Pro-Inflammatory Stimuli Influence Expression of Intercellular Adhesion Molecule 1 in Human Anulus Fibrosus Cells through FAK/ERK/GSK3 and PKCδ Signaling Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
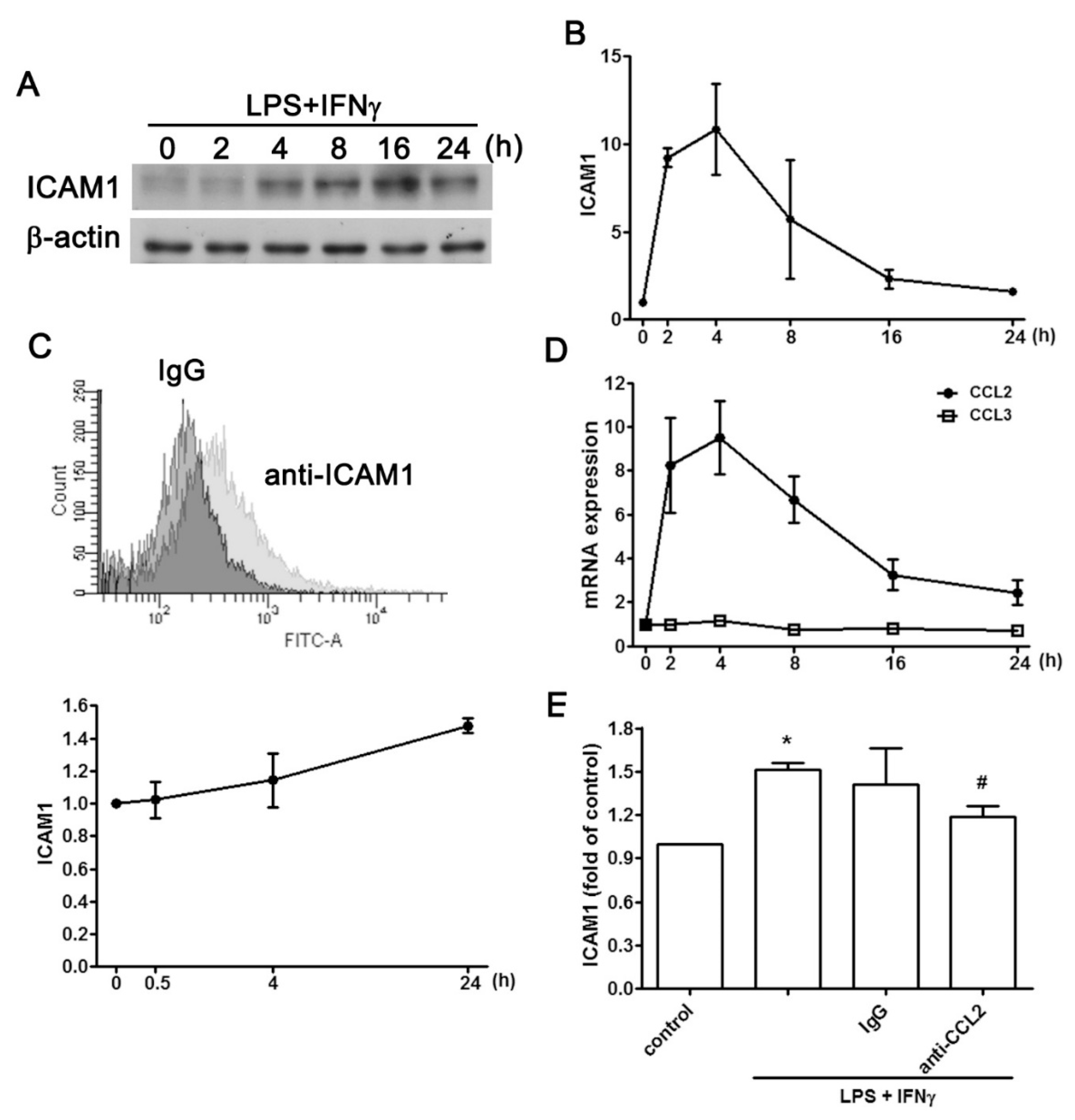
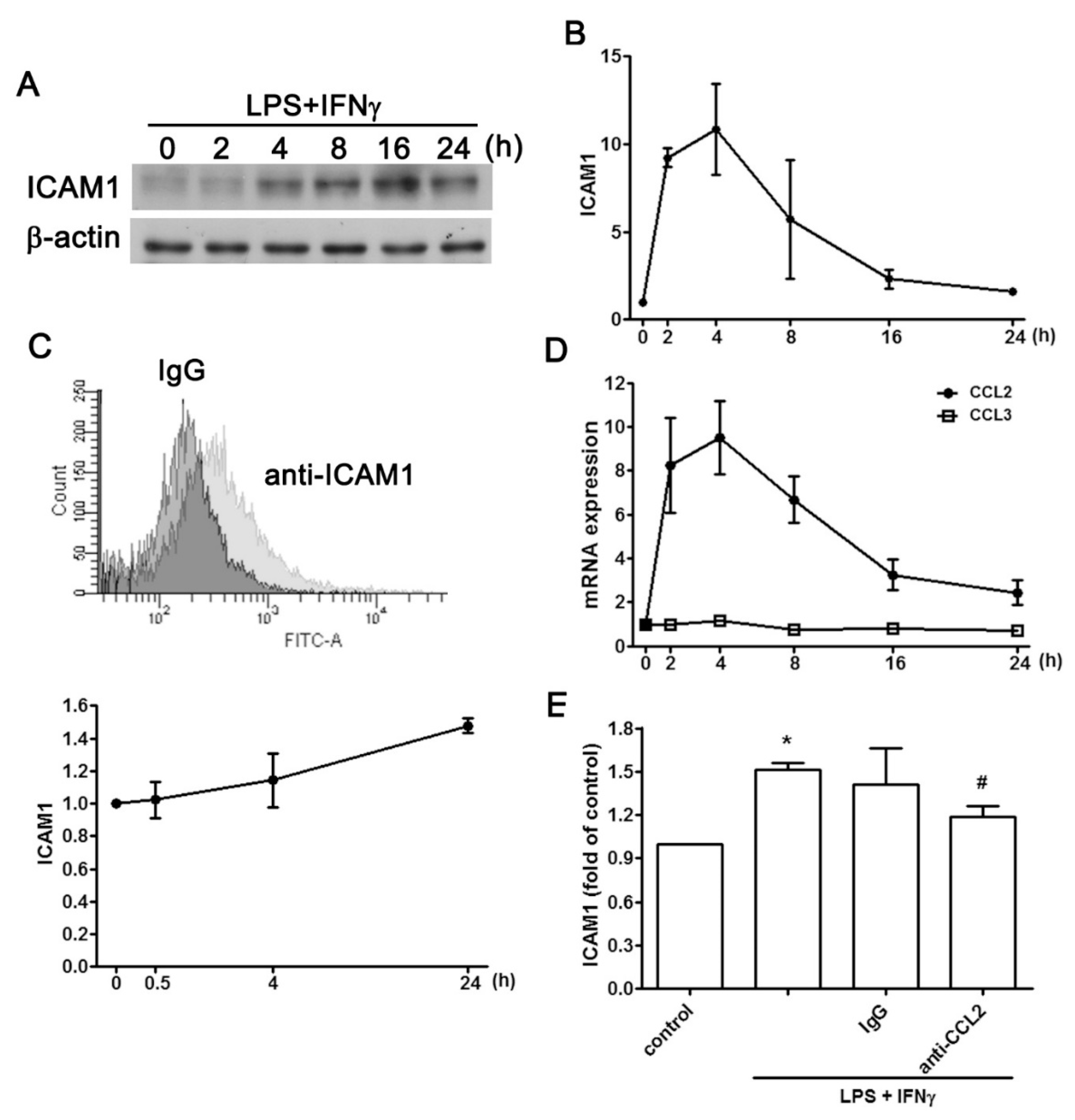
2.1. Regulatory Effect of CCL2 on ICAM1 Expression in IVD Cells
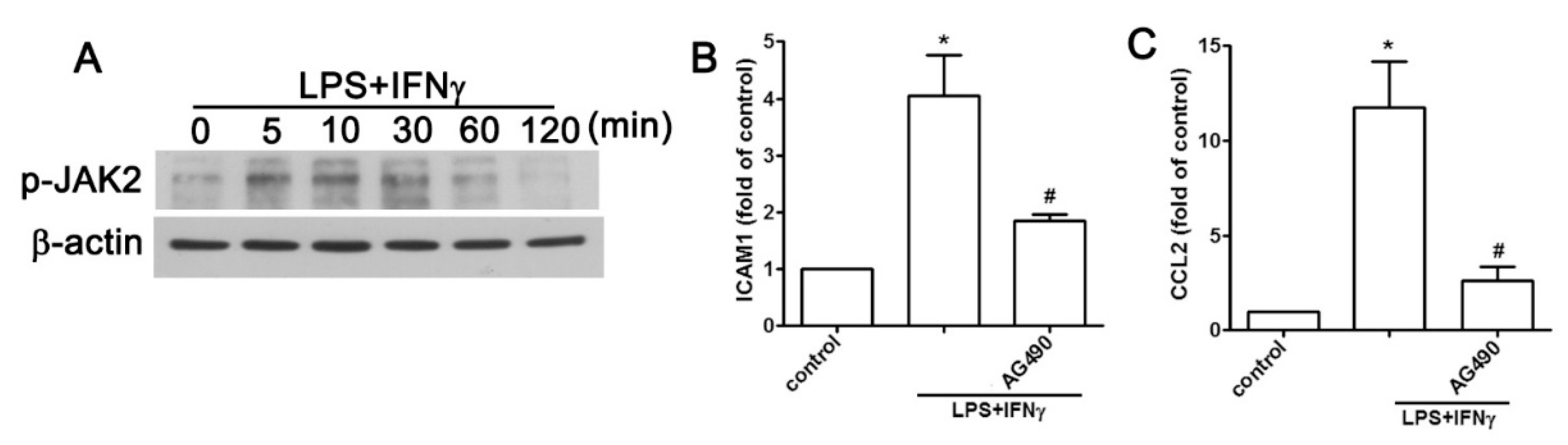
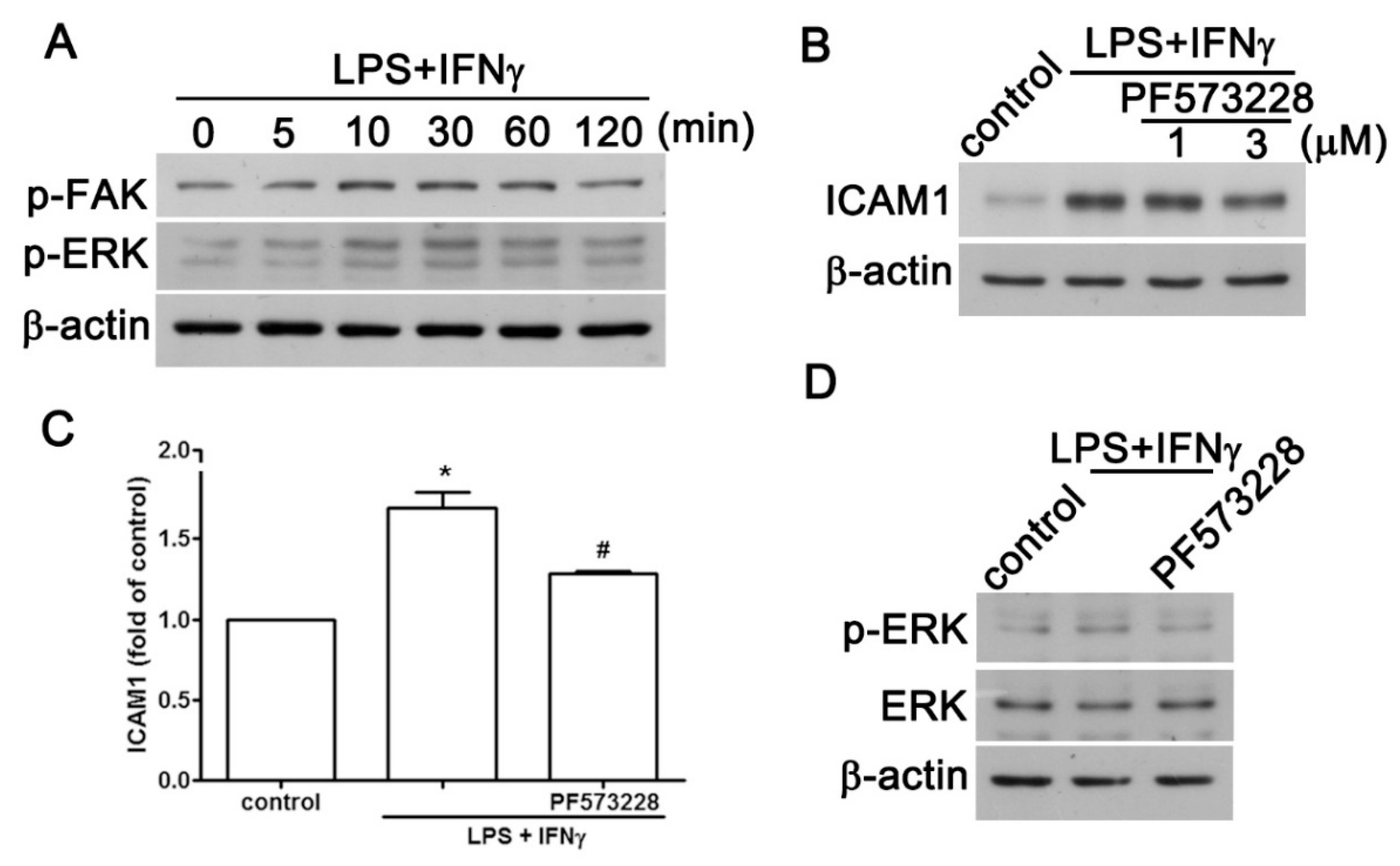
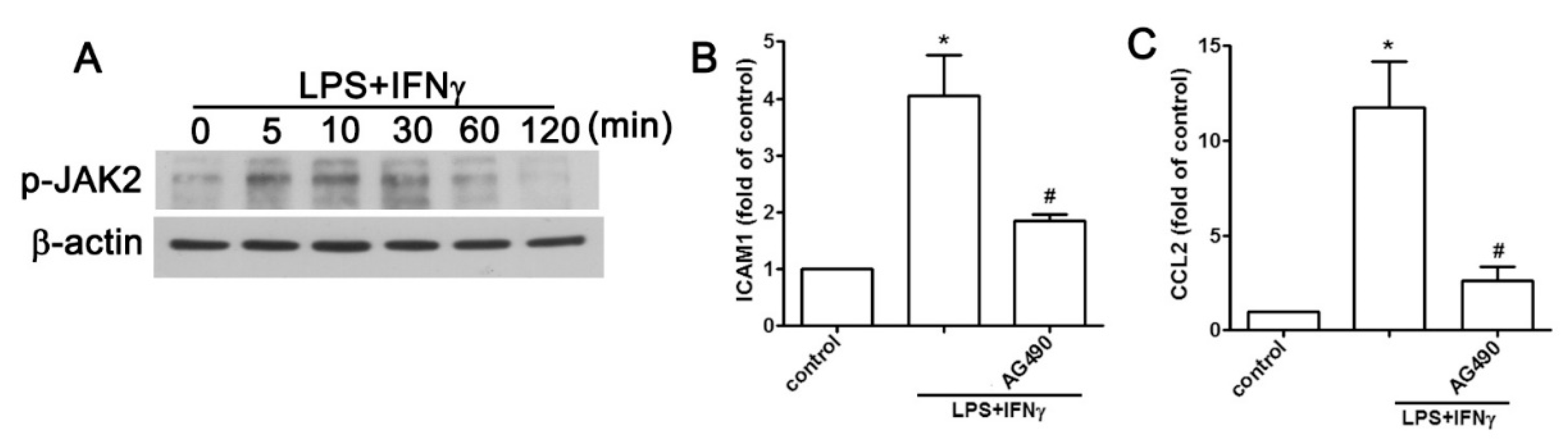
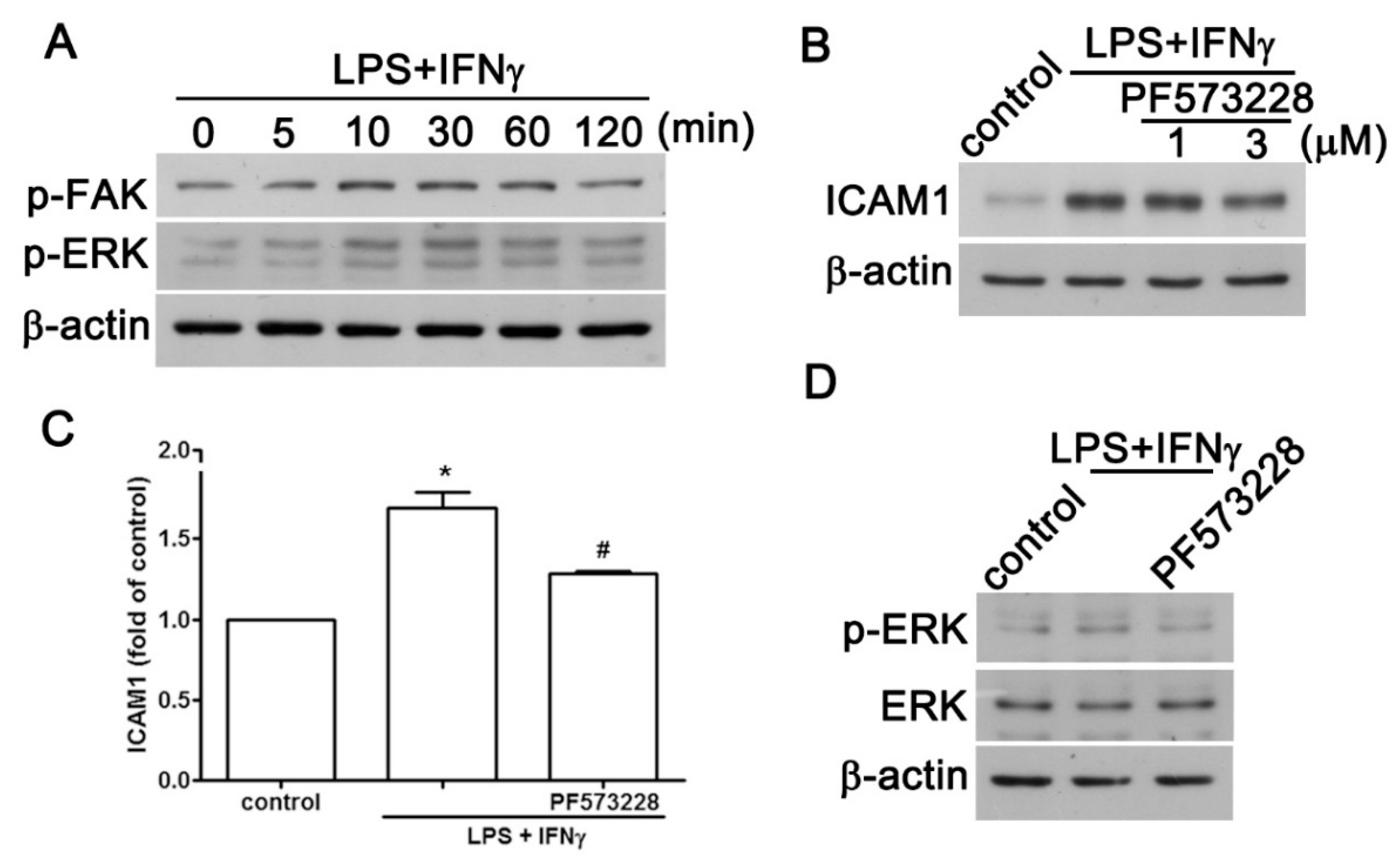
2.2. JAK2 and FAK/ERK Pathways Regulate ICAM1 and CCL2 Expression
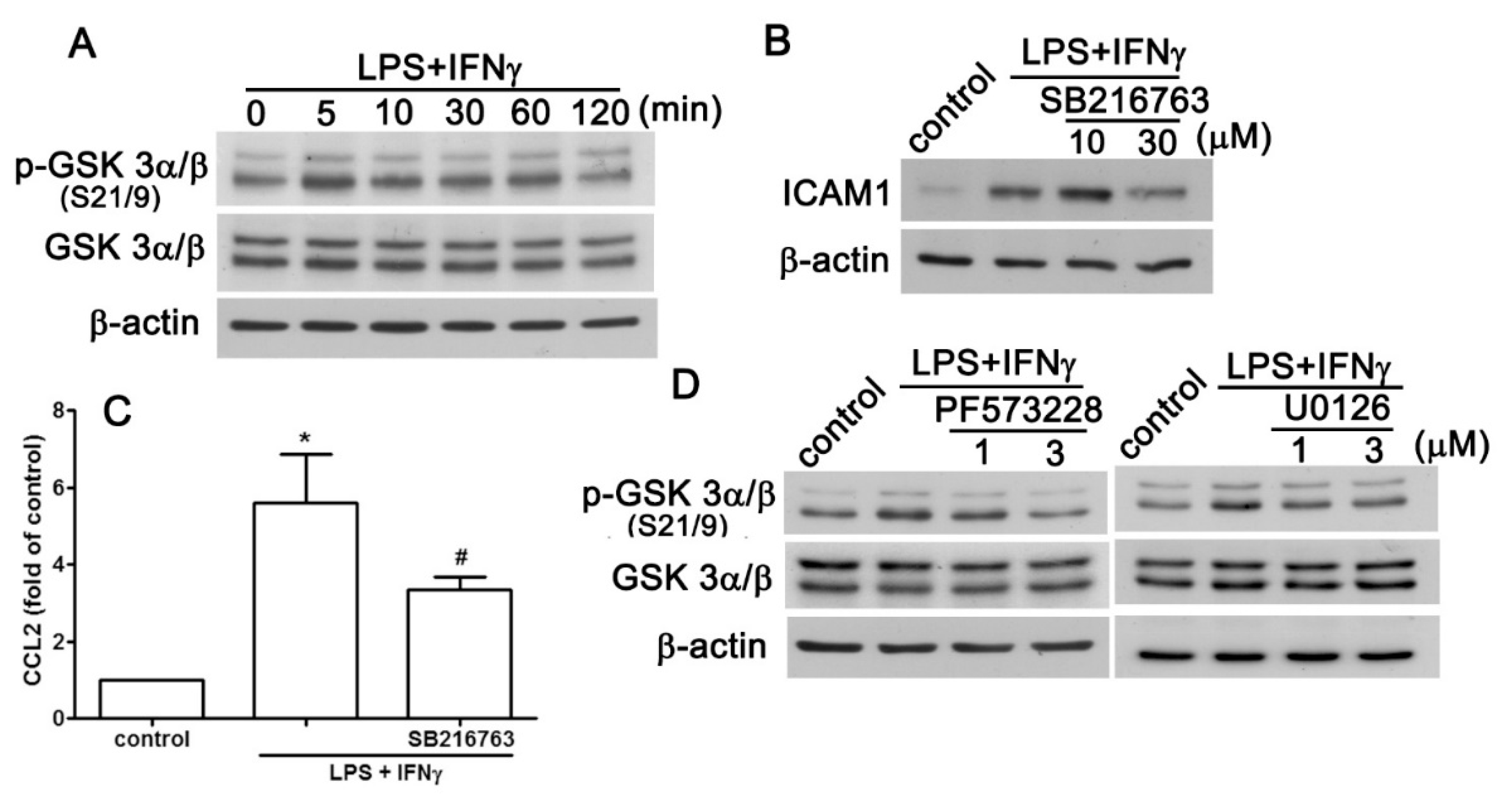
2.3. GSK3 Signaling Pathway is Involved in the Enhancement of CCL2/ICAM1
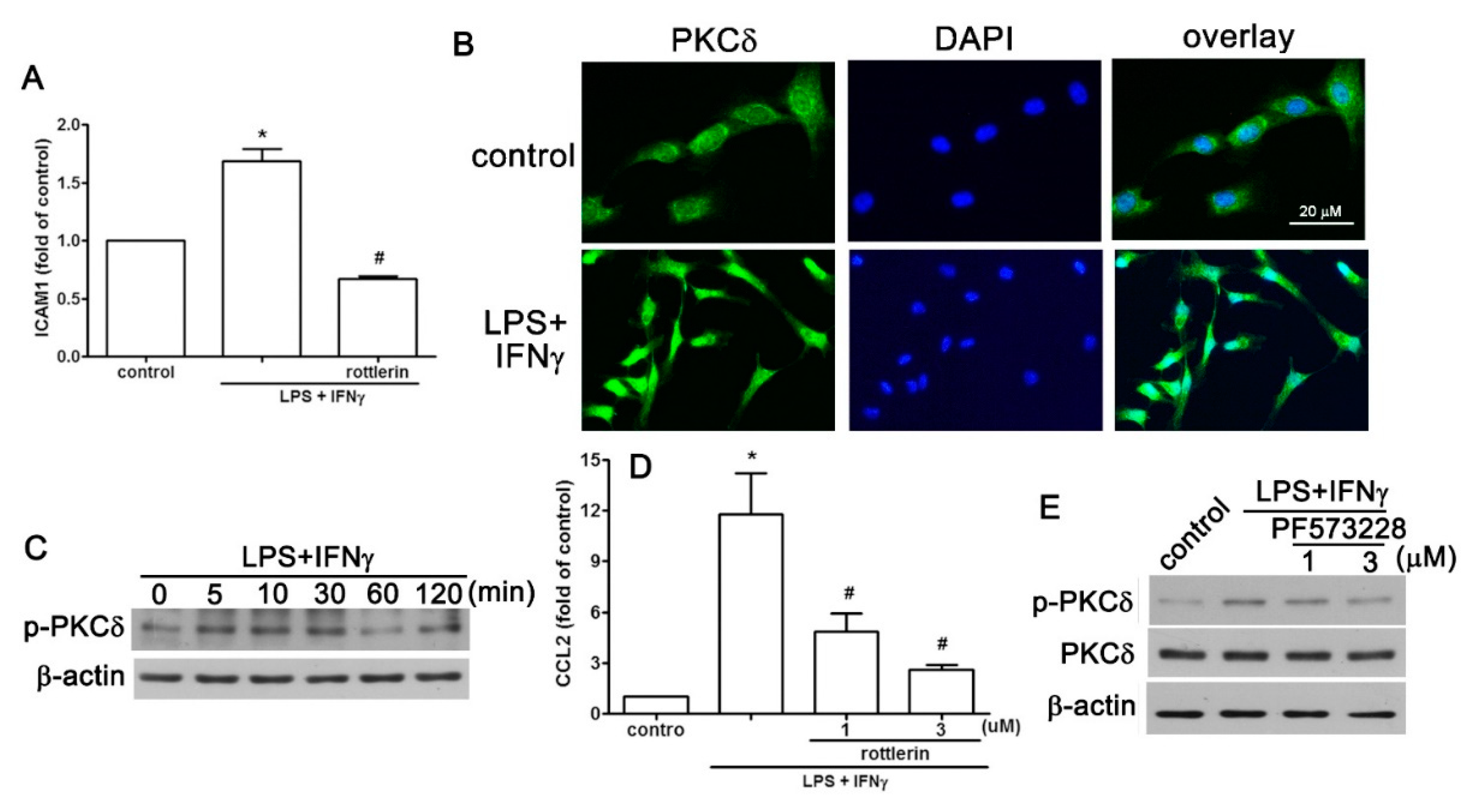
2.4. PKCδ is Involved in LPS Plus IFNγ-Induced Expression of CCL2/ICAM1 in AF Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Human AF Cells
4.3. Western Blot Analysis
4.4. RNA Extraction and Quantitative Real-Time PCR
4.5. Immunocytofluorescence Staining
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coppes, M.H.; Marani, E.; Thomeer, R.T.; Groen, G.J. Innervation of “painful” lumbar discs. Spine 1997, 22, 2342–2349. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.A.; McNally, D.S.; Dolan, P. ‘Stress’ distributions inside intervertebral discs. The effects of age and degeneration. J. Bone Jt. Surg. Br. 1996, 78, 965–972. [Google Scholar] [CrossRef]
- Khan, A.N.; Jacobsen, H.E.; Khan, J.; Filippi, C.G.; Levine, M.; Lehman, R.A., Jr.; Riew, K.D.; Lenke, L.G.; Chahine, N.O. Inflammatory biomarkers of low back pain and disc degeneration: A review. Ann. N. Y. Acad. Sci. 2017, 1410, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Le Maitre, C.L.; Hoyland, J.A.; Freemont, A.J. Catabolic cytokine expression in degenerate and herniated human intervertebral discs: IL-1beta and TNFalpha expression profile. Arthritis Res. Ther. 2007, 9, R77. [Google Scholar] [CrossRef] [PubMed]
- Mulleman, D.; Mammou, S.; Griffoul, I.; Watier, H.; Goupille, P. Pathophysiology of disk-related sciatica. I.--Evidence supporting a chemical component. Jt. Bone Spine 2006, 73, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Olmarker, K.; Rydevik, B.; Nordborg, C. Autologous nucleus pulposus induces neurophysiologic and histologic changes in porcine cauda equina nerve roots. Spine 1993, 18, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Shamji, M.F.; Setton, L.A.; Jarvis, W.; So, S.; Chen, J.; Jing, L.; Bullock, R.; Isaacs, R.E.; Brown, C.; Richardson, W.J. Proinflammatory cytokine expression profile in degenerated and herniated human intervertebral disc tissues. Arthritis Rheum. 2010, 62, 1974–1982. [Google Scholar]
- Cooper, R.G.; Freemont, A.J. TNF-alpha blockade for herniated intervertebral disc-induced sciatica: A way forward at last? Rheumatology 2004, 43, 119–121. [Google Scholar] [CrossRef]
- Takahashi, H.; Suguro, T.; Okazima, Y.; Motegi, M.; Okada, Y.; Kakiuchi, T. Inflammatory cytokines in the herniated disc of the lumbar spine. Spine 1996, 21, 218–224. [Google Scholar] [CrossRef]
- Kobayashi, S.; Meir, A.; Kokubo, Y.; Uchida, K.; Takeno, K.; Miyazaki, T.; Yayama, T.; Kubota, M.; Nomura, E.; Mwaka, E.; et al. Ultrastructural analysis on lumbar disc herniation using surgical specimens: Role of neovascularization and macrophages in hernias. Spine 2009, 34, 655–662. [Google Scholar] [CrossRef]
- Diamond, M.S.; Staunton, D.E.; de Fougerolles, A.R.; Stacker, S.A.; Garcia-Aguilar, J.; Hibbs, M.L.; Springer, T.A. ICAM-1 (CD54): A counter-receptor for Mac-1 (CD11b/CD18). J. Cell Biol. 1990, 111 Pt 2, 3129–3139. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A. Adhesion receptors of the immune system. Nature 1990, 346, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, J.F.; Sun, Z.Y.; Tian, J.W. Bioinformatics analysis of the gene expression profiles in human intervertebral disc degeneration associated with inflammatory cytokines. J. Neurosurg. Sci. 2018, 62, 16–23. [Google Scholar] [PubMed]
- Doita, M.; Kanatani, T.; Harada, T.; Mizuno, K. Immunohistologic study of the ruptured intervertebral disc of the lumbar spine. Spine 1996, 21, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Kulbe, H.; Levinson, N.R.; Balkwill, F.; Wilson, J.L. The chemokine network in cancer--much more than directing cell movement. Int. J. Dev. Biol. 2004, 48, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Karli, P.; Martle, V.; Bossens, K.; Summerfield, A.; Doherr, M.G.; Turner, P.; Vandevelde, M.; Forterre, F.; Henke, D. Dominance of chemokine ligand 2 and matrix metalloproteinase-2 and -9 and suppression of pro-inflammatory cytokines in the epidural compartment after intervertebral disc extrusion in a canine model. Spine J. 2014, 14, 2976–2984. [Google Scholar] [CrossRef]
- Zhu, X.; Cao, S.; Zhu, M.D.; Liu, J.Q.; Chen, J.J.; Gao, Y.J. Contribution of chemokine CCL2/CCR2 signaling in the dorsal root ganglion and spinal cord to the maintenance of neuropathic pain in a rat model of lumbar disc herniation. J. Pain 2014, 15, 516–526. [Google Scholar] [CrossRef]
- Wang, J.; Tian, Y.; Phillips, K.L.; Chiverton, N.; Haddock, G.; Bunning, R.A.; Cross, A.K.; Shapiro, I.M.; Le Maitre, C.L.; Risbud, M.V. Tumor necrosis factor alpha- and interleukin-1beta-dependent induction of CCL3 expression by nucleus pulposus cells promotes macrophage migration through CCR1. Arthritis Rheum. 2013, 65, 832–842. [Google Scholar] [CrossRef]
- Ni, L.; Zheng, Y.; Gong, T.; Xiu, C.; Li, K.; Saijilafu; Li, B.; Yang, H.; Chen, J. Proinflammatory macrophages promote degenerative phenotypes in rat nucleus pulpous cells partly through ERK and JNK signaling. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Rannou, F. Treatment of degenerative disk disease: Fact or fiction? Jt. Bone Spine Rev. Rhum. 2009, 76, 619–620. [Google Scholar] [CrossRef]
- Sung, Y.Y.; Kim, H.K. Illicium verum extract suppresses IFN-gamma-induced ICAM-1 expression via blockade of JAK/STAT pathway in HaCaT human keratinocytes. J. Ethnopharmacol. 2013, 149, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Duzagac, F.; Inan, S.; Ela Simsek, F.; Acikgoz, E.; Guven, U.; Khan, S.A.; Rouhrazi, H.; Oltulu, F.; Aktug, H.; Erol, A.; et al. JAK/STAT pathway interacts with intercellular cell adhesion molecule (ICAM) and vascular cell adhesion molecule (VCAM) while prostate cancer stem cells form tumor spheroids. J. Buon 2015, 20, 1250–1257. [Google Scholar]
- Chuang, J.Y.; Chang, P.C.; Shen, Y.C.; Lin, C.; Tsai, C.F.; Chen, J.H.; Yeh, W.L.; Wu, L.H.; Lin, H.Y.; Liu, Y.S.; et al. Regulatory effects of fisetin on microglial activation. Molecules 2014, 19, 8820–8839. [Google Scholar] [CrossRef]
- Liu, S.Q.; Su, Y.J.; Qin, M.B.; Mao, Y.B.; Huang, J.A.; Tang, G.D. Sphingosine kinase 1 promotes tumor progression and confers malignancy phenotypes of colon cancer by regulating the focal adhesion kinase pathway and adhesion molecules. Int. J. Oncol. 2013, 42, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, M.P.; Madrigal, J.L.; Pujol, A.; Galea, E. JNK/ERK/FAK mediate promigratory actions of basic fibroblast growth factor in astrocytes via CCL2 and COX2. Neurosignals 2012, 20, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.R.; Chang, P.C.; Yeh, W.L.; Lee, C.H.; Tsai, C.F.; Lin, C.; Lin, H.Y.; Liu, Y.S.; Wu, C.Y.; Ko, P.Y.; et al. Anti-neuroinflammatory effects of the calcium channel blocker nicardipine on microglial cells: Implications for neuroprotection. PLoS ONE 2014, 9, e91167. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Lin, H.Y.; Chen, J.H.; Tseng, W.P.; Ko, P.Y.; Liu, Y.S.; Yeh, W.L.; Lu, D.Y. Effects of paeonol on anti-neuroinflammatory responses in microglial cells. Int. J. Mol. Sci. 2015, 16, 8844–8860. [Google Scholar] [CrossRef]
- Tsoyi, K.; Jang, H.J.; Nizamutdinova, I.T.; Park, K.; Kim, Y.M.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chang, K.C. PTEN differentially regulates expressions of ICAM-1 and VCAM-1 through PI3K/Akt/GSK-3beta/GATA-6 signaling pathways in TNF-alpha-activated human endothelial cells. Atherosclerosis 2010, 213, 115–121. [Google Scholar] [CrossRef]
- Chen, P.C.; Lin, T.H.; Cheng, H.C.; Tang, C.H. CCN3 increases cell motility and ICAM-1 expression in prostate cancer cells. Carcinogenesis 2012, 33, 937–945. [Google Scholar] [CrossRef] [Green Version]
- Minhajuddin, M.; Bijli, K.M.; Fazal, F.; Sassano, A.; Nakayama, K.I.; Hay, N.; Platanias, L.C.; Rahman, A. Protein kinase C-delta and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-kappaB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells. J. Biol. Chem. 2009, 284, 4052–4061. [Google Scholar] [CrossRef]
- Song, S.; Choi, K.; Ryu, S.W.; Kang, S.W.; Choi, C. TRAIL promotes caspase-dependent pro-inflammatory responses via PKCdelta activation by vascular smooth muscle cells. Cell Death Dis. 2011, 2, e223. [Google Scholar] [CrossRef] [PubMed]
- Luoma, K.; Riihimaki, H.; Luukkonen, R.; Raininko, R.; Viikari-Juntura, E.; Lamminen, A. Low back pain in relation to lumbar disc degeneration. Spine 2000, 25, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.S.; Carlin, D.; Mulholland, R.C. Lumbar disc high-intensity zone: The value and significance of provocative discography in the determination of the discogenic pain source. Eur. Spine J. 2000, 9, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Fairbank, J. Clinical importance of the intervertebral disc, or back pain for biochemists. Biochem. Soc. Trans. 2002, 30 Pt 6, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Luoma, K.; Vehmas, T.; Raininko, R.; Luukkonen, R.; Riihimaki, H. Lumbosacral transitional vertebra: Relation to disc degeneration and low back pain. Spine 2004, 29, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Creemers, L. Mechanisms of intervertebral disk degeneration/injury and pain: A review. Glob. Spine J. 2013, 3, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Awad, J.N.; Moskovich, R. Lumbar disc herniations: Surgical versus nonsurgical treatment. Clin. Orthop. Relat. Res. 2006, 443, 183–197. [Google Scholar] [CrossRef]
- Huang, B.R.; Tsai, C.F.; Lin, H.Y.; Tseng, W.P.; Huang, S.S.; Wu, C.R.; Lin, C.; Yeh, W.L.; Lu, D.Y. Interaction of inflammatory and anti-inflammatory responses in microglia by Staphylococcus aureus-derived lipoteichoic acid. Toxicol. Appl. Pharmacol. 2013, 269, 43–50. [Google Scholar] [CrossRef]
- Ohtori, S.; Miyagi, M.; Eguchi, Y.; Inoue, G.; Orita, S.; Ochiai, N.; Kishida, S.; Kuniyoshi, K.; Nakamura, J.; Aoki, Y.; et al. Efficacy of epidural administration of anti-interleukin-6 receptor antibody onto spinal nerve for treatment of sciatica. Eur. Spine J. 2012, 21, 2079–2084. [Google Scholar] [CrossRef] [Green Version]
- Ohtori, S.; Miyagi, M.; Eguchi, Y.; Inoue, G.; Orita, S.; Ochiai, N.; Kishida, S.; Kuniyoshi, K.; Nakamura, J.; Aoki, Y.; et al. Epidural administration of spinal nerves with the tumor necrosis factor-alpha inhibitor, etanercept, compared with dexamethasone for treatment of sciatica in patients with lumbar spinal stenosis: A prospective randomized study. Spine 2012, 37, 439–444. [Google Scholar] [CrossRef]
- Genevay, S.; Viatte, S.; Finckh, A.; Zufferey, P.; Balague, F.; Gabay, C. Adalimumab in severe and acute sciatica: A multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010, 62, 2339–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korhonen, T.; Karppinen, J.; Paimela, L.; Malmivaara, A.; Lindgren, K.A.; Bowman, C.; Hammond, A.; Kirkham, B.; Jarvinen, S.; Niinimaki, J.; et al. The treatment of disc-herniation-induced sciatica with infliximab: One-year follow-up results of FIRST II, a randomized controlled trial. Spine 2006, 31, 2759–2766. [Google Scholar] [CrossRef] [PubMed]
- Virri, J.; Sikk, S.; Gronblad, M.; Tolonen, J.; Seitsalo, S.; Kankare, J.; Karaharju, E.O. Concomitant immunocytochemical study of macrophage cells and blood vessels in disc herniation tissue. Eur. Spine J. 1994, 3, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Koike, Y.; Uzuki, M.; Kokubun, S.; Sawai, T. Angiogenesis and inflammatory cell infiltration in lumbar disc herniation. Spine 2003, 28, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar] [PubMed]
- Gronblad, M.; Virri, J.; Tolonen, J.; Seitsalo, S.; Kaapa, E.; Kankare, J.; Myllynen, P.; Karaharju, E.O. A controlled immunohistochemical study of inflammatory cells in disc herniation tissue. Spine 1994, 19, 2744–2751. [Google Scholar] [CrossRef] [PubMed]
- Rajan, N.E.; Bloom, O.; Maidhof, R.; Stetson, N.; Sherry, B.; Levine, M.; Chahine, N.O. Toll-Like Receptor 4 (TLR4) expression and stimulation in a model of intervertebral disc inflammation and degeneration. Spine 2013, 38, 1343–1351. [Google Scholar] [CrossRef]
- Chin, J.E.; Winterrowd, G.E.; Krzesicki, R.F.; Sanders, M.E. Role of cytokines in inflammatory synovitis. The coordinate regulation of intercellular adhesion molecule 1 and HLA class I and class II antigens in rheumatoid synovial fibroblasts. Arthritis Rheum. 1990, 33, 1776–1786. [Google Scholar] [CrossRef]
- Gabr, M.A.; Jing, L.; Helbling, A.R.; Sinclair, S.M.; Allen, K.D.; Shamji, M.F.; Richardson, W.J.; Fitch, R.D.; Setton, L.A.; Chen, J. Interleukin-17 synergizes with IFNgamma or TNFalpha to promote inflammatory mediator release and intercellular adhesion molecule-1 (ICAM-1) expression in human intervertebral disc cells. J. Orthop. Res. 2011, 29, 1–7. [Google Scholar] [CrossRef]
- Phillips, K.L.; Chiverton, N.; Michael, A.L.; Cole, A.A.; Breakwell, L.M.; Haddock, G.; Bunning, R.A.; Cross, A.K.; Le Maitre, C.L. The cytokine and chemokine expression profile of nucleus pulposus cells: Implications for degeneration and regeneration of the intervertebral disc. Arthritis Res. Ther. 2013, 15, R213. [Google Scholar] [CrossRef]
- Langert, K.A.; Von Zee, C.L.; Stubbs, E.B., Jr. Tumour necrosis factor alpha enhances CCL2 and ICAM-1 expression in peripheral nerve microvascular endoneurial endothelial cells. ASN Neuro 2013, 5, e00104. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.N.; Norden, D.M.; Faw, T.D.; Deibert, R.; Wohleb, E.S.; Sheridan, J.F.; Godbout, J.P.; Basso, D.M. Lumbar Myeloid Cell Trafficking into Locomotor Networks after Thoracic Spinal Cord Injury. Exp. Neurol. 2016, 282, 86–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, N.; Lachnit, N.; Streppel, M.; Witter, B.; Neiss, W.F.; Guntinas-Lichius, O.; Angelov, D.N. Increased expression of ICAM-1, VCAM-1, MCP-1, and MIP-1 alpha by spinal perivascular macrophages during experimental allergic encephalomyelitis in rats. BMC Immunol. 2002, 3, 11. [Google Scholar] [CrossRef]
- Takahashi, M.; Miyazaki, H.; Furihata, M.; Sakai, H.; Konakahara, T.; Watanabe, M.; Okada, T. Chemokine CCL2/MCP-1 negatively regulates metastasis in a highly bone marrow-metastatic mouse breast cancer model. Clin. Exp. Metastasis 2009, 26, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Kaidoh, M.; Yokoyama, Y.; Sano, K.; Ohhashi, T. Chemokine CCL2 facilitates ICAM-1-mediated interactions of cancer cells and lymphatic endothelial cells in sentinel lymph nodes. Cancer Sci. 2009, 100, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C., Jr.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stambolic, V.; Woodgett, J.R. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 1994, 303 Pt 3, 701–704. [Google Scholar] [CrossRef]
- Lakshmanan, J.; Zhang, B.; Nweze, I.C.; Du, Y.; Harbrecht, B.G. Glycogen Synthase Kinase 3 Regulates IL-1beta Mediated iNOS Expression in Hepatocytes by Down-Regulating c-Jun. J. Cell. Biochem. 2015, 116, 133–141. [Google Scholar] [CrossRef]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef]
- Cheng, Y.L.; Wang, C.Y.; Huang, W.C.; Tsai, C.C.; Chen, C.L.; Shen, C.F.; Chi, C.Y.; Lin, C.F. Staphylococcus aureus induces microglial inflammation via a glycogen synthase kinase 3beta-regulated pathway. Infect. Immun. 2009, 77, 4002–4008. [Google Scholar] [CrossRef]
- Cremer, T.J.; Shah, P.; Cormet-Boyaka, E.; Valvano, M.A.; Butchar, J.P.; Tridandapani, S. Akt-mediated proinflammatory response of mononuclear phagocytes infected with Burkholderia cenocepacia occurs by a novel GSK3beta-dependent, IkappaB kinase-independent mechanism. J. Immunol. 2011, 187, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Jing, D.; Bai, H.; Yin, S. Renoprotective effects of emodin against diabetic nephropathy in rat models are mediated via PI3K/Akt/GSK-3beta and Bax/caspase-3 signaling pathways. Exp. Ther. Med. 2017, 14, 5163–5169. [Google Scholar] [PubMed]
- Lappas, M. GSK3beta is increased in adipose tissue and skeletal muscle from women with gestational diabetes where it regulates the inflammatory response. PLoS ONE 2014, 9, e115854. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Tsai, C.H.; Lin, C.; Yeh, W.L.; Tsai, C.F.; Chang, P.C.; Wu, L.H.; Lu, D.Y. Cobalt Protoporphyrin Upregulates Cyclooxygenase-2 Expression Through a Heme Oxygenase-Independent Mechanism. Mol. Neurobiol. 2016, 53, 4497–4508. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Forman, L.W.; Williams, R.M.; Faller, D.V. Protein kinase C-delta inactivation inhibits the proliferation and survival of cancer stem cells in culture and in vivo. BMC Cancer 2014, 14, 90. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.C.; Jeon, W.K.; Hong, H.Y.; Jeon, K.B.; Hahn, J.H.; Kim, Y.M.; Numazawa, S.; Yosida, T.; Park, E.H.; Lim, C.J. The anti-inflammatory activity of Phellinus linteus (Berk. & M.A. Curt.) is mediated through the PKCdelta/Nrf2/ARE signaling to up-regulation of heme oxygenase-1. J. Ethnopharmacol. 2007, 113, 240–247. [Google Scholar] [PubMed]
- Han, Y.S.; Zheng, W.H.; Bastianetto, S.; Chabot, J.G.; Quirion, R. Neuroprotective effects of resveratrol against beta-amyloid-induced neurotoxicity in rat hippocampal neurons: Involvement of protein kinase C. Br. J. Pharmacol. 2004, 141, 997–1005. [Google Scholar] [CrossRef]
- Wu, L.H.; Lin, C.; Lin, H.Y.; Liu, Y.S.; Wu, C.Y.; Tsai, C.F.; Chang, P.C.; Yeh, W.L.; Lu, D.Y. Naringenin Suppresses Neuroinflammatory Responses Through Inducing Suppressor of Cytokine Signaling 3 Expression. Mol. Neurobiol. 2016, 53, 1080–1091. [Google Scholar] [CrossRef]
- Ellman, M.B.; Kim, J.S.; An, H.S.; Kroin, J.S.; Li, X.; Chen, D.; Yan, D.; Buechter, D.D.; Nakayama, K.; Liu, B.; et al. The pathophysiologic role of the protein kinase Cdelta pathway in the intervertebral discs of rabbits and mice: In vitro, ex vivo, and in vivo studies. Arthritis Rheum. 2012, 64, 1950–1959. [Google Scholar] [CrossRef]
- Yokoyama, K.; Hiyama, A.; Arai, F.; Nukaga, T.; Sakai, D.; Mochida, J. C-Fos regulation by the MAPK and PKC pathways in intervertebral disc cells. PLoS ONE 2013, 8, e73210. [Google Scholar] [CrossRef]
- Huang, B.R.; Chen, T.S.; Bau, D.T.; Chuang, I.C.; Tsai, C.F.; Chang, P.C.; Lu, D.Y. EGFR is a pivotal regulator of thrombin-mediated inflammation in primary human nucleus pulposus culture. Sci. Rep. 2017, 7, 8578. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, B.-R.; Bau, D.-T.; Chen, T.-S.; Chuang, I.-C.; Tsai, C.-F.; Chang, P.-C.; Hsu, H.-C.; Lu, D.-Y. Pro-Inflammatory Stimuli Influence Expression of Intercellular Adhesion Molecule 1 in Human Anulus Fibrosus Cells through FAK/ERK/GSK3 and PKCδ Signaling Pathways. Int. J. Mol. Sci. 2019, 20, 77. https://doi.org/10.3390/ijms20010077
Huang B-R, Bau D-T, Chen T-S, Chuang I-C, Tsai C-F, Chang P-C, Hsu H-C, Lu D-Y. Pro-Inflammatory Stimuli Influence Expression of Intercellular Adhesion Molecule 1 in Human Anulus Fibrosus Cells through FAK/ERK/GSK3 and PKCδ Signaling Pathways. International Journal of Molecular Sciences. 2019; 20(1):77. https://doi.org/10.3390/ijms20010077
Chicago/Turabian StyleHuang, Bor-Ren, Da-Tian Bau, Tzu-Sheng Chen, I-Chen Chuang, Cheng-Fang Tsai, Pei-Chun Chang, Horng-Chaung Hsu, and Dah-Yuu Lu. 2019. "Pro-Inflammatory Stimuli Influence Expression of Intercellular Adhesion Molecule 1 in Human Anulus Fibrosus Cells through FAK/ERK/GSK3 and PKCδ Signaling Pathways" International Journal of Molecular Sciences 20, no. 1: 77. https://doi.org/10.3390/ijms20010077