Metformin Results in Diametrically Opposed Effects by Targeting Non-Stem Cancer Cells but Protecting Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma

Abstract
:1. Introduction
2. Results
2.1. Metformin Mitigates Cisplatin-Mediated Cell Death in HNSCC CSCs but Reduces Cell Proliferation in Non-Stem HNSCC Cells
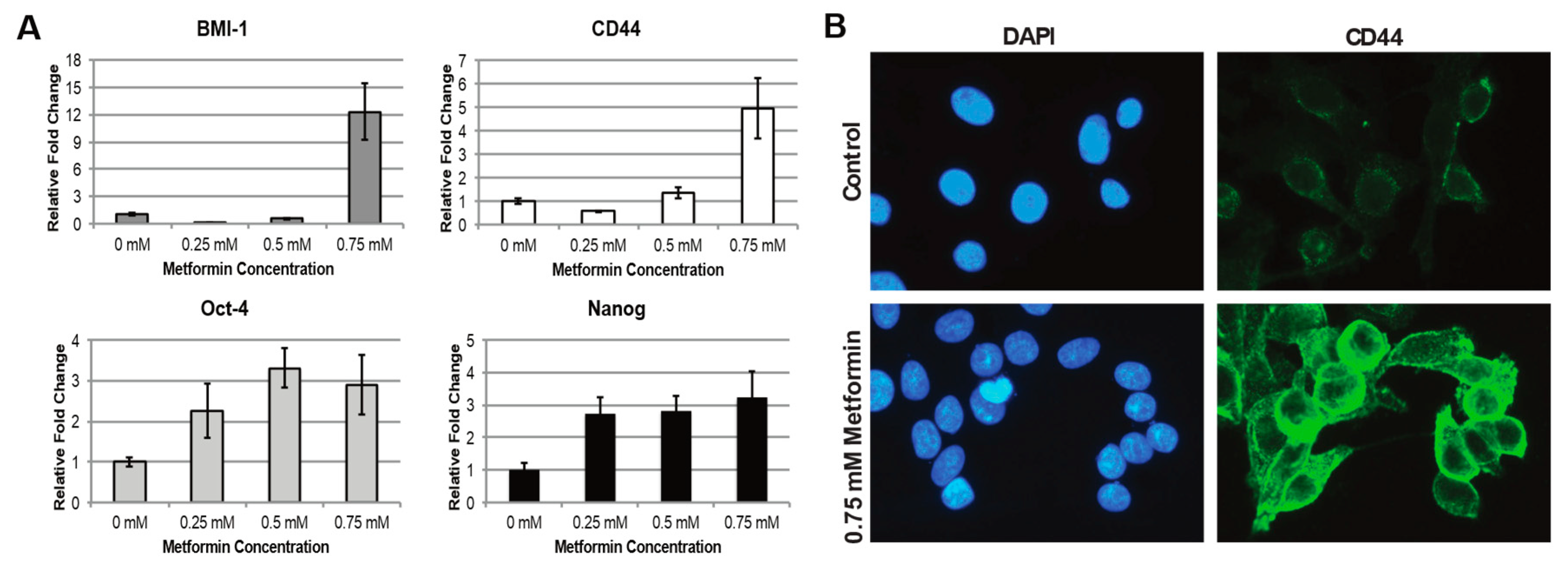
2.2. Metformin Increases Stem Cell Characteristics in HNSCC CSC Population
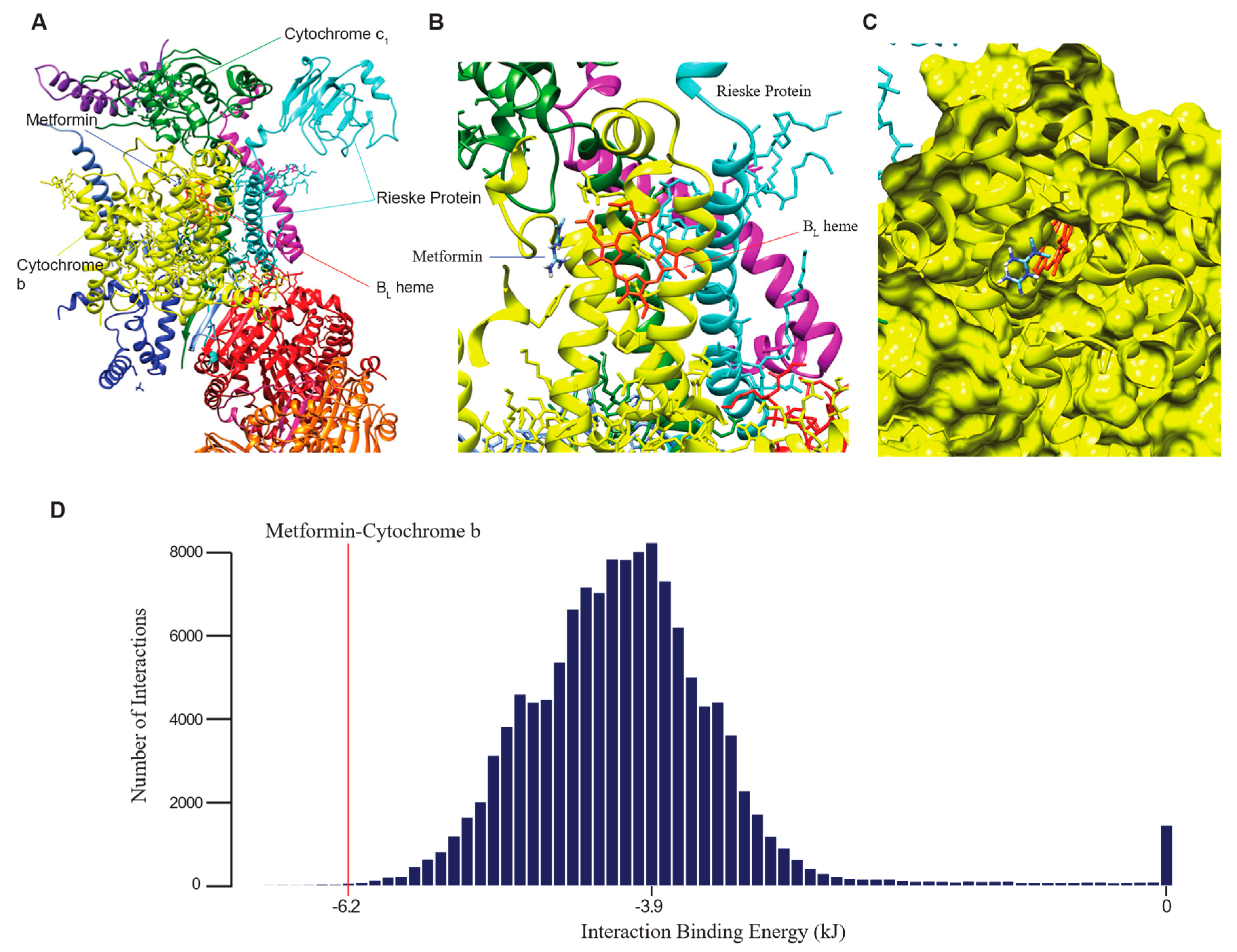
2.3. Computational Binding Analysis Reveals Strong Interaction of Metformin with Mitochondrial Complex III
2.4. Expressions of Complex III Genes Correlate with Clinical Variables and Stem Cell Marker Expressions
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cultures
4.2. FACS Identification of ALDH+ and ALDH- Cell Populations
4.3. Cell Proliferation Assay
4.4. TUNEL Assay
4.5. Western Blot Analysis
4.6. Quantitative Real-Time PCR and siRNA Knockdown
4.7. Immunofluorescence
4.8. Computational Prediction of Metformin Binding Energy
4.9. Correlation of Complex III Gene Expressions to Survival and Histologic Grade
4.10. Correlation of Complex III Gene Expressions to Expressions of Stem Cell Markers Using TCGA Data
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CSCs | Cancer stem cells |
| HNSCC | Head and neck squamous cell carcinoma |
| ROS | Reactive oxygen species |
References
- Dowling, R.J.O.; Niraula, S.; Stambolic, V.; Goodwin, P.J. Metformin in cancer: Translational challenges. J. Mol. Endocrinol. 2012, 48, R31–R43. [Google Scholar] [CrossRef] [PubMed]
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.-C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 2009, 27, 3297–3302. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Shank, J.J.; Yang, K.; Ghannam, J.; Cabrera, L.; Johnston, C.J.; Reynolds, R.K.; Buckanovich, R.J. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol. Oncol. 2012, 127, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Bednar, F.; Simeone, D.M. Metformin and cancer stem cells: Old drug, new targets. Cancer Prev. Res. 2012, 5, 351–354. [Google Scholar] [CrossRef]
- Iglesias-Bartolome, R.; Martin, D.; Gutkind, J.S. Exploiting the head and neck cancer oncogenome: Widespread PI3K-mTOR pathway alterations and novel molecular targets. Cancer Discov. 2013, 3, 722–725. [Google Scholar] [CrossRef]
- Fontaine, E. Metformin and respiratory chain complex I: The last piece of the puzzle? Biochem. J. 2014, 463, e3–e5. [Google Scholar] [CrossRef]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta-Bioenerg. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [Green Version]
- Batandier, C.; Guigas, B.; Detaille, D.; El-Mir, M.-Y.; Fontaine, E.; Rigoulet, M.; Leverve, X.M. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J. Bioenerg. Biomembr. 2006, 38, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Algire, C.; Moiseeva, O.; Deschênes-Simard, X.; Amrein, L.; Petruccelli, L.; Birman, E.; Viollet, B.; Ferbeyre, G.; Pollak, M.N. Metformin Reduces Endogenous Reactive Oxygen Species and Associated DNA Damage. Cancer Prev. Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, C.I.; Suda, T. Regulation of reactive oxygen species in stem cells and cancer stem cells. J. Cell. Physiol. 2012, 227, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [Green Version]
- Schumacker, P.T. Reactive oxygen species in cancer: A dance with the devil. Cancer Cell 2015, 27, 156–157. [Google Scholar] [CrossRef]
- Kuo, S.Z.; Blair, K.J.; Rahimy, E.; Kiang, A.; Abhold, E.; Fan, J.-B.; Wang-Rodriguez, J.; Altuna, X.; Ongkeko, W.M. Salinomycin induces cell death and differentiation in head and neck squamous cell carcinoma stem cells despite activation of epithelial-mesenchymal transition and Akt. BMC Cancer 2012, 12, 556. [Google Scholar] [CrossRef]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007. [Google Scholar] [CrossRef]
- Clay, M.R.; Tabor, M.; Owen, J.H.; Carey, T.E.; Bradford, C.R.; Wolf, G.T.; Wicha, M.S.; Prince, M.E. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck 2010, 32, 1195–1201. [Google Scholar] [CrossRef] [Green Version]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Loh, Y.H.; Wu, Q.; Chew, J.L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Mohanta, S.; Siddappa, G.; Valiyaveedan, S.G.; Dodda Thimmasandra Ramanjanappa, R.; Das, D.; Pandian, R.; Khora, S.S.; Kuriakose, M.A.; Suresh, A. Cancer stem cell markers in patterning differentiation and in prognosis of oral squamous cell carcinoma. Tumor Biol. 2017, 39, 1010428317703656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Filho, M.S.; Nör, J.E. The biology of head and neck cancer stem cells. Oral Oncol. 2012, 48, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Major, A.G.; Pitty, L.P.; Farah, C.S. Cancer stem cell markers in head and neck squamous cell carcinoma. Stem Cells Int. 2013, 2013, 319489. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A.; Powers, M.A.; Dellavalle, R.P. Therapeutic potential of the anti-diabetic agent metformin in targeting the skin cancer stem cell diaspora. Exp. Dermatol. 2014, 23, 345–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rêgo, D.F.; Elias, S.T.; Amato, A.A.; Canto, G.D.L.; Guerra, E.N.S. Anti-tumor effects of metformin on head and neck carcinoma cell lines: A systematic review. Oncol. Lett. 2017, 13, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Sikka, A.; Kaur, M.; Agarwal, C.; Deep, G.; Agarwal, R. Metformin suppresses growth of human head and neck squamous cell carcinoma via global inhibition of protein translation. Cell Cycle 2012, 11, 1374–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.S.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J.; et al. V HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 2007, 12, 230–238. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Gormsen, L.C.; Sundelin, E.I.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Hougaard Christensen, M.M.; Brosen, K.; Frokiaer, J.; Jessen, N. In Vivo Imaging of Human 11C-Metformin in Peripheral Organs: Dosimetry, Biodistribution, and Kinetic Analyses. J. Nucl. Med. 2016, 57, 1920–1926. [Google Scholar] [CrossRef]
- Schäfer, G. Site-specific uncoupling and inhibition of oxidative phosphorylation by biguanides. II. Biochim. Biophys. Acta 1969, 172, 334–337. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Terazawa, R.; Kojima, K.; Nakane, K.; Deguchi, T.; Ando, M.; Tsukamasa, Y.; Ito, M.; Nozawa, Y. Cisplatin induces production of reactive oxygen species via NADPH oxidase activation in human prostate cancer cells. Free Radic. Res. 2011, 45, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial Complex II Can Generate Reactive Oxygen Species at High Rates in Both the Forward and Reverse Reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [Green Version]
- Abhold, E.L.; Kiang, A.; Rahimy, E.; Kuo, S.Z.; Wang-Rodriguez, J.; Lopez, J.P.; Blair, K.J.; Yu, M.A.; Haas, M.; Brumund, K.T.; et al. EGFR kinase promotes acquisition of stem cell-like properties: A potential therapeutic target in head and neck squamous cell carcinoma stem cells. PLoS ONE 2012. [Google Scholar] [CrossRef]
- Amporndanai, K.; Johnson, R.M.; O’Neill, P.M.; Fishwick, C.W.G.; Jamson, A.H.; Rawson, S.; Muench, S.P.; Samar Hasnain, S.; Antonyuk, S.V. X-ray and cryo-EM structures of inhibitor-bound cytochrome bc 1 complexes for structure-based drug discovery. IUCrJ 2018. [Google Scholar] [CrossRef]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tufféry, P. Fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Towns, J.; Cockerill, T.; Dahan, M.; Foster, I.; Gaither, K.; Grimshaw, A.; Hazlewood, V.; Lathrop, S.; Lifka, D.; Peterson, G.D.; et al. XSEDE: Accelerating scientific discovery. Comput. Sci. Eng. 2014. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD44 | forward: | 5′-AGAAGAAAGCCAGTGCGTCT-3′ |
| CD44 | reverse: | 5′-TGACCTAAGACGGAGGGAGG-3′ |
| GAPDH | forward: | 5′-TTCTTTTGCGTCGCCAGCC-3′ |
| GAPDH | reverse: | 5′-CGTTCTCAGCCTTGACGGTG-3′ |
| BMI1 | forward: | 5′-CGAGACAATGGGGATGTGGG-3′ |
| BMI1 | reverse: | 5′-AAATGAATGCGAGCCAAGCG-3′ |
| ALDH1A1 | forward: | 5′-CACGCCAGACTTACCTGTCC-3′ |
| ALDH1A1 | reverse: | 5′-TTGTACGGCCCTGGATCTTG-3′ |
| NANOG | forward: | 5′-AATGGTGTGACGCAGGGATG-3′ |
| NANOG | reverse: | 5′-ACCTCGCTGATTAGGCTCCA-3′ |
| POU5F1 | forward: | 5′-TCCCGAATGGAAAGGGGAGA-3′ |
| POU5F1 | reverse: | 5′-GGCTGAATACCTTCCCAAATAGA-3′ |
| ABCG2 | forward: | 5′-TTACGCACAGAGCAAAGCCA-3′ |
| ABCG2 | reverse: | 5′-GCAAGGGGCTAGAAGAAGGG-3′ |
| PROM1 | forward: | 5′-GAATCCTTTCCATTACGGCGG-3′ |
| PROM1 | reverse: | 5′-CCTGAAAAGGAGTTCCCGCA-3′ |
| LGR5 | forward: | 5′-GGAGTTACGTCTTGCGGGAA-3′ |
| LGR5 | reverse: | 5′-CAGGCCACTGAAACAGCTTG-3′. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, S.Z.; Honda, C.O.; Li, W.T.; Honda, T.K.; Kim, E.; Altuna, X.; Abhold, E.; Wang-Rodriguez, J.; Ongkeko, W.M. Metformin Results in Diametrically Opposed Effects by Targeting Non-Stem Cancer Cells but Protecting Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 193. https://doi.org/10.3390/ijms20010193
Kuo SZ, Honda CO, Li WT, Honda TK, Kim E, Altuna X, Abhold E, Wang-Rodriguez J, Ongkeko WM. Metformin Results in Diametrically Opposed Effects by Targeting Non-Stem Cancer Cells but Protecting Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2019; 20(1):193. https://doi.org/10.3390/ijms20010193
Chicago/Turabian StyleKuo, Selena Z., Christine O. Honda, Wei Tse Li, Thomas K. Honda, Elizabeth Kim, Xabier Altuna, Eric Abhold, Jessica Wang-Rodriguez, and Weg M. Ongkeko. 2019. "Metformin Results in Diametrically Opposed Effects by Targeting Non-Stem Cancer Cells but Protecting Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma" International Journal of Molecular Sciences 20, no. 1: 193. https://doi.org/10.3390/ijms20010193
APA StyleKuo, S. Z., Honda, C. O., Li, W. T., Honda, T. K., Kim, E., Altuna, X., Abhold, E., Wang-Rodriguez, J., & Ongkeko, W. M. (2019). Metformin Results in Diametrically Opposed Effects by Targeting Non-Stem Cancer Cells but Protecting Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma. International Journal of Molecular Sciences, 20(1), 193. https://doi.org/10.3390/ijms20010193