zhang, B. High-throughput deep sequencing shows that microRNAs play important roles in switchgrass responses to drought and salinity stress. Plant Biotechnol. J. 2014, 12, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Au, K.F.; Underwood, J.G.; Lee, L.; Wong, W.H. Improving PacBio Long Read Accuracy by Short Read Alignment. PLoS ONE 2012, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhu, G.Z.; Du, L.; Shang, X.G.; Cheng, C.Z.; Yang, B.; Hu, Y.; Cai, C.P.; Guo, W.Z. Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci. Rep. 2016, 6, 15. [Google Scholar] [CrossRef]
- Takahashi, F.; Tilbrook, J.; Trittermann, C.; Berger, B.; Roy, S.J.; Seki, M.; Shinozaki, K.; Tester, M. Comparison of Leaf Sheath Transcriptome Profiles with Physiological Traits of Bread Wheat Cultivars under Salinity Stress. PLoS ONE 2015, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.D.; Wang, J.Q.; Yang, N.; Dong, Y.Y.; Liu, L.; Wang, F.W.; Wang, N.; Chen, H.; Liu, W.C.; Sun, Y.P.; et al. Gene expression profiling of soybean leaves and roots under salt, saline-alkali and drought stress by high-throughput Illumina sequencing. Gene 2013, 512, 392–402. [Google Scholar] [CrossRef]
- Negrao, S.; Almadanim, M.C.; Pires, I.S.; Abreu, I.A.; Maroco, J.; Courtois, B.; Gregorio, G.B.; McNally, K.L.; Oliveira, M.M. New allelic variants found in key rice salt-tolerance genes: An association study. Plant Biotechnol. J. 2013, 11, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.J.; Pan, X.T.; Zhang, J.; Ma, Y.Y.; He, S.L.; Zheng, Y.Q.; Ma, Y.T. Effect of salt-stress on gene expression in citrus roots revealed by RNA-seq. Funct. Integr. Genom. 2018, 18, 155–173. [Google Scholar] [CrossRef]
- Long, R.C.; Li, M.N.; Li, X.; Gao, Y.L.; Zhang, T.J.; Sun, Y.; Kang, J.M.; Wang, T.H.; Cong, L.L.; Yang, Q.C. A Novel miRNA Sponge Form Efficiently Inhibits the Activity ofmiR393 and Enhances the Salt Tolerance and ABA Insensitivity in Arabidopsis thaliana. Plant Mol. Biol. Rep. 2017, 35, 409–415. [Google Scholar] [CrossRef]
- Feng, K.W.; Nie, X.J.; Cui, L.C.; Deng, P.C.; Wang, M.X.; Song, W.N. Genome-Wide Identification and Characterization of Salinity Stress-Responsive miRNAs in Wild Emmer Wheat (Triticum turgidum ssp dicoccoides). Genes 2017, 8, 18. [Google Scholar] [CrossRef]
- Li, Y.S.; Li, C.H.; Bai, L.Q.; He, C.X.; Yu, X.C. MicroRNA and target gene responses to salt stress in grafted cucumber seedlings. Acta Physiol. Plant. 2016, 38, 12. [Google Scholar] [CrossRef]
- Deng, P.C.; Wang, L.; Cui, L.C.; Feng, K.W.; Liu, F.Y.; Du, X.H.; Tong, W.; Nie, X.J.; Ji, W.Q.; Weining, S. Global Identification of MicroRNAs and Their Targets in Barley under Salinity Stress. PLoS ONE 2015, 10, 20. [Google Scholar] [CrossRef]
- Fu, R.; Zhang, M.; Zhao, Y.C.; He, X.C.; Ding, C.Y.; Wang, S.K.; Feng, Y.; Song, X.L.; Li, P.; Wang, B.H. Identification of Salt Tolerance-related microRNAs and Their Targets in Maize (Zea mays L.) Using High-throughput Sequencing and Degradome Analysis. Front. Plant Sci. 2017, 8, 13. [Google Scholar] [CrossRef]
- Yu, M.; Zhao, J. The cytological changes of tobacco zygote and proembryo cells induced by beta-glucosyl Yariv reagent suggest the involvement of arabinogalactan proteins in cell division and cell plate formation. BMC Plant Biol. 2012, 12, 16. [Google Scholar] [CrossRef]
- Yin, F.; Gao, J.; Liu, M.; Qin, C.; Zhang, W.; Yang, A.; Xia, M.; Zhang, Z.; Shen, Y.; Lin, H.; et al. Genome-wide analysis of Water-stress-responsive microRNA expression profile in tobacco roots. Funct. Integr. Genom. 2014, 14, 319–332. [Google Scholar] [CrossRef]
- Sierro, N.; Battey, J.N.D.; Ouadi, S.; Bakaher, N.; Bovet, L.; Willig, A.; Goepfert, S.; Peitsch, M.C.; Ivanov, N.V. The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 2014, 5, 9. [Google Scholar] [CrossRef]
- Bombarely, A.; Rosli, H.G.; Vrebalov, J.; Moffett, P.; Mueller, L.A.; Martin, G.B. A Draft Genome Sequence of Nicotiana benthamiana to Enhance Molecular Plant-Microbe Biology Research. Mol. Plant-Microbe Interact. 2012, 25, 1523–1530. [Google Scholar] [CrossRef]
- Frazier, T.P.; Sun, G.L.; Burklew, C.E.; Zhang, B.H. Salt and Drought Stresses Induce the Aberrant Expression of microRNA Genes in Tobacco. Mol. Biotechnol. 2011, 49, 159–165. [Google Scholar] [CrossRef]
- Aysin, F.; Erson-Bensan, A.; Eyidogan, F.; Oktem, H.A. Generating salt-tolerant Nicotiana tabacum and identification of stress-responsive miRNAs in transgenics. Turk. J. Bot. 2015, 39, 757–768. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytologist 2018, 217, 523–539. [Google Scholar] [CrossRef]
- Cellier, F.; Conejero, G.; Ricaud, L.; Luu, D.T.; Lepetit, M.; Gosti, F.; Casse, F. Characterization of AtCHX17, a member of the cation/H+ exchangers, CHX family, from Arabidopsis thaliana suggests a role in K+ homeostasis. Plant J. 2004, 39, 834–846. [Google Scholar] [CrossRef]
- Obata, T.; Kitamoto, H.K.; Nakamura, A.; Fukuda, A.; Tanaka, Y. Rice Shaker potassium channel OsKAT1 confers tolerance to salinity stress on yeast and rice cells. Plant Physiol. 2007, 144, 1978–1985. [Google Scholar] [CrossRef]
- Trowitzsch, S.; Tempe, R. ABC Transporters in Dynamic Macromolecular Assemblies. J. Mol. Biol. 2018, 430, 4481–4495. [Google Scholar] [CrossRef]
- Panikashvili, D.; Savaldi-Goldstein, S.; Mandel, T.; Yifhar, T.; Franke, R.B.; Hoefer, R.; Schreiber, L.; Chory, J.; Aharoni, A. The Arabidopsis DESPERADO/AtWBC11 transporter is required for cutin and wax secretion. Plant Physiol. 2007, 145, 1345–1360. [Google Scholar] [CrossRef]
- Afzal, Z.; Howton, T.C.; Sun, Y.L.; Mukhtar, M.S. The Roles of Aquaporins in Plant Stress Responses. J. Dev. Biol. 2016, 4. [Google Scholar] [CrossRef]
- Kong, W.L.; Yang, S.Z.; Wang, Y.L.; Bendahmane, M.; Fu, X.P. Genome-wide identification and characterization of aquaporin gene family in Beta vulgaris. Peerj 2017, 5. [Google Scholar] [CrossRef]
- Hu, W.; Yuan, Q.Q.; Wang, Y.; Cai, R.; Deng, X.M.; Wang, J.; Zhou, S.Y.; Chen, M.J.; Chen, L.H.; Huang, C.; et al. Overexpression of a Wheat Aquaporin Gene, TaAQP8, Enhances Salt Stress Tolerance in Transgenic Tobacco. Plant Cell Physiol. 2012, 53, 2127–2141. [Google Scholar] [CrossRef] [Green Version]
- Lienard, D.; Durambur, G.; Kiefer-Meyer, M.C.; Nogue, F.; Menu-Bouaouiche, L.; Charlot, F.; Gomord, V.; Lassalles, J.P. Water transport by aquaporins in the extant plant Physcomitrella patens. Plant Physiol. 2008, 146, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Yasmeen, T.; Riaz, M.; Arif, M.S.; Ali, S.; Raza, S.H. Proteus mirabilis alleviates zinc toxicity by preventing oxidative stress in maize (Zea mays) plants. Ecotox. Environ. Saf. 2014, 110, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.Z.; Zheng, G.Y.; Li, W.Z.; Wang, Y.Q.; Hu, B.; Wang, H.R.; Wu, H.K.; Qian, Y.W.; Zhu, X.G.; Tan, D.X.; et al. Melatonin delays leaf senescence and enhances salt stress tolerance in rice. J. Pineal Res. 2015, 59, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Valerio, C.; Costa, A.; Marri, L.; Issakidis-Bourguet, E.; Pupillo, P.; Trost, P.; Sparla, F. Thioredoxin-regulated beta-amylase (BAM1) triggers diurnal starch degradation in guard cells, and in mesophyll cells under osmotic stress. J. Exp. Bot. 2011, 62, 545–555. [Google Scholar] [CrossRef]
- Timabud, T.; Yin, X.J.; Pongdontri, P.; Komatsu, S. Gel-free/label-free proteomic analysis of developing rice grains under heat stress. J. Proteomics 2016, 133, 1–19. [Google Scholar] [CrossRef]
- Dejardin, A.; Sokolov, L.N.; Kleczkowski, L.A. Sugar/osmoticum levels modulate differential abscisic acid-independent expression of two stress-responsive sucrose synthase genes in Arabidopsis. Biochem. J. 1999, 344, 503–509. [Google Scholar] [CrossRef]
- Islam, M.O.; Kato, H.; Shima, S.; Tezuka, D.; Matsui, H.; Imai, R. Functional identification of a rice trehalase gene involved in salt stress tolerance. Gene 2019, 685, 42–49. [Google Scholar] [CrossRef]
- Liu, H.-H.; Tian, X.; Li, Y.-J.; Wu, C.-A.; Zheng, C.-C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 2008, 14, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Xu, L.; Wang, Y.; Yu, R.; Zhu, X.; Luo, X.; Gong, Y.; Wang, R.; Limera, C.; Zhang, K.; et al. Identification of novel and salt-responsive miRNAs to explore miRNA-mediated regulatory network of salt stress response in radish (Raphanus sativus L.). Bmc Genom. 2015, 16, 197. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, G.; Yuan, H.; Cheng, L.; Zhao, D.; Huang, W.; Zhang, S.; Zhang, L.; Chen, H.; Zhang, J.; et al. Identification and characterization of miRNAs and targets in flax (Linum usitatissimum) under saline, alkaline, and saline-alkaline stresses. BMC Plant Biol. 2016, 16, 124. [Google Scholar] [CrossRef]
- Cao, C.; Long, R.; Zhang, T.; Kang, J.; Wang, Z.; Wang, P.; Sun, H.; Yu, J.; Yang, Q. Genome-Wide Identification of microRNAs in Response to Salt/Alkali Stress in Medicago truncatula through High-Throughput Sequencing. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.; Gruber, M.Y.; Wall, K.; Hannoufa, A. An Insight into microRNA156 Role in Salinity Stress Responses of Alfalfa. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, Q.L.; Zhang, B.H. Response of miRNAs and their targets to salt and drought stresses in cotton (Gossypium hirsutum L.). Gene 2013, 530, 26–32. [Google Scholar] [CrossRef]
- Si, J.-N.; Zhou, T.; Xu, F.; Bo, W.-H.; Wu, R.-L. Salt-responsive MicroRNAs in Populus euphratica by Deep Sequencing. Bull. Bot. Res. 2015, 35, 836–842. [Google Scholar]
- Sathik, M.B.M. Identification and Validation of Cold Responsive MicroRNAs of Hevea brasiliensis Using High Throughput Sequencing. J. Crop Sci. Biotechnol. 2017, 20, 369–377. [Google Scholar]
- Akdogan, G.; Tufekci, E.D.; Uranbey, S.; Unver, T. miRNA-based drought regulation in wheat. Funct. Integr. Genom. 2016, 16, 221–233. [Google Scholar] [CrossRef]
- Tang, S.; Wang, Y.; Li, Z.; Gui, Y.; Xiao, B.; Xie, J.; Zhu, Q.-H.; Fan, L. Identification of wounding and topping responsive small RNAs in tobacco (Nicotiana tabacum). BMC Plant Biol. 2012, 12. [Google Scholar] [CrossRef]
- Bukhari, S.A.H.; Shang, S.; Zhang, M.; Zheng, W.; Zhang, G.; Wang, T.-Z.; Shamsi, I.H.; Wu, F. Genome-wide identification of chromium stress-responsive micro RNAs and their target genes in tobacco (Nicotiana tabacum) roots. Environ. Toxicol. Chem. 2015, 34, 2573–2582. [Google Scholar] [CrossRef]
- Long, R.-C.; Li, M.-N.; Kang, J.-M.; Zhang, T.-J.; Sun, Y.; Yang, Q.-C. Small RNA deep sequencing identifies novel and salt-stress-regulated microRNAs from roots of Medicago sativa and Medicago truncatula. Physiologia Plantarum 2015, 154, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Guo, W.; Zhang, B. Genome-wide identification and characterization of SPL transcription factor family and their evolution and expression profiling analysis in cotton. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Hong, G.; Li, L.; Zhang, X.; Kong, Y.; Sun, Z.; Li, J.; Chen, J.; He, Y. miR156/SPL9 Regulates Reactive Oxygen Species Accumulation and Immune Response in Arabidopsis thaliana. Phytopathology 2019, PHYTO08180306R. [Google Scholar] [CrossRef]
- Tao, Y.; Yuan, F.H.; Leister, R.T.; Ausubel, F.M.; Katagiri, F. Mutational analysis of the Arabidopsis nucleotide binding site-leucine-rich repeat resistance gene RPS2. Plant Cell 2000, 12, 2541–2554. [Google Scholar] [PubMed]
- Zhu, Q.-H.; Fan, L.; Liu, Y.; Xu, H.; Llewellyn, D.; Wilson, I. miR482 Regulation of NBS-LRR Defense Genes during Fungal Pathogen Infection in Cotton. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Suzuki, N.; Rivero, R.M.; Shulaev, V.; Blumwald, E.; Mittler, R. Abiotic and biotic stress combinations. New Phytologist 2014, 203, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foyer, C.H.; Rasool, B.; Davey, J.W.; Hancock, R.D. Cross-tolerance to biotic and abiotic stresses in plants: A focus on resistance to aphid infestation. J. Exp. Bot. 2016, 67, 2025–2037. [Google Scholar] [CrossRef]
- Chen, Q.; Li, M.; Zhang, Z.; Tie, W.; Chen, X.; Jin, L.; Zhai, N.; Zheng, Q.; Zhang, J.; Wang, R.; et al. Integrated mRNA and microRNA analysis identifies genes and small miRNA molecules associated with transcriptional and post-transcriptional-level responses to both drought stress and re-watering treatment in tobacco. BMC Genom. 2017, 18. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protocols 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11. [Google Scholar] [CrossRef]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [Green Version]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. Bmc Bioinform. 2012, 13. [Google Scholar] [CrossRef]
- Friedlaender, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Wu, H.-J.; Ma, Y.-K.; Chen, T.; Wang, M.; Wang, X.-J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L.; et al. Integrated Profiling of MicroRNAs and mRNAs: MicroRNAs Located on Xq27.3 Associate with Clear Cell Renal Cell Carcinoma. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Raw Reads | Clean Reads | Clean Bases | Q30 (%) | Mapped Reads |
|---|---|---|---|---|---|
| CK_1 | 55,946,150 | 54,753,170 | 8.21 G | 93.32 | 51,560,453 (94.17%) |
| CK_2 | 52,426,666 | 50,127,414 | 7.52 G | 93.82 | 47,158,934 (94.08%) |
| CK_3 | 43,620,248 | 42,722,964 | 6.41 G | 93.71 | 40,181,708 (94.05%) |
| SS_1 | 51,641,948 | 50,378,090 | 7.56 G | 93.54 | 47,605,747 (94.5%) |
| SS_2 | 61,792,742 | 60,177,334 | 9.03 G | 93.74 | 56,855,737 (94.48%) |
| SS_3 | 65,172,280 | 64,106,196 | 9.62 G | 93.64 | 60,509,819 (94.39%) |
| AS_1 | 64,693,340 | 63,648,474 | 9.55 G | 93.49 | 59,519,456 (93.51%) |
| AS_2 | 58,326,842 | 57,264,752 | 8.59 G | 93.49 | 53,875,061 (94.08%) |
| AS_3 | 57,919,408 | 57,037,200 | 8.56 G | 93.24 | 53,759,723 (94.25%) |
| Treatment | Raw Reads | Clean Reads | Mature miRNAs | Novel miRNAs |
|---|---|---|---|---|
| CK_1 | 15,192,456 | 14,706,127 | 79 | 99 |
| CK_2 | 16,160,064 | 15,672,009 | 74 | 99 |
| CK_3 | 13,289,928 | 12,949,487 | 67 | 99 |
| SS_1 | 13,583,796 | 13,079,201 | 80 | 100 |
| SS_2 | 12,296,168 | 11,973,197 | 72 | 97 |
| SS_3 | 14,031,804 | 13,667,590 | 76 | 100 |
| AS_1 | 13,551,741 | 13,090,227 | 80 | 99 |
| AS_2 | 15,333,030 | 14,915,331 | 78 | 98 |
| AS_3 | 13,272,983 | 12,863,432 | 81 | 101 |
| miRNA | log2FC | p-Value | Mature Sequence | Regulated |
|---|---|---|---|---|
| nta-miR156a | −0.64 | 2.56 × 10−2 | UGACAGAAGAGAGUGAGCAC | SS |
| nta-miR396a | 0.64 | 1.07 × 10−3 | UUCCACAGCUUUCUUGAACUG | SS |
| nta-miR482d | 0.78 | 4.42 × 10−4 | UUCCCGACUCCCCCCAUACCAC | SS |
| nta-miR6145a | −0.55 | 4.91 × 10−2 | CAUUUUCACAUGUAGCACUGAC | SS |
| nta-miR6149a | 0.75 | 1.51 × 10−3 | UUGAUACGCACCUGAAUCGGC | SS |
| miR-novel_31 | −0.93 | 4.56 × 10−4 | AAGGACUGCUAUUUAGAAAUUAGC | SS |
| miR-novel_59 | −0.57 | 2.21 × 10−2 | ACGGGCUGCUAUUUAAGAAUUAGC | SS |
| miR-novel_61 | 1.15 | 1.82 × 10−5 | UUGAAGUGUUUGGGGGAACUC | SS |
| miR-novel_64 | −0.98 | 1.62 × 10−3 | CUCCGGUCAAGAUCUUCCAUC | SS |
| miR-novel_127 | −0.71 | 3.30 × 10−2 | ACGUGUCGGAUCCUCUAAAAGU | SS |
| miR-novel_163 | −0.81 | 1.48 × 10−2 | AUGGCGUGGCUUCAGAUCUCUGAU | SS |
| miR-novel_216 | −1.86 | 3.39 × 10−8 | AGGACGAUCUCUAUGACUCUUUGG | SS |
| miR-novel_228 | −0.88 | 9.24 × 10−3 | ACGUGGACUGGGCUGGGCUAGCCU | SS |
| miR-novel_237 | −0.65 | 3.14 × 10−2 | AGGACUGCUAUUUAGAAAUUAGGC | SS |
| miR-novel_258 | −0.84 | 7.63 × 10−3 | ACUCCGAUUUGUGUAUGACUUGAC | SS |
| nta-miR156a | 0.71 | 2.82 × 10−2 | UGACAGAAGAGAGUGAGCAC | AS |
| nta-miR164a | 0.84 | 2.40 × 10−2 | UGGAGAAGCAGGGCACGUGCA | AS |
| nta-miR168a | 0.83 | 3.65 × 10−4 | UCGCUUGGUGCAGGUCGGGAC | AS |
| nta-miR168d | 0.73 | 2.59 × 10−2 | UCGCUUGGUGCAGGUCGGGAA | AS |
| nta-miR171c | 1.00 | 8.94 × 10−3 | UGAUUGAGCCGUGCCAAUAUC | AS |
| nta-miR394 | 0.85 | 1.83 × 10−2 | UUGGCAUUCUGUCCACCUCC | AS |
| nta-miR398 | −0.70 | 2.68 × 10−2 | UGUGUUCUCAGGUCGCCCCUG | AS |
| nta-miR408 | −0.70 | 1.94 × 10−2 | UGCACUGCCUCUUCCCUGGCU | AS |
| nta-miR482b-3p | 0.85 | 1.61 × 10−2 | UCUUGCCAAUGCCAUCCAUUCC | AS |
| nta-miR6020b | 1.12 | 2.31 × 10−3 | AAAUGUUCUUCGAGUAUCUUC | AS |
| nta-miR6145e | 0.54 | 4.90 × 10−2 | AUUGUUACAUGUAGCACUGGC | AS |
| nta-miR6149a | 0.69 | 2.27 × 10−2 | UUGAUACGCACCUGAAUCGGC | AS |
| nta-miR6159 | 0.72 | 1.57 × 10−2 | UAGCAUAGAAUUCUCGCACCUA | AS |
| nta-miR827 | 0.64 | 3.92 × 10−2 | UUAGAUGAACAUCAACAAACA | AS |
| miR-novel_43 | −1.08 | 5.83 × 10−5 | UUGGGCGUGCACAAGUAGGC | AS |
| miR-novel_64 | −1.17 | 1.38 × 10−3 | CUCCGGUCAAGAUCUUCCAUC | AS |
| miR-novel_93 | 1.18 | 1.42 × 10−3 | ACGCAGGAGAGAUGAUGCUGGA | AS |
| miR-novel_156 | −0.91 | 1.26 × 10−2 | ACCGAUGUGGGACUCUGUCUAAAG | AS |
| miR-novel_167 | −0.91 | 9.95 × 10−3 | AAAGAGCUCUAUAUAUAAAAAUGU | AS |
| miR-novel_193 | −0.81 | 2.60 × 10−2 | AAGUCCGGUAACAUUUUGAAGAGU | AS |
| miR-novel_215 | −1.29 | 1.11 × 10−4 | ACGUGGGCUGGGCUGAGCUAACCC | AS |
| miR-novel_216 | −1.31 | 4.12 × 10−4 | AGGACGAUCUCUAUGACUCUUUGG | AS |
| Gene ID | CK (FPKM) | SS (FPKM) | AS (FPKM) | Log2FC (SS/CK) | Log2FC (AS/CK) | Regulation | Annotation |
|---|---|---|---|---|---|---|---|
| Ion transport | |||||||
| 107775281 | 0.13 | 0 | 0 | −inf | −inf | Down | cation/H(+) antiporter 18 |
| 107803899 | 0.02 | 0.18 | 0.13 | 3.05 | 2.46 | Up | cation/H(+) antiporter 15 |
| 107814619 | 0.02 | 0.19 | 0.67 | 2.83 | 4.61 | Up | potassium channel AKT1 |
| 107774409 | 0.04 | 0.28 | 9.04 | 2.68 | 7.62 | Up | ABC transporter G family member 11 |
| 107800765 | 1.53 | 0.40 | 0.20 | −2.22 | −3.26 | Down | ABC transporter B family member 19 |
| Aquaporins | |||||||
| 107762778 | 12.65 | 1.79 | 1.38 | −3.12 | −3.56 | Down | aquaporin PIP2-1 |
| 107782948 | 42.89 | 6.99 | 6.57 | −2.92 | −3.07 | Down | aquaporin PIP2-1 |
| 107786743 | 39.82 | 7.32 | 4.89 | −2.74 | −3.39 | Down | aquaporin PIP2-7 |
| 107789972 | 95.21 | 15.78 | 12.69 | −2.89 | −3.27 | Down | aquaporin PIP2-7 |
| 107807226 | 75.86 | 8.04 | 6.26 | −3.54 | −3.96 | Down | aquaporin TIP1-3 |
| 107814785 | 34.86 | 2.09 | 2.89 | −4.36 | −3.95 | Down | aquaporin TIP2-1 |
| 107820018 | 31.08 | 7.70 | 6.70 | −2.31 | −2.58 | Down | aquaporin TIP1-3 |
| 107821662 | 4.55 | 1.28 | 0.39 | −2.13 | −3.92 | Down | aquaporin TIP2-1 |
| Antioxidant activity | |||||||
| 107819573 | 0.08 | 0.48 | 0.71 | 2.23 | 2.74 | Up | superoxide dismutase family |
| 107832827 | 0.25 | 1.71 | 2.38 | 2.45 | 2.86 | Up | superoxide dismutase family |
| 107781013 | 2.73 | 17.09 | 16.50 | 2.34 | 2.23 | Up | catalase family |
| 107758994 | 1.20 | 0.11 | 0 | −3.74 | −inf | Down | peroxidase 19 |
| 107766849 | 0.28 | 0 | 0 | −inf | −inf | Down | peroxidase 10 |
| 107777661 | 0.40 | 3.98 | 2.17 | 3.04 | 2.08 | Up | peroxidase P7 |
| 107793453 | 0.48 | 3.42 | 5.06 | 2.56 | 3.05 | Up | peroxidase P7 |
| 107813838 | 0.24 | 2.62 | 1.34 | 3.17 | 2.13 | Up | peroxidase 12 |
| 107814038 | 0.14 | 0 | 0 | −inf | −inf | Down | peroxidase 3 |
| 107820597 | 0.81 | 4.34 | 5.04 | 2.13 | 2.28 | Up | probable glutathione peroxidase 8 |
| 107777621 | 6.99 | 40.69 | 87.80 | 2.24 | 3.29 | Up | glutathione transferase GST 23 |
| 107764067 | 1.12 | 14.98 | 74.03 | 3.44 | 5.68 | Up | glutathione transferase GST 23 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Chen, Q.; Liu, P.; Jia, W.; Chen, Z.; Xu, Z. Integration of mRNA and miRNA Analysis Reveals the Molecular Mechanism Underlying Salt and Alkali Stress Tolerance in Tobacco. Int. J. Mol. Sci. 2019, 20, 2391. https://doi.org/10.3390/ijms20102391
Xu J, Chen Q, Liu P, Jia W, Chen Z, Xu Z. Integration of mRNA and miRNA Analysis Reveals the Molecular Mechanism Underlying Salt and Alkali Stress Tolerance in Tobacco. International Journal of Molecular Sciences. 2019; 20(10):2391. https://doi.org/10.3390/ijms20102391
Chicago/Turabian StyleXu, Jiayang, Qiansi Chen, Pingping Liu, Wei Jia, Zheng Chen, and Zicheng Xu. 2019. "Integration of mRNA and miRNA Analysis Reveals the Molecular Mechanism Underlying Salt and Alkali Stress Tolerance in Tobacco" International Journal of Molecular Sciences 20, no. 10: 2391. https://doi.org/10.3390/ijms20102391