Introduction
Transition-metal carbides and nitrides, commonly referred to as refractory hard metals, possess an unusual combination of physical and chemical properties[
1] which make them attractive from both a fundamental and technological point of view. They have high melting points and extreme hardness, properties which are typical of covalent crystals, as well as metallic conductivities comparable to those of pure transition metals. These materials have good corrosion resistance and some of them have shown high superconducting transition temperatures.[
2] Also they have attracted considerable interest as potential catalysts.[
3]
Most of investigations on carbides and nitrides compounds have been aimed to study their bulk properties, while the works devoted to the study of their surfaces are relatively scarce. The interest for the structural properties of these surfaces was stimulated when an analysis[
4] of the diffraction pattern of TaC revealed the existence of a rippled relaxation on the (001) surface. Similar relaxation characteristics on the same surface was also observed in HfC[
5] and VN.[
6] To generalize these surface observations and state that they are representative for this whole class of materials is however premature since only two carbide and one nitride of transition-metal have been reported. Such surface relaxations will be analyzed for a series of nitrides in a forthcoming paper.[
7]
From a theoretical point of view several works have been devoted to the analysis of the electronic structure of carbides and nitrides.[
8] Focusing in particular on TiN, band structure calculations have been reported using a wide variety of theoretical approaches.[
9,
10] Properties like lattice constants, and bulk modulus have also been theoretically estimated.[
11,
12,
13] Concerning the TiN surface, two recent works have been reported both of them using density functional theory based methods. The electronic structure of the (001) face was carefully examined by Li et al.[
13] from periodic slab calculations (Crystal95 [
14]) in which the surface was kept fixed. The structure of the low index (001), (110) and (111) surfaces including relaxation effects were analyzed by Marlo et al.[
12] from DFT calculations including generalized gradient approximation (GGA) and using plane waves (CASTEP [
15]) to expand the electronic states.
Because of their very special properties, these materials have also been considered as support to deposit metal transition clusters, both for technological purposes (sensors, electronic devices,…) and for catalysts. However, as far as we can ascertain, no theoretical results concerning transition metal deposition on TiN surfaces have been reported.
In this paper we report on a theoretical analysis of the Au/TiN interface based on DFT periodic GGA calculations. We have selected gold as transition metal due to the recently catalytic properties reported for this material.[
16]
Computational Details
In order to model the extended nature of the Au/TiN system, periodic 3D DFT calculations were carried out using the VASP 4.4.3 code.[
17,
18] In these calculations, the energy was obtained using the GGA implementation of DFT proposed by Perdew et al.[
19] Ultrasoft pseudopotentials[
20] were employed to remove the core electrons from the calculation and a plane-wave basis set was used. The cutoff energy for the plane waves was 225 eV and the Monkhorst-Pack set of four k points was used.
Forces on the ions are calculated through the Hellmann-Feyman theorem as the partial derivatives of the free energy with respect to the atomic position, including the Harris-Foulkes[
21] like correction to forces. This calculation of the forces allows a geometry optimization using the conjugate-gradient scheme. Iterative relaxation of atomic positions was stopped when the change in total energy between successive steps was less than 0.001 eV. With this criterion, forces on the atoms were generally less than 0.1 eV/ Å.
Titanium nitride is an instance of a diatomic solid AB having the rock salt lattice, with a bulk experimental parameter of cell of 2.119 Å.[
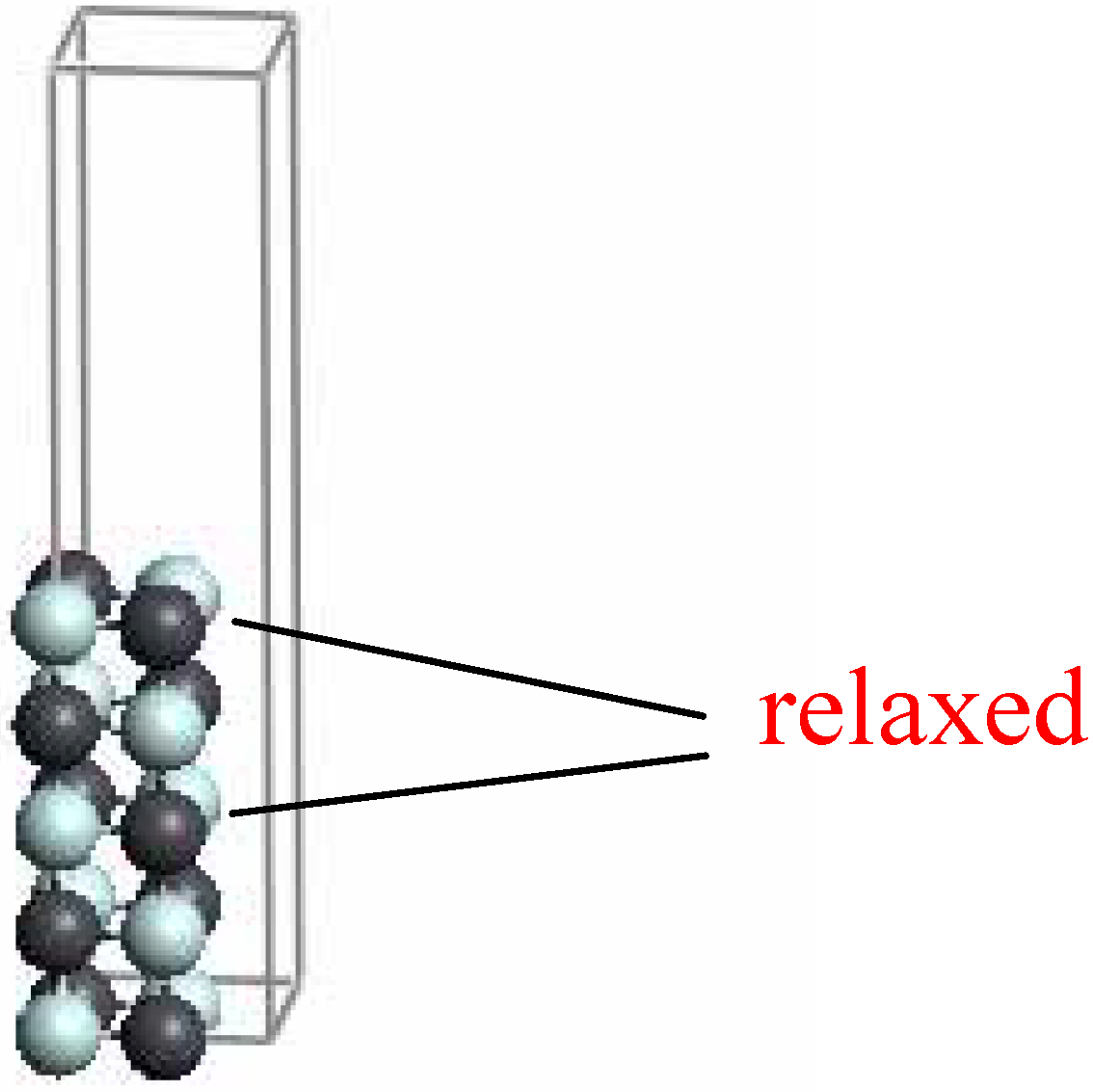
22] In order to test the reliability of our approach, the first step was to perform an optimization of the cell parameter. For this purpose a cube containing 4 Ti atoms and 4 N atoms was selected as supercell. The optimization of such a cell gives a lattice parameter of 4.253 Å, in agreement with the experimental value (4.238 Å). The nitride surface was then represented by a 2x2 supercell 5 layers thick on which periodic 3D boundary conditions were applied, allowing a vacuum space of 20 Å between the slabs (
Figure 1). In the optimization of both the surface and surface + gold adatoms, only the three outermost layers were allowed to relax. This choice, as well as that concerning the thickness of the slab, will be justified later.
Figure 1.
View of the 2 x 2 cell five layer thick used to represent the TiN (001) surface.
Figure 1.
View of the 2 x 2 cell five layer thick used to represent the TiN (001) surface.
Results and discussion
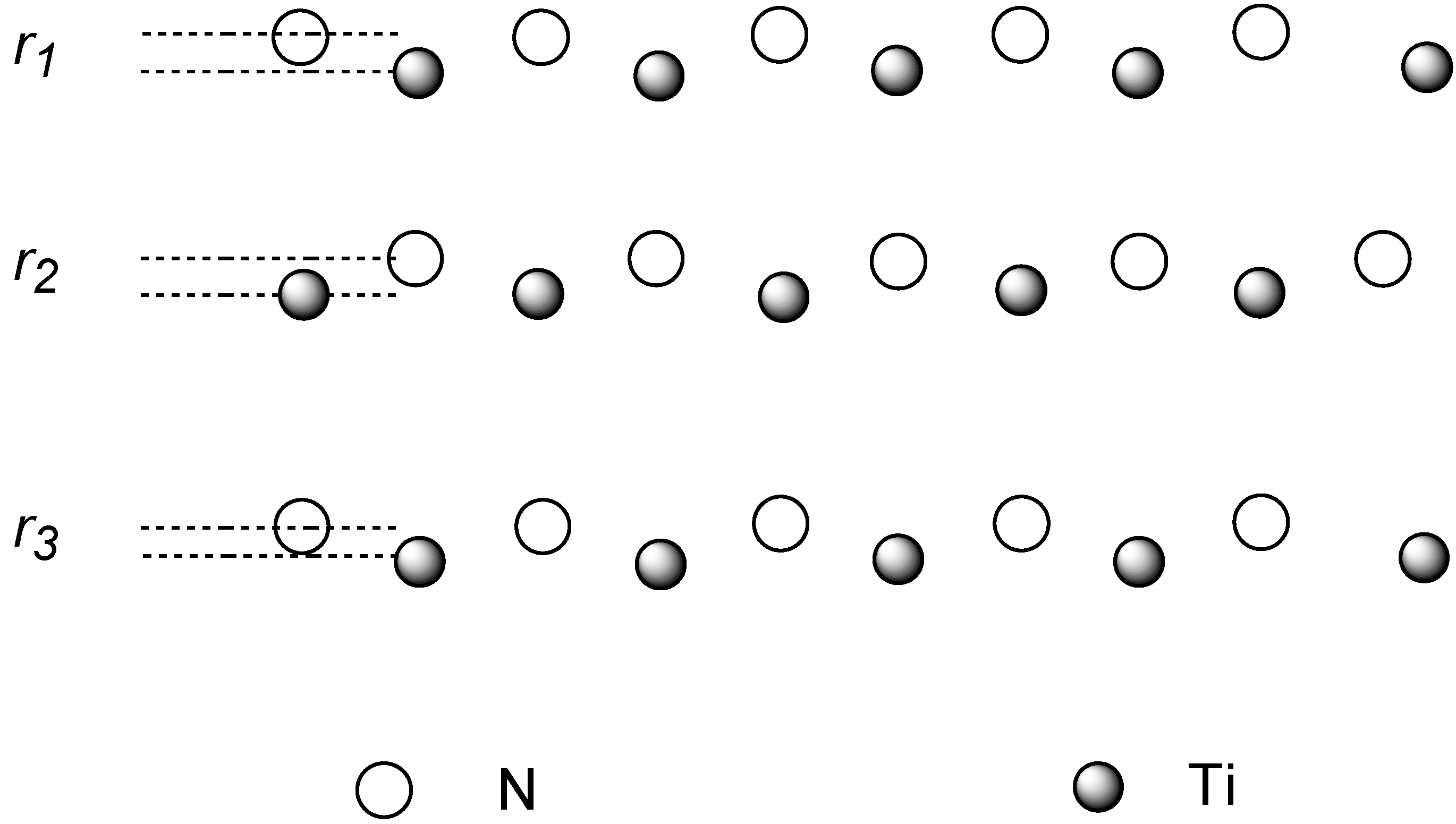
Relaxation of the isolated surface will be first considered. In a binary material the surface charge distribution is not the same for both species and therefore the ionic cores feature different forces in the surface region. In consequence, the surfaces tend to rumple in more or less extent. The relaxation involves an outwards displacement of <011> rows of a species and inwards movements of the other. This rippled relaxation has been experimentally observed in some metal carbides[
5] and nitrides[
6] where the nonmetal atoms are found to move outwards and the metal atom rows inwards with respect to a bulk-truncated layer. Our calculations agree with this kind of surface relaxation as shown in
Figure 2, where a schematic description of the phenomenon is depicted.
Figure 2.
Schematic view of the rippled relaxation.
Figure 2.
Schematic view of the rippled relaxation.
In
Table I the rippling parameter for the first layers computed using several surface models are reported. In these models some of the layers are allowed to relax (free) and some others are kept fixed (fix). As can be seen the rippling parameter
r for the first layer ranges between 0.178 and 0.193 Å. The actual value oscillates when the model becomes larger. It starts at 0.178 Å for the 2/2 model, growths for the 3/2 and 3/3 models, smoothly converging to 0.188 Å for the 5/5 super cell. Rippling for the inner layers is also observed, but within an order of magnitude smaller. It is also worth to note that for the 5/5 model the deepest rippling parameter is practically null (0.002 Å), i.e., it almost reaches the bulk value, indicating that the rippled relaxation is almost completely attenuated beyond the fifth layer.
Table I.
Rippling parameter r (Å) for the TiN (001) surface computed from periodic GGA DFT calculations and different slab models.
Table I.
Rippling parameter r (Å) for the TiN (001) surface computed from periodic GGA DFT calculations and different slab models.
| Model | 2 free/2 fix | 3 free/2 fix | 3 free/3 fix | 4 free/4 fix | 5 free/5 fix |
| r1 | 0.178 | 0.188 | 0.193 | 0.191 | 0.188 |
| r2 | 0.014 | 0.013 | 0.032 | 0.018 | 0.026 |
| r3 | | -0.034 | -0.061 | -0.035 | -0.034 |
| r4 | | | | 0.033 | 0.024 |
| r5 | | | | | 0.002 |
When the different models are compared, it turns out that, as far as the first layer rippling parameter is concerned, the surface model consisting of 3 free layers and 2 fixed ones, gives a suitable representation at a moderate size, and therefore that is why this surface model has been chosen for the calculations in this work.
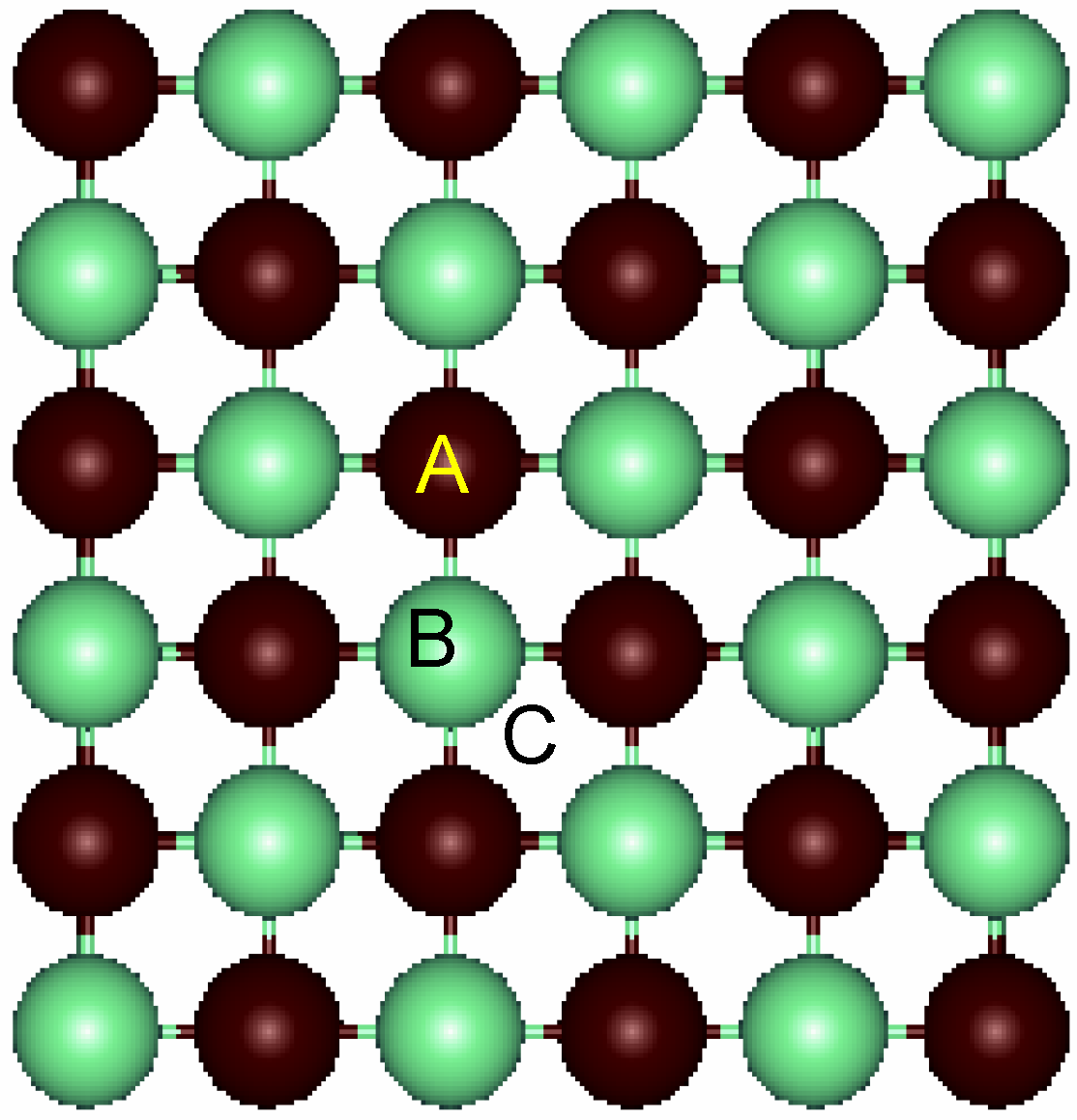
Figure 3.
Top view of the relaxed surface. The labels for the three different sites for adsorption are A: on-top of Ti atoms; B: on-top of N atoms and C: fourfold hollow.
Figure 3.
Top view of the relaxed surface. The labels for the three different sites for adsorption are A: on-top of Ti atoms; B: on-top of N atoms and C: fourfold hollow.
Next, deposition of Au atoms on the surface for a coverage of 0.25 (i.e. one Au per cell) was performed. Three sites were considered as depicted in
Figure 3. Sites A and B correspond to on-top of Ti and N atoms, respectively. Model C refers to the fourfold hollow site in which the Au atom is in the vertical of the center of the square ring. An additional site corresponding to the center of the Ti-N bond was also considered, but our calculations showed it to be unstable.
Adsorption energies, E
ads, and distances of gold atoms to the surface computed from spin-polarized DFT GGA calculations are reported in
Table II (because of the rumpling, surface here represent the outermost surface atoms, i.e. the nitrogen subplane). Our calculations show that the preferred sites are on-top of surface Ti atoms, with an adsorption energy of -1.92 eV. The on-top N sites are less favorable by about 0.4 eV. Furthermore, adsorption energy on hollow sites (-1.90 eV), appear to be virtually identical to that of A sites, suggesting that for low coverage, Au atoms could migrate easily between neighbor Ti surface atoms, providing a possible mechanism for surface diffusion of gold atoms. Compared to other gold/surface systems the interaction energy for the Au/TiN interface appears to be relatively low. Thus, for the Au/TiO
2(110) interface, the adsorption energy is computed to be 0.99 eV from ab initio embedded cluster DFT calculations, i.e. almost half that of the Au/TiN system.[
23] These results suggest a significant larger binding of gold atoms on TiN even taking into account the different theoretical approaches used. (Actually, for Cu/TiO2 (110) system, the adsorption energies computed from ab initio embedded cluster DFT calculations and periodic GGA DFT calculations are 2.85 and 2.39 eV respectively.[
23])
Table II.
Adsorption energies (eV) and Au - surface distances (Å) computed from periodic GGA DFT calculations for coverage of 0.25 ML.
Table II.
Adsorption energies (eV) and Au - surface distances (Å) computed from periodic GGA DFT calculations for coverage of 0.25 ML.
| Site | -Eads | Distance to the surface |
| A | 1.92 | 2.496 |
| B | 1.54 | 2.518 |
| C | 1.90 | 2.368 |
With the aim to understand the type of metal – surface interaction an analysis of the density of states (DOS) as well as the electron density maps for the most stable site was carried out:
Figure 4 and
Figure 5.
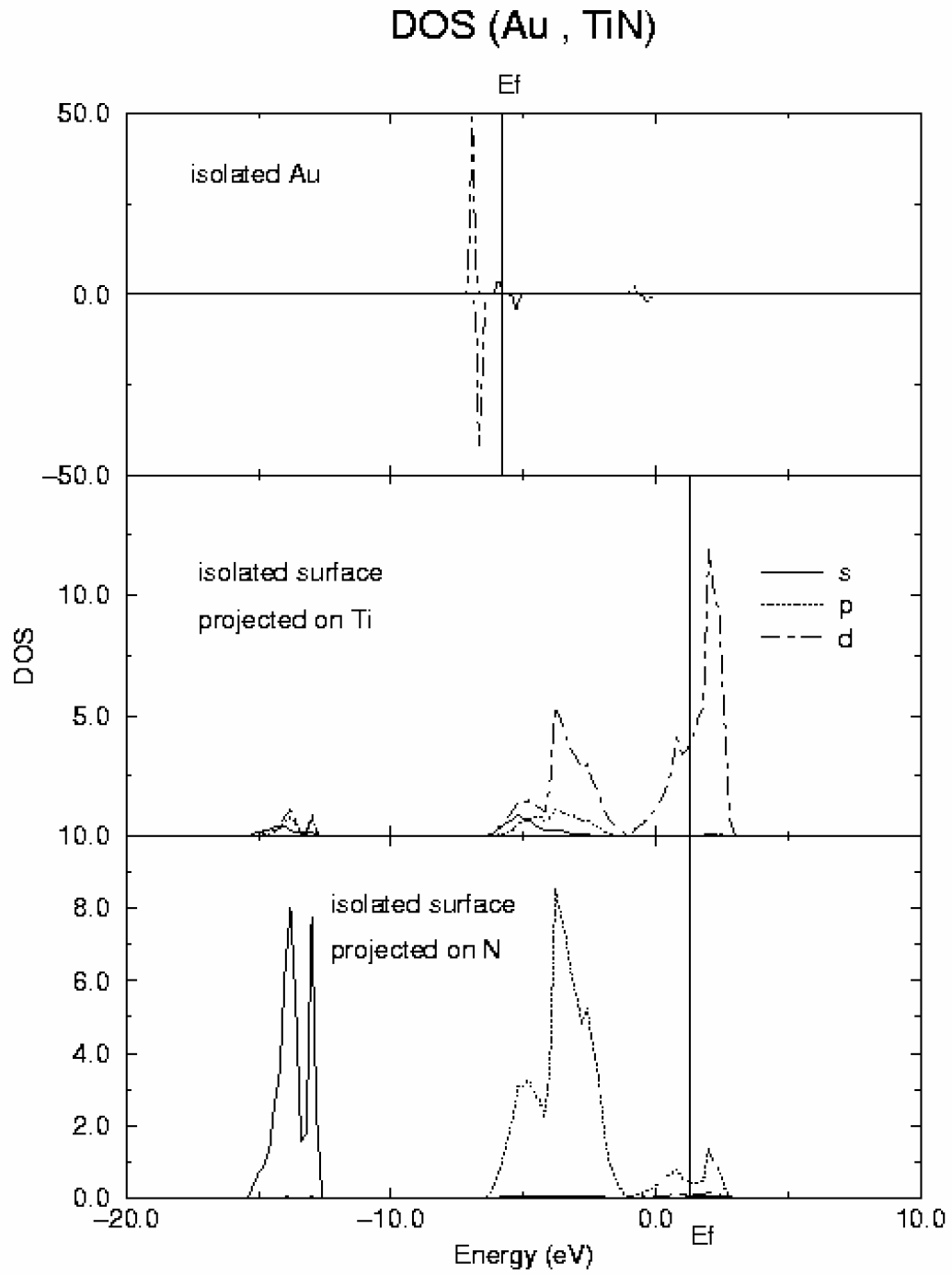
Figure 4.
DOS curves for Au atom (top) and a slab of TiN (001) surface (middle and bottom).
Figure 4.
DOS curves for Au atom (top) and a slab of TiN (001) surface (middle and bottom).
In
Figure 4 DOS curves for both isolated Au and surface are represented (in these draws, as usual, alpha spin is represented positive and beta spin negative). DOS for isolated Au atom (top of the figure) clearly shows the d
10s
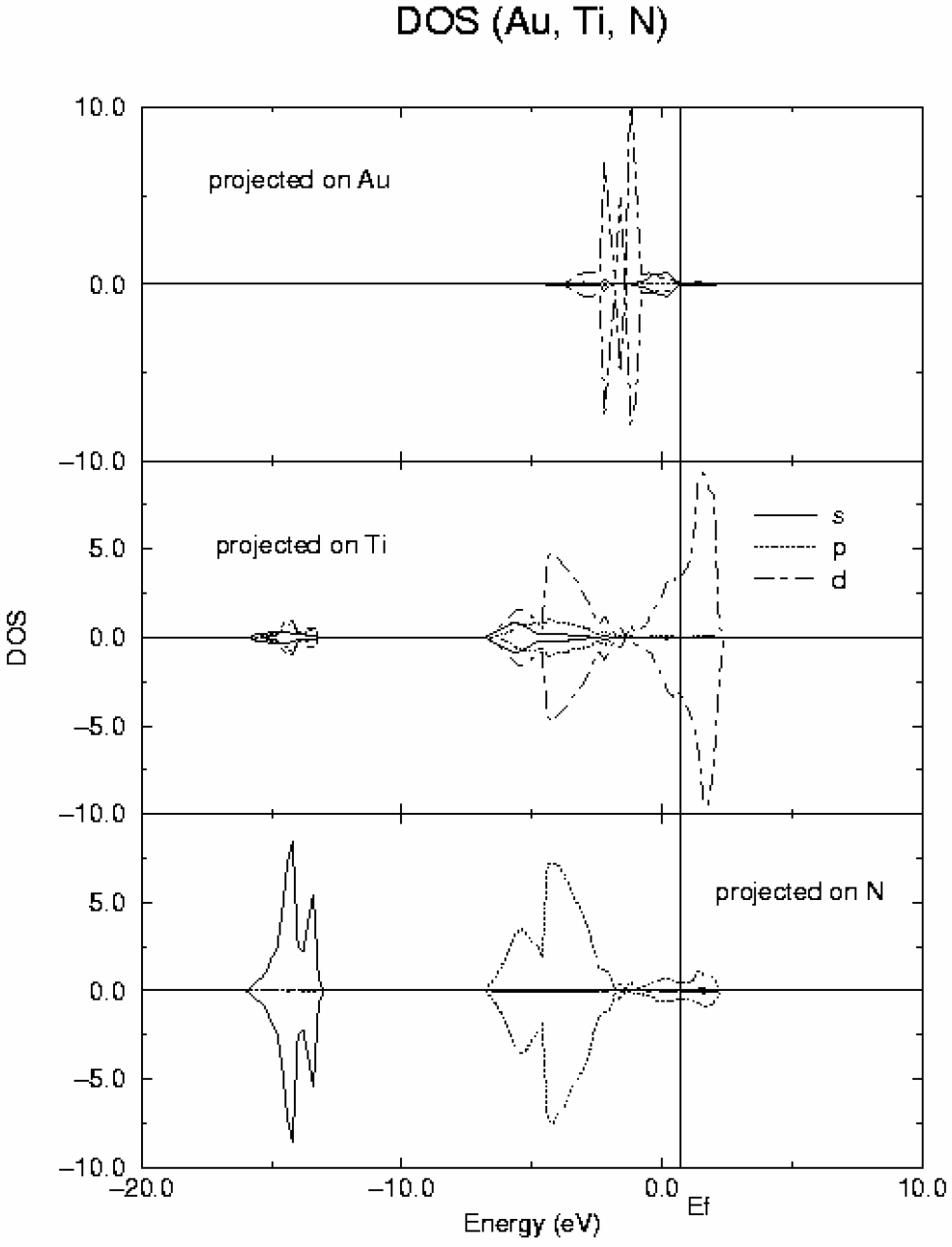
1 configuration of the gold ground state. In the case of TiN surface, the most relevant feature is the metallic character of this material in agreement with previous calculations and experiment. The covalent contribution to the Ti-N bond is reflected by the strong mix of Ti d and N p orbitals clearly observed in the DOS. After adsorption, the gold d band splits into four main components whose energies coincide with those of new peaks appearing in the d component of the DOS projected on the surface Ti atoms as shown in
Figure 5.
Figure 5.
DOS curves for Au adsorbed on-top of Ti surface atoms obtained from periodic GGA DFT calculations. Top: projected on Au; middle: projected on Ti; bottom: projected on N.
Figure 5.
DOS curves for Au adsorbed on-top of Ti surface atoms obtained from periodic GGA DFT calculations. Top: projected on Au; middle: projected on Ti; bottom: projected on N.
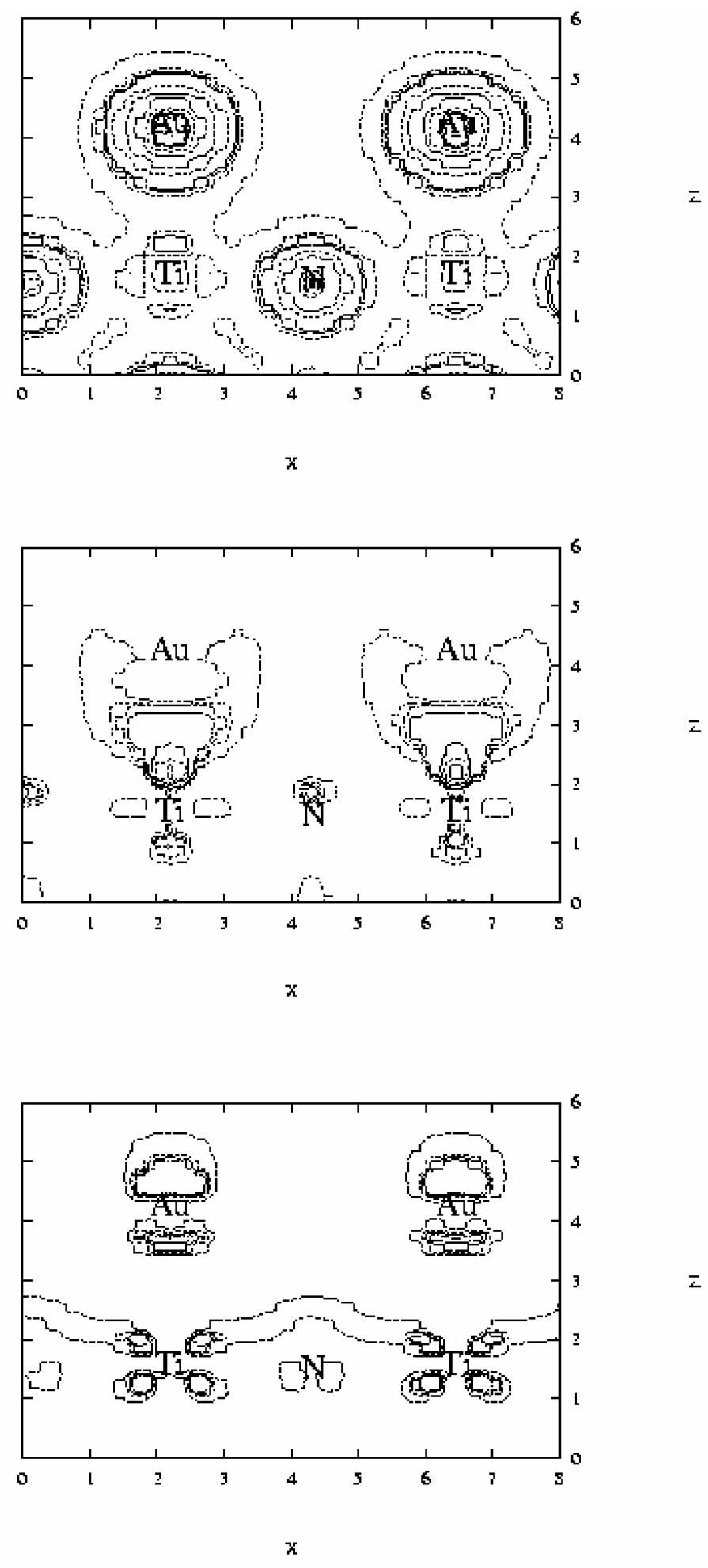
The analysis of electron density maps reported in
Figure 6 appears to be more eloquent. These maps correspond to the plane (100) containing Ti, N and Au atoms. The total electron density, reported at the top of the figure, shows that density around N atoms is larger than near Ti centers. Taking into account that the core of both N and Ti atoms is described through a pseudopotential, the number of valence electrons for these centers is 5 and 4 respectively. Therefore this larger electron density around N atoms reveals that the Ti-N bond is considerably polarized, and therefore some ionic character on this bond still is present. Of special interest in this kind of analysis is the evaluation of the difference between the total electron density of the Au/TiN system and that of the isolated fragments. This difference is reported in the middle and bottom of
Figure 6. The isolines of the middle correspond to the region where the difference is positive, and therefore there is an increment of the electron density. The isolines at the bottom are negative and show the regions where there is a lowering of electron density. The analysis shows that there is an electron density increment in the Au-Ti interatomic region indicating that some covalent bond is formed. When these maps are compared one can observe that for Ti atom the increment of density along the z direction involves a population of the d
z2 orbital, which is accompanied by a lowering of that of the d
xz orbital. A similar repolarization is found for gold atom. For this center there is a lowering in the inner electron density (d
z2 orbital) with an increasing in the outer region falling between the Ti and Au atoms, likely also involving some hybridization of 6s and 6p orbitals. Both repolarizations gives raise to a reinforcement of the overlap between the two transition metal atoms. Finally, a careful analysis of these maps do not make evident if some net charge transfer between the surface and the deposited transition metal takes place. We conclude, therefore, that the main contributions to the metal-surface adsorption energy arise from polarization and covalent interactions.
Figure 6.
Electron density maps for Au adsorbed on-top of Ti surface atoms obtained from periodic GGA DFT calculations. Top: total density; levels: 100, 300, 400, 500, 1000, 1500, 2000. Difference between the total density and those of the fragments, i.e. (Au+TiN)-(Au)-(TiN), is reported at the middle (positive) and at the bottom (negative). For density difference the positive levels are: 10, 30, 40, 50, 100, 150, 200, and the negative: –100, -50, -40, -30, and –10.
Figure 6.
Electron density maps for Au adsorbed on-top of Ti surface atoms obtained from periodic GGA DFT calculations. Top: total density; levels: 100, 300, 400, 500, 1000, 1500, 2000. Difference between the total density and those of the fragments, i.e. (Au+TiN)-(Au)-(TiN), is reported at the middle (positive) and at the bottom (negative). For density difference the positive levels are: 10, 30, 40, 50, 100, 150, 200, and the negative: –100, -50, -40, -30, and –10.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}