Reaction Mechanism and Control of Selectivity in Catalysis by Oxides: Some Challenges and Open Questions


Department of Industrial Chemistry and Engineering of Materials, University of Messina, Salita Sperone 31, 98166 Messina, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2001, 2(5), 183-196; https://doi.org/10.3390/i2050183
Submission received: 15 October 2001
/
Accepted: 30 October 2001
/
Published: 13 November 2001
(This article belongs to the Special Issue Elementary Steps in Heterogeneous Catalysis: Theory and Experiment (Papers from Euroconference on Molecular Mechanism of Heterogeneous Catalysis Held in San Feliu de Guixols, Spain, in June 2001))
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Some aspects of the reaction mechanisms in multistep selective (amm)oxidation reactions over oxide surfaces are discussed evidencing some of the challenges for surface science and theory in describing these reactions, and for applied catalysis in order to have a more in deep identification of the key factors governing surface reactivity and which may be used to improve catalytic performances. In particular, the role of chemisorbed species in the modification of the surface reactivity and the presence of multiple pathways of reaction are evidenced by comparing the behavior of V-based catalysts in C3-C4 alkanes and alkene oxidation.
Introduction
Oxide-based catalysts are widely used in chemical processes in several applications ranging from selective oxidation to total oxidation, hydrogenation, dehydrogenation and environmental applications (NOx conversion, etc.). More than one third of the worldwide catalyst production is based on oxide-type catalysts, apart from their use as support for other active components. Selective oxidation can be considered the most typical example of the use of oxide-type materials as heterogeneous catalysts [1]. Around one quarter of the main organic chemicals are produced by selective oxidation processes and furthermore the recent report “Technology Vision 2020: The Chemical Industry” (Catalysis Report) [2] indicates selective oxidation as one of the top areas of application of catalyst technology able to combine scientific challenges to economical benefits. In particular, for selective oxidation the following technical targets necessary for achieving the Vision 2020 goals have been identified: (i) characterization of the different types of oxygen present on oxides surfaces and their role in alkane activation and subsequent oxidation, (ii) identification of the factors controlling the selectivity in selective oxidation and oxidative dehydrogenation of alkanes, (iii) the selective oxidation of olefins and aromatics and (iv) identification of novel methods for activating O2 [2]. Therefore, the technical challenge of improving performances in selective oxidation processes requires a better understanding of the surface reaction mechanisms at oxide surfaces as the critical step.
A characteristic of oxide-type catalysts for selective oxidation reaction is their multifunctionality which allows making in one single "formal" step very complex reactions over the catalyst surface. A typical example is the synthesis of maleic anhydride from n-butane over vanadyl pyrophosphate catalyst [3,4]. This product (worldwide production of several millions tons per year) is synthesized industrially with high selectivities (up to over 80% at low conversion) without desorption of any reaction intermediate from the catalyst surface. The reaction is a 14 e- oxidation, with abstraction of 8 H atoms from butane molecule and insertion of 3 O atoms, plus 4 other oxygen atoms necessary for desorbing as water molecules the H atoms abstracted from n-butane. Different catalyst functional properties (active sites) are required for the complete selective oxidation transformation, such as sites for abstracting paraffinic H atoms, sites for abstracting allylic H atoms, sites for coordinating olefinic-type intermediate as π-allyl complex, sites for insertion of O into allylic position and sites for insertion of O in 1,4 dienic position, etc [4]. Not only the presence of such a type of sites, but also their relative turnover frequency is important, because a selective transformation implies that all the consecutive elementary steps must occur in a concerted way [5]. Otherwise, the occurrences of side reactions lowering the selectivity are possible. A volcano-type dependence of the activity as a function of the catalyst properties (for example, M-O bond strength) is the basis of the concept of catalysis (Sabatier principle), because the optimum behavior is the compromise between the rate and strength of chemisorption. This concept applies to the rate limiting step which is the alkane activation in n-butane selective oxidation on vanadyl pyrophosphate. The original concept discussed in the cited reference [5] is instead that for complex reactions such as n-butane oxidation the selectivity passes through a maximum when the catalyst properties are varied and this is a consequence of a change of the relative rates in the competitive pathways of reaction after the rate limiting step. A kinetic model based on this assumption correctly describes the experimental data [5].
Therefore, the understanding of catalyst multifunctionality and how to tune its properties in order to have a concerted sequence of elementary steps are two important challenges for a better design of these catalysts and make possible the development of new complex catalytic transformations over solid catalysts. The latter is a relevant objective for a new sustainable chemistry. This concept may be demonstrated using again the example of n-butane oxidation to maleic anhydride. This process has substituted the former industrial process starting from benzene, when a catalyst (vanadyl pyrophosphate) able to selectively oxidative an alkane (n-butane) was discovered [3,4]. The substitution of the process has lead to considerable benefits in terms of sustainable (green) chemistry [6], because (i) the new process use non toxic reactants, (ii) there is an improvement in atom economy with respect to benzene process, (iii) the transformation is made in one single step, without solvents, and (iv) waste formation is minimized.
There is a need of catalysis for a sustainable chemistry, because one-step synthesis of complex transformations over solid catalysts reduces the process complexity, avoids solvents and decreases risks and energy use. Furthermore, high (super) selectivity avoids byproducts and improves atom economy. The development of next-generation chemical industry thus requires an integrated effort in understanding how to control catalyst properties in order to make possible a selective multistep process in a single passage over a solid catalyst surface. The understanding of the reaction mechanisms and the control of selectivity in catalysis by oxides is a relevant passage towards this more general scope.
This understanding requires, however, an integrated effort between theory, modeling, surface characterization and reactivity testing. A remarkable effort has been put in bridging the gap between surface science and applied catalysis in recent years, especially regarding catalytic transformation at oxide surface [7,8]. There is a definitive trend in going from metal to metal-oxide surface science. Mainly relatively simple catalytic transformations such as CO or NH3 oxidation and model catalysts have been addressed up to now, although some more complex reaction such as methanol oxidation was also studied [9]. Metal-oxide catalysts show a number of additional characteristics quite relevant for catalysis with respect to metals which indicates the intrinsic higher complexity of arriving to a full understanding of the surface catalytic chemistry. Some of these relevant aspects of the metal-oxide catalytic chemistry are listed below:
- Easy stabilization of defect structures
- Pronounced surface relaxation
- Surface heterogeneity (different oxygen atoms, acid and redox sites, possible multiple cations, etc.)
- Reducibility and easy reconstruction (aggregated defects, shear planes, etc.)
- Easy bulk diffusion (cation, oxygen, etc) which derive from the presence of an open structures; often this diffusion is anisotropic and different for the different crystal planes
- Easy intergrowth structures
- Anisotropic electron transfer
Therefore, the metal-oxide catalytic chemistry is characterized from a high degree of complexity. In bridging the gap between surface science and applied catalysis there is the need to combine the surface science bottom-up approach [modeling from (almost) first principle and step-by-step increase complexity] to the top-down approach of applied catalysis: use complex (industrial and model) materials and identify key aspects in the reactivity and surface structure. In order to bridge this gap, not only the materials and pressure gap should be considered, but also the complexity gap [3]. The analysis of this question from the applied catalysis point of view (identification of critical questions in the analysis of the reactions mechanisms) can be thus a pulling factor towards a progress in bridging this gap.
The aim of this paper is to analyze some of these critical questions about the reaction mechanisms over oxide surfaces (mainly regarding selective oxidation reactions), in order to contribute to identify some of the challenges for surface science and theory in describing these reactions, but also for applied catalysis in order to have a more in deep identification of the key factors governing surface reactivity and which may be used to improve catalytic performances.
The Kinetic Dilemma
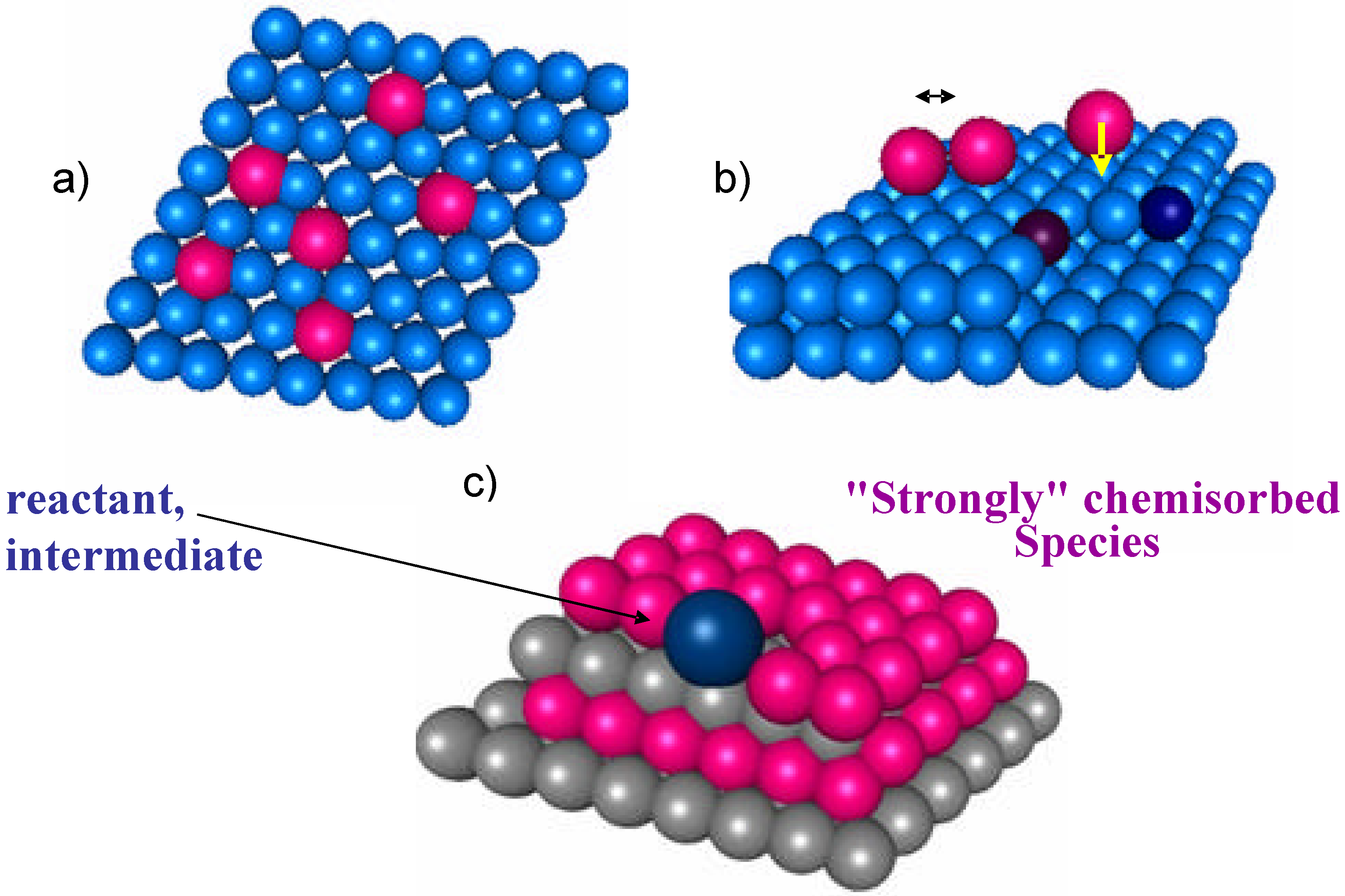
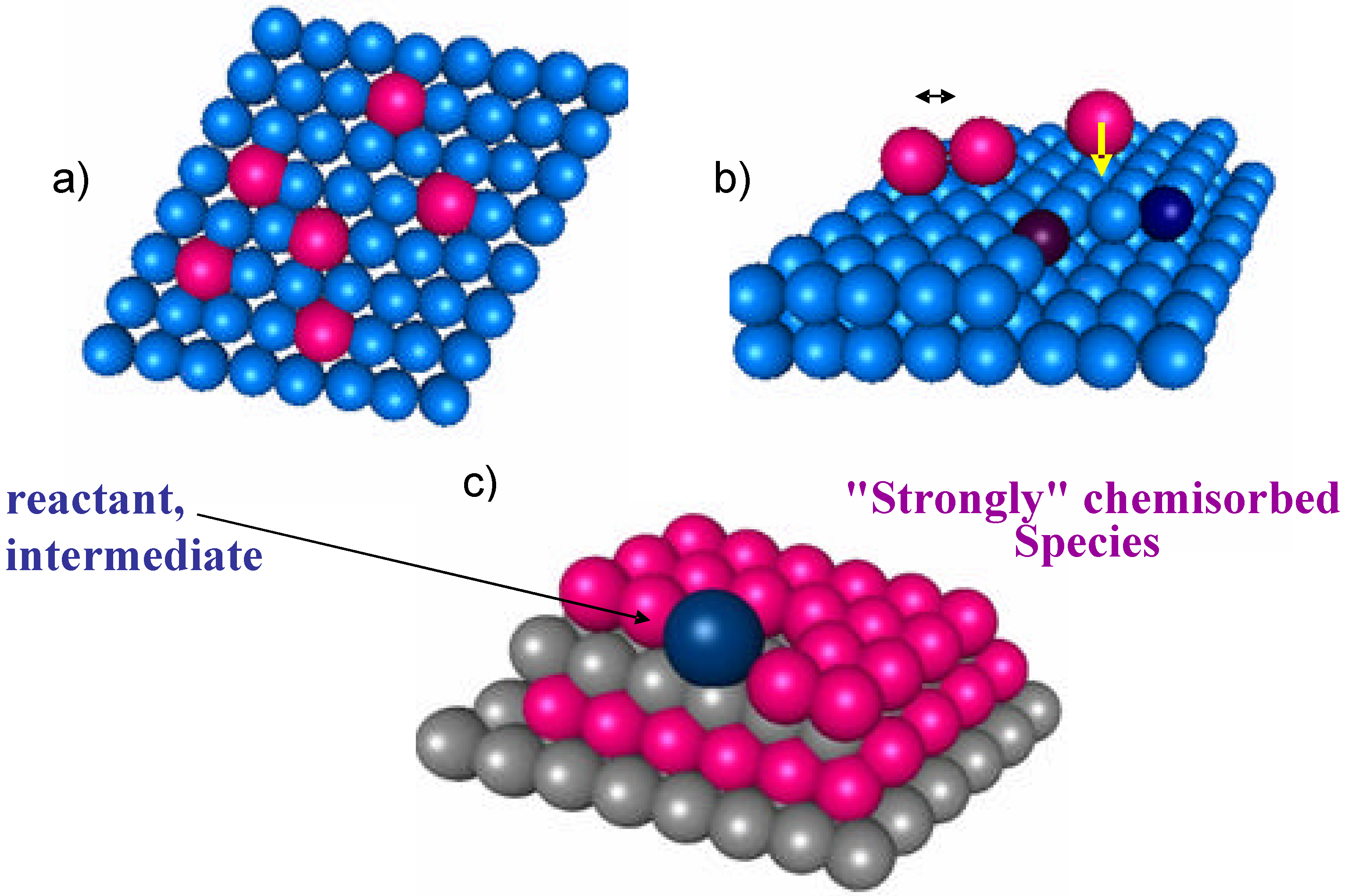
A central question in analyzing the problem of bridging the gap between surface science and applied catalysis approaches is the verification of the possibility of describing the macrokinetic behavior using rate equations and constants derived from the analysis of the kinetics of the single elementary steps. Impressive results have been obtained in this direction [10]. The fitting of macrokinetic data on the basis of a microkinetic model is usually considered the best demonstration of the applicability of the suggested reaction mechanism under real working conditions. Is it this true also for more complex and multifunctional reactions? In order to replay to this question it is good to briefly recall the principles of the Langmuir description of the catalytic reaction on an "ideal" surface, because this is the basis of most of the macrokinetic models for deriving the reaction rates over catalytic surfaces. On the ideal Langmuir surface there is one type only of active sites, the energetic of chemisorption is independent on the coverage, and there is no interaction between substrate (see Figure 1a). Real surfaces are far from the ideal Langmuir surface, as schematically represented in Figure 1b, due to the presence of surface heterogeneities (the simplest is the presence of steps and kinks, but as mentioned in the introduction the situation is far more complex on oxides) which imply a distribution in the energetic of interaction, and the presence of interaction between the adsorbates. A chemical bond of a molecule with a surface site implies the donation of electrons to or from the surface with thus a modification of the catalyst conduction band, band gap, etc.; therefore, the energetic of interaction of molecule with the surface is not independent from the surface coverage, and the adsorption of "spectator" species, i.e. species which do not play a direct role in the reaction mechanism, should be also considered. This problem is even more accentuated in selective oxidation reactions where a multielectron transfer occurs, as mentioned before.
It may be thus expected that kinetic models based on Langmuir approach do not correctly fit macrokinetic data, but on the contrary it is a common experience that these models are well suited to describe the macrokinetic behavior. How can this dilemma be solved? One possible interpretation, which is schematically shown in Figure 1c, is that during catalytic reaction the largest part of the catalyst surface is covered by "strongly chemisorbed" species ("strongly" chemisorbed species indicates in this context species characterized by a surface lifetimes much longer than that of reaction intermediates) and therefore reactant or intermediate seen a local situation similar to "ideal" surface.This pointed out that the concept of "clean" catalyst surface, i.e. of reaction of a molecule at single specific active sites without considering the modification of the surface reactivity induced by the presence of other coadsorbates (reactants, intermediates, "spectator" species), may lead to a not correct description of the "real" working catalyst surface and reaction mechanism, especially when complex, multisteps reactions (selective oxidation reactions, for example) are considered.
Figure 1.
Schematic model of the "ideal" Langmuir surface (a), of a simple model of real surface with kinks and steps, surface interaction between adsorbates and electron donation of a chemisorbed molecule to catalyst conduction band (b), and of a working catalyst surface with the largest part of the surface occupied from "strongly" chemisorbed species (c).
Figure 1.
Schematic model of the "ideal" Langmuir surface (a), of a simple model of real surface with kinks and steps, surface interaction between adsorbates and electron donation of a chemisorbed molecule to catalyst conduction band (b), and of a working catalyst surface with the largest part of the surface occupied from "strongly" chemisorbed species (c).
Modification of the Surface Reactivity by Chemisorbed Species
The discussion regarding the applicability of the "ideal" Langmuir model in describing the macrokinetic behavior has evidenced the importance of the proper consideration of the role of chemisorbed species in the modification of the surface reactivity [11,12]. A demonstration of this concept and the necessity of its use in order to correctly describe the catalytic behavior of oxide-type catalysts for selective oxidation reactions can be shown by comparing the behavior of alkanes and alkenes on the same catalyst. A first example is shown in the analysis of the catalytic behavior of VSbO4+Sb2O4 catalyst (Sb:V = 2) in the ammoxidation of propane and propene to acrylonitrile, a large scale chemical produced worldwide in an amount of about 5 million tons per year. V-Sb-oxides have been extensively patented by, among others, BP America and ICI-Katalco [13,14]. These catalysts are selective in the formation of acrylonitrile from both the C3 alkane and alkene. Propane and propene, due to their different chemical properties, in propene allylic hydrogens and a π-bond system, possess different chemical properties of chemisorption and activation. The comparison of the catalytic behavior of VSbO4+Sb2O4 catalyst (Sb:V = 2) in propane and propene ammoxidation is thus a useful system to understand how the catalytic properties of the catalyst are influenced by the chemisorption of the reactants on the catalyst surface.
Propane and propene ammoxidation using the same catalyst and reaction temperature are compared in Figure 2 which reports the dependence of the selectivity to acrylonitrile on the conversion of the hydrocarbon at 480°C.
Figure 2.
Comparison of the selectivity dependence on acrylonitrile in propane and propene ammoxidation at 480°C on a VSbO4+Sb2O4 catalysts (Sb:V = 2).
Figure 2.
Comparison of the selectivity dependence on acrylonitrile in propane and propene ammoxidation at 480°C on a VSbO4+Sb2O4 catalysts (Sb:V = 2).

Acrylonitrile can be further oxidized to give carbon oxides and thus it is reasonable that at high conversion of the hydrocarbon its selectivity decreases while that to carbon oxides increases. However, the data in Figure 2 point out the contradiction that on the same catalyst, and at the same reaction temperature, acrylonitrile converts faster to carbon oxides when formed from propane than when formed from propene, even though propene is the reaction intermediate from propane [13]. In fact, the maximum in acrylonitrile formation from propane is observed at much lower conversions than in the case of propene and as a consequence the maximum yield is significantly lower. This fact has clear negative implications on the industrial development of a process of acrylonitrile synthesis from propane instead of from propene, and therefore it is of critical importance to understand the reasons for this phenomenon. We shall see that the difference in the behavior can be associated with the different modifications of the surface reactivity when feeding alkane or alkene. This is, however, a more general phenomenon not only restricted to the case of V-Sb-oxides.
Another example of how the reactant induces a change in the surface properties in given in Figure 3 which reports the effect of the change in the oxygen to alkane ratio in the feed on the selectivity to propene from propane on the (VO)2P2O7 catalyst for two initial concentrations of propane in the feed.
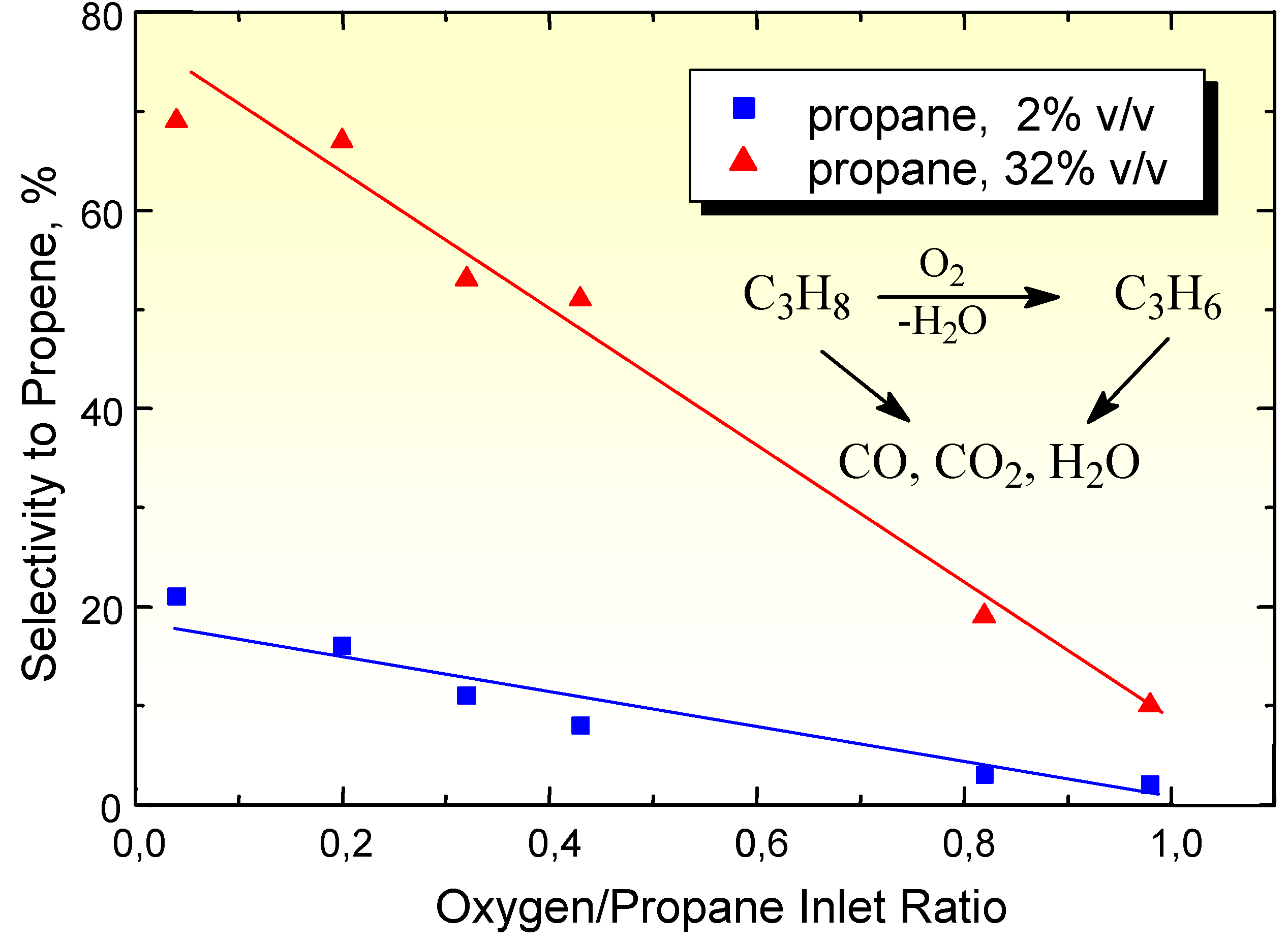
Figure 3.
Selectivity in propene formation from propane on (VO)2P2O7 as a function of the inlet ratio between oxygen and propane concentrations, for two initial concentrations of propane in the feed. Reaction temperatures: 322°C.
Figure 3.
Selectivity in propene formation from propane on (VO)2P2O7 as a function of the inlet ratio between oxygen and propane concentrations, for two initial concentrations of propane in the feed. Reaction temperatures: 322°C.
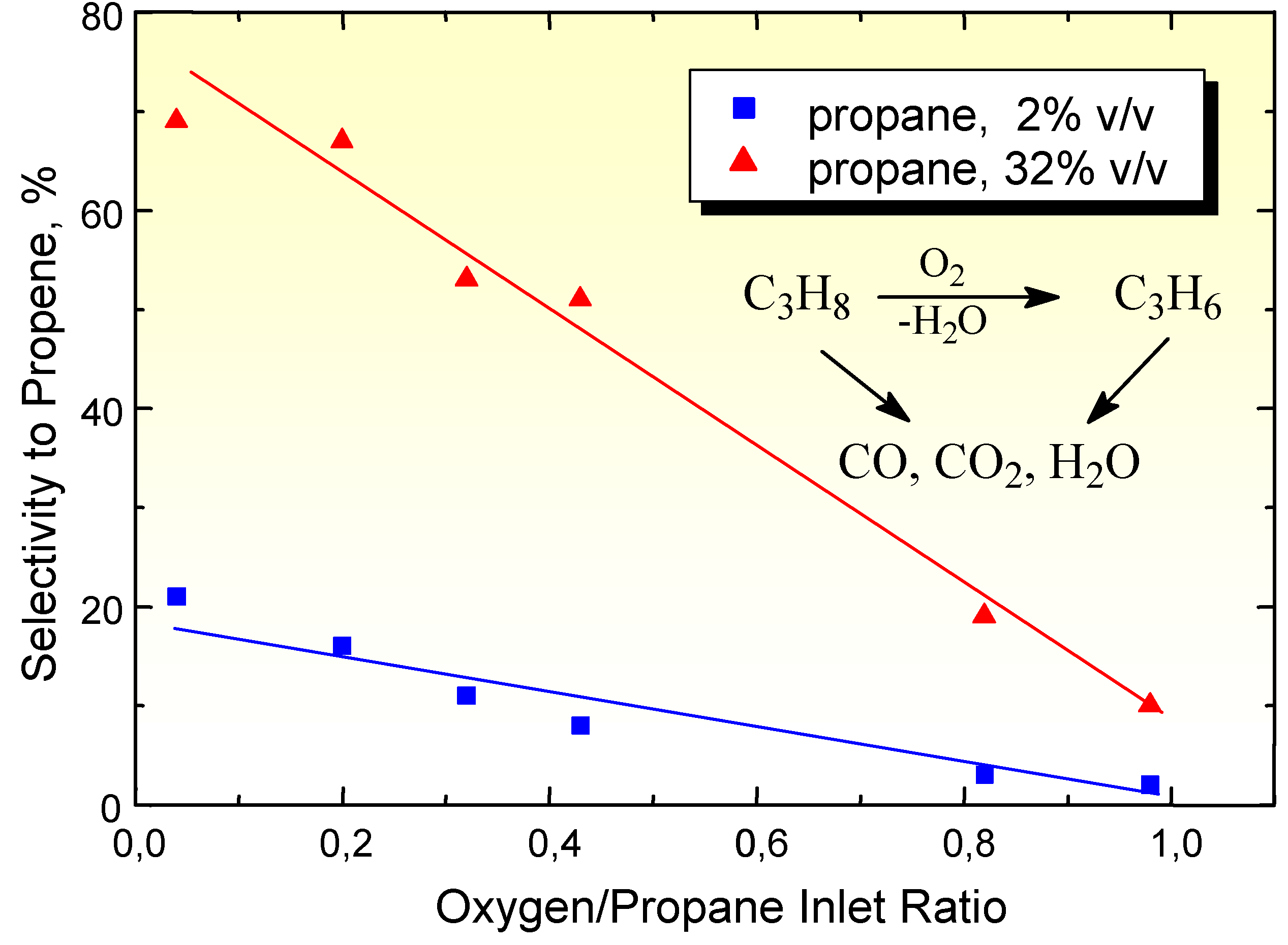
(VO)2P2O7 is the active phase in the industrial catalysts for the selective oxidation of n-butane to maleic anhydride [3,4]. This catalyst is very selective in n-butane oxidation, but when propane is fed instead of n-butane, only carbon oxides and traces of other products (propene mainly) are detected. A first question is thus why the decrease in length of the carbon chain produces a so drastic change in the selectivity on the same catalyst. The answer can be that from n-butane a stable product against consecutive oxidation (maleic anhydride) forms, but not in the case of propane oxidation. However, this answer responds to only part of the problem. The data reported in Figure 2 have already pointed out that the sensitivity of the reaction product against consecutive oxidation is not the only factor that determines the rate of consecutive oxidation to carbon oxides. Data in Figure 3 further evidence this problem. In fact, it is expected that a decrease in the oxygen to alkane ratio may increase the selectivity to the partial oxidation product (propene in the case of propane), because the formation of carbon oxides requires a larger number of oxygen atoms (propane oxidation to CO2 + H2O requires 5 O2 molecules, whereas propene formation from propane requires ten time less oxygen). However, data in Figure 3 show that this is possible only for high initial concentrations of propane in the feed, whereas for lower initial concentrations of propane the increase in propene selectivity decreasing the O2/propane inlet ratio is much less remarkable. A comparison between the two cases points out that the differences arise from the different formation of alkene product, which is about ten times larger using the higher inlet concentration of propane in the feed. This indicates that when the formation of propene is higher, the rate of its consecutive oxidation to carbon oxides also decreases and thus the selectivity increases. The same type of phenomenon has also been observed by changing the O2/hydrocarbon ratio in n-butane oxidation on (VO)2P2O7, although in this case a wider range of intermediate products was detected [4].
The formation of alkenes thus induces a self-modification of the surface reactivity. This in confirmed from the analysis of the kinetics of but-1-ene oxidation on (VO)2P2O7, the analysis of the transient reactivity as well as spectroscopic studies [4]. This evidences that without considering this self-modification of the surface reactivity induced by the reaction intermediates itself it is not possible to correctly describe the kinetic of the surface reactions.
Both (VO)2P2O7 and VSbO4+Sb2O4 catalysts are characterized from the presence of coordinatively unsaturated V4+=O surface sites which act as strong Lewis acid sites and which play a relevant role in the mechanism of alkane activation [3,15]. Alkenes through their π-bond system can chemisorb on these sites forming relatively stable chemisorbed species, although they may be considered "spectator" species, because are not directly involved in the mechanism of further selective oxidation of these alkene intermediates. Oxygen also strongly chemisorbs on the surface Lewis acid sites forming thermal stable species (they desorb above 450-500°C), but which play a relevant role in the mechanism of oxidation [3]. When the surface concentration of the intermediate alkenes in alkane oxidation is as high as to limit the amount of chemisorbed oxygen, due to this competitive chemisorption, it is thus possible to control the population of oxygen adspecies by this mechanism. This explains the considerable promotion of selectivity to partial oxidation products by increasing alkane inlet concentration (Figure 3) and, on the other hand, explains also the apparent contradiction of the different rate of acrylonitrile consecutive oxidation when forms from propene instead that from propane (Figure 2).
Although propene forms from propane as a reaction intermediate, its concentration is clearly higher when it is fed directly and thus its effect in limiting the concentration of surface oxygen species is present even for higher conversions of the hydrocarbon. Therefore, the maximum in the formation of acrylonitrile is observed at higher hydrocarbon conversions when the alkene is fed instead of the alkane on the same catalyst (Figure 2).
Multiple Surface Pathways of Transformation
The chemisorption of reactants or intermediates may influence not only the rate of the selective vs. unselective oxidation, but also the rate of competitive pathways of transformation. Often only a reaction pathway of surface transformation is considered to exist. On the contrary, multiple pathways of surface transformation are possible also leading to the same final product of reaction. The chemisorption of reactants or intermediates may influence the rates of these multiple pathways of transformation which are characterized by a different intrinsic selectivity. This concept is exemplified studying the behavior of (VO)2P2O7 and VSbO4+Sb2O4 catalysts in propane ammoxidation to acrylonitrile.
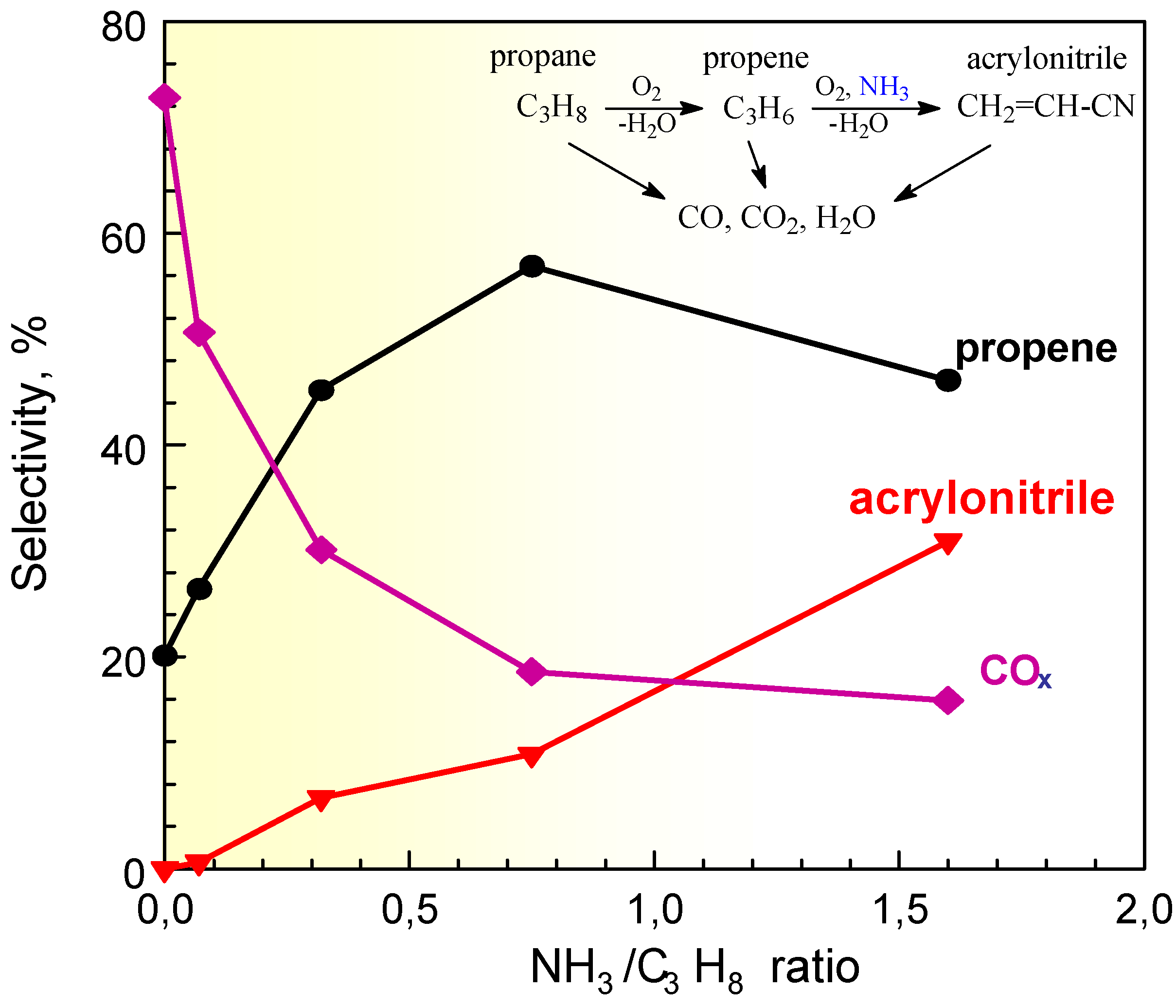
An interesting effect is noted studying the effect of the addition of ammonia to a propane/air feed on a VSbO4+Sb2O4 catalyst [16]. Ammonia is a reactant necessary to form acrylonitrile from propane. When ammonia is not fed and the propane concentration is low, but high the O2/propane concentration, carbon oxides are the only products of reaction. It may thus be expected that when ammonia is added to the feed, the formation of carbon oxides will decrease and the formation of acrylonitrile will increase. However, small doses of ammonia in the feed do not cause the increase in the formation of acrylonitrile with a parallel decrease of carbon oxides, but instead determine a drastic jump of the formation of the intermediate propene (Figure 4). At low levels ammonia thus does not promote the formation of the N-containing product, but only strong depress the unselective conversion to carbon oxides of the intermediate propene.
Figure 4.
Selectivity to products in propane (amm)oxidation on VSbO4+Sb2O4 as a function of the inlet ratio between ammonia and propane concentrations. Reaction temperatures: 480°C; 1% v/v propane, 15% O2.
Figure 4.
Selectivity to products in propane (amm)oxidation on VSbO4+Sb2O4 as a function of the inlet ratio between ammonia and propane concentrations. Reaction temperatures: 480°C; 1% v/v propane, 15% O2.
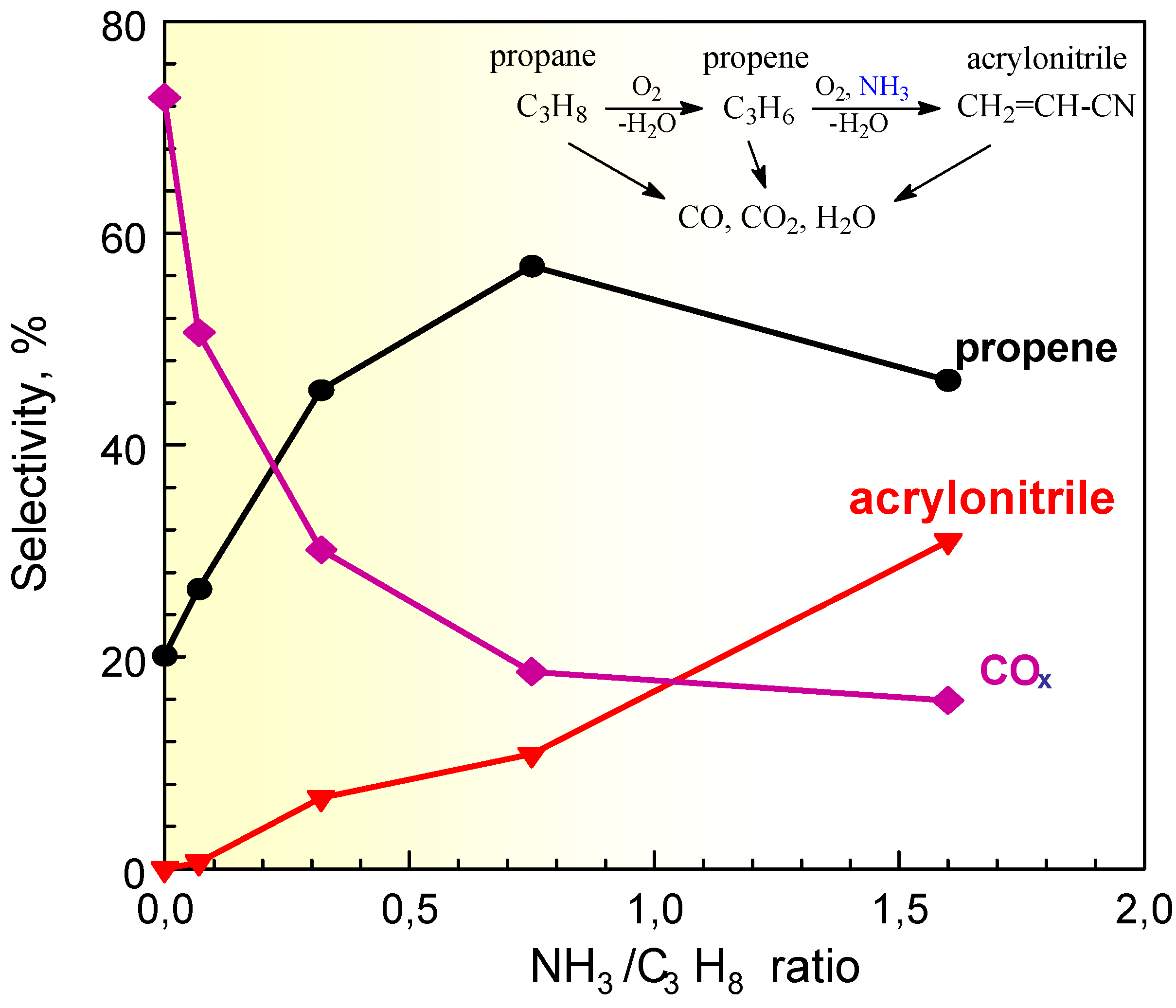
FT-IR data [16,17] show that ammonia interacts with the Brönsted acid sites of the catalyst forming ammonium ions, the stability of which, however, is weak and evacuation at temperatures of about 200°C is enough for their nearly complete removal. This suggests that at the typical reaction temperatures for the catalytic tests of propane ammoxidation (400-500°C) the interaction of ammonia with these surface Brönsted acid sites may be considered negligible. Ammonia remains instead strongly chemisorbed as such on surface Lewis acid sites, up to evacuation temperatures of about 400°C. However, in the presence of water vapor the situation is different and it was observed that ammonium ions form at high temperature from ammonia coordinated on Lewis acid sites. Water is one of the main products of reaction in hydrocarbon oxidation and thus all considerations on the stability of surface species must take into account its presence. Furthermore, desorption of ammonia adspecies is an equilibrium reaction with ammonia in the gas phase. When NH3 is present in the gas phase, Brönsted acid sites are nearly completely converted to ammonium ions even at high temperature. It is thus reasonable that during the catalytic reaction of propane conversion on VSbO4+Sb2O4 in the presence of ammonia, the larger part of the surface Brönsted acid sites is transformed to ammonium ions and consequently the reactivity of these Brönsted acid sites is inhibited.
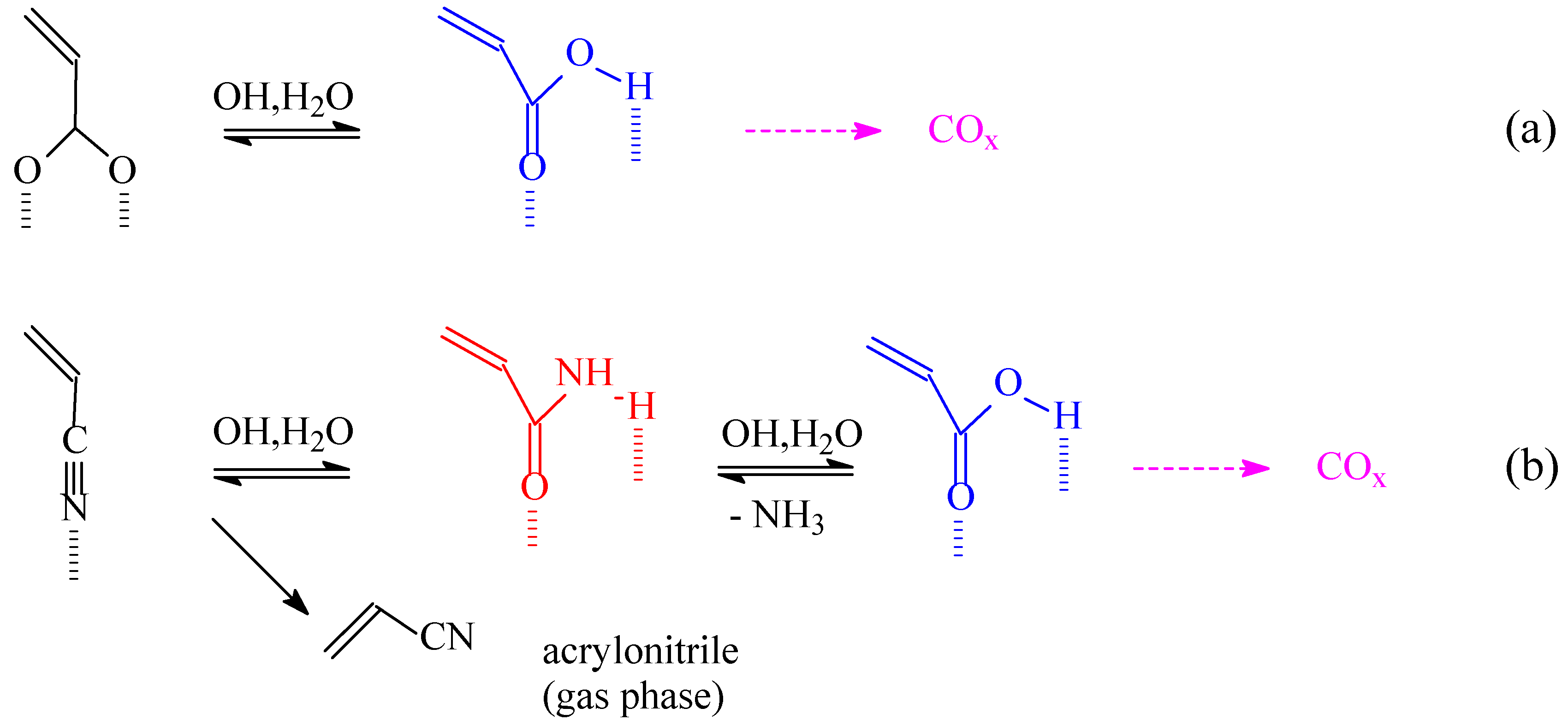
Brönsted acid sites catalyze various side reactions. In particular, it has been observed [16,17] that acrylic acid forms stable and relatively inert acrylate species with VSbO4+Sb2O4 catalyst up to high temperatures of evacuation. In the presence of Brönsted acid sites and water, however, this species transforms to the corresponding free acid form weakly bound to the surface and easily susceptible to consecutive decarboxylation and further total oxidation (Scheme 1a). The strong interaction of acrylate with the surface explains why even when formed from propane and propene (as detected by FT-IR spectroscopy) acrylic acid is detected only in traces as a product of C3 hydrocarbon oxidation on VSbO4+Sb2O4. The interaction with the surface of the corresponding species formed in the presence of ammonia (acrylonitrile) is weaker and thus the desorption is easier (Scheme 1.b). Acrylonitrile may also be transformed to the corresponding amide by the action of Brönsted acid sites and water (Scheme 1.b), which more easily further transforms to the free acid and then finally to carbon oxides. The self inhibition of ammonia on the activity of Brönsted acid sites converting them to ammonium ions, however, inhibits this pathway.
Scheme 1.
Reaction schemes of acrylate (a) and acrylonitrile (b) oxidation to carbon oxides.
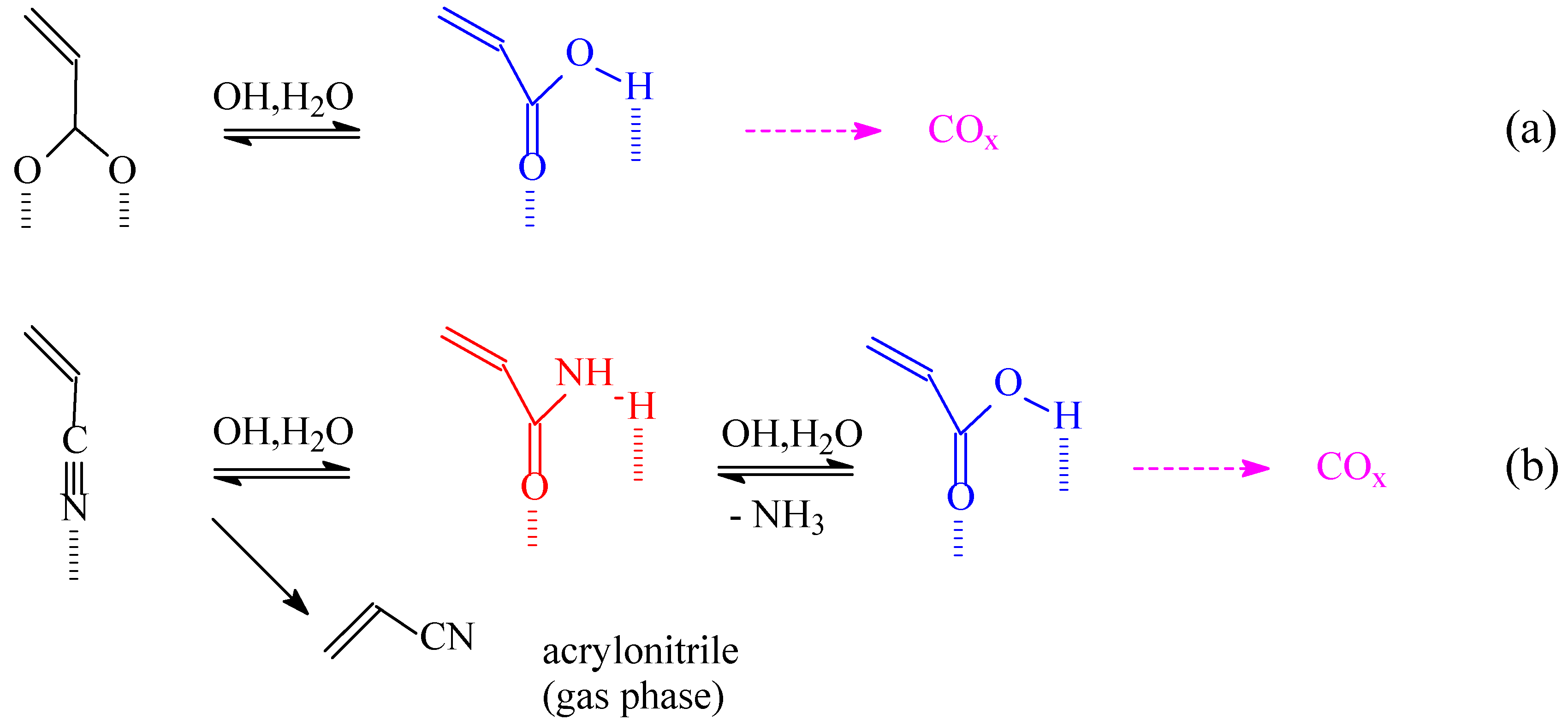
The reasons for the much higher selectivity in propane ammoxidation than in propane oxidation on the same VSbO4+Sb2O4catalyst thus derive from the two combined effects:
- Less interaction of acrylonitrile with the surface with respect to acrylic acid.
- Self inhibition of ammonia on the degradation of the adsorbed intermediate catalyzed by Brönsted acid sites and water.
When small amounts of ammonia are added to the feed, the primary effect of ammonia is to block the reactivity of Brönsted acid sites forming ammonium ions and thus an inhibition of the further transformation of acrylate. The latter species, however, strongly interacts with the surface sites and does not desorb, if not minimally, and thus tends to block the surface reactivity, as evidenced by IR spectroscopy. As a consequence, propane transformation stops at the first intermediate which can relatively more easily desorb to the gas phase (propene). This explains why the addition of small amounts of ammonia to the propane/air feed considerably promotes the selectivity to partial oxidation products, but mainly to propene rather than to other products, including acrylonitrile. Weaker coordinated ammonia species also form which react with propene or its conversion products to give acrylonitrile only for higher amounts of ammonia in the feed. Ammonium ions thus do not play a direct role as reaction intermediates, but they determine the catalytic behavior by their positive influence on the surface reactivity.
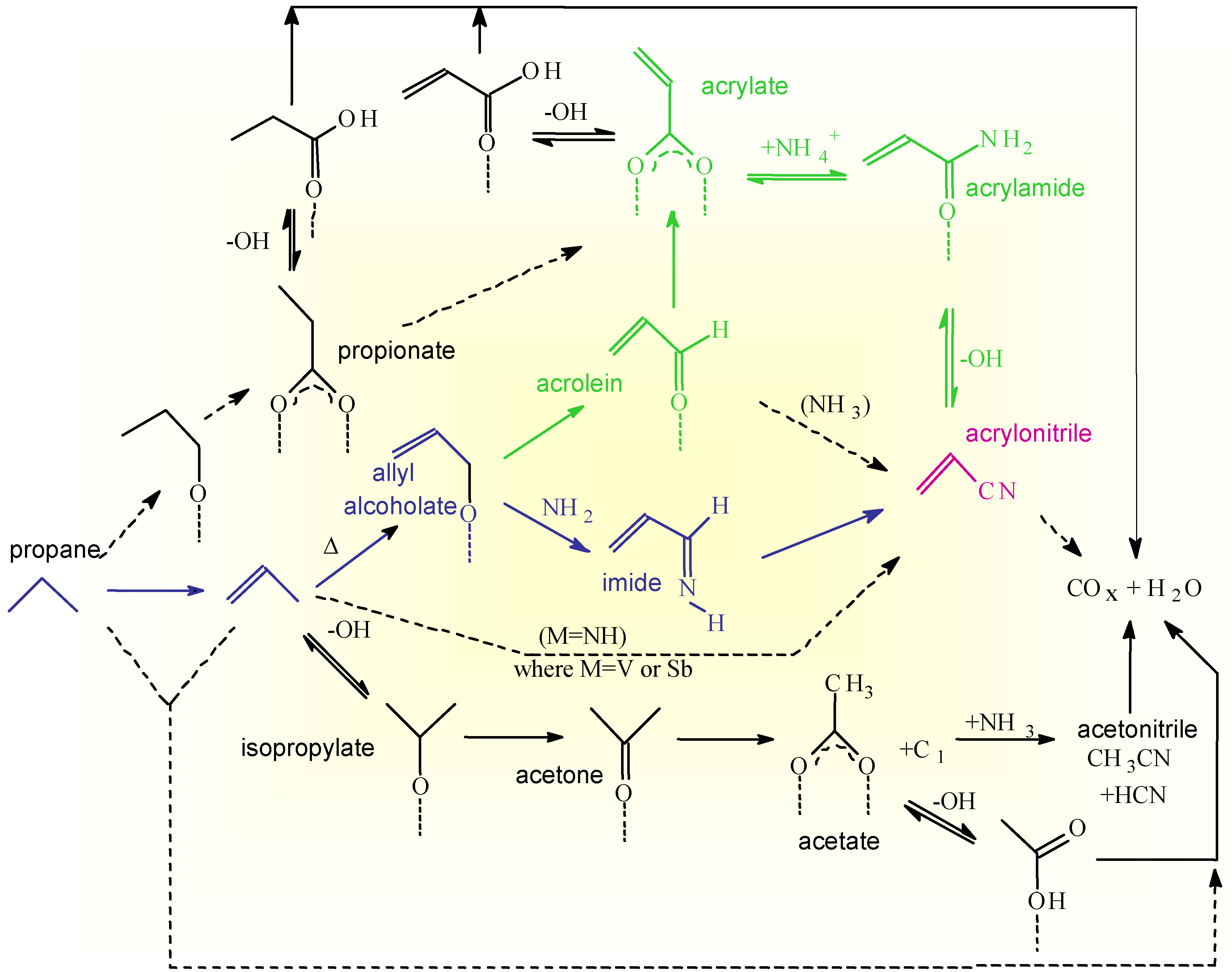
The overall surface reaction network for propane ammoxidation as derived from combined IR and transient reactivity studies [11,12,13,14] is reported in Scheme 2.
Scheme 2.
Surface reaction network for propane ammoxidation on VSbO4+Sb2O4.
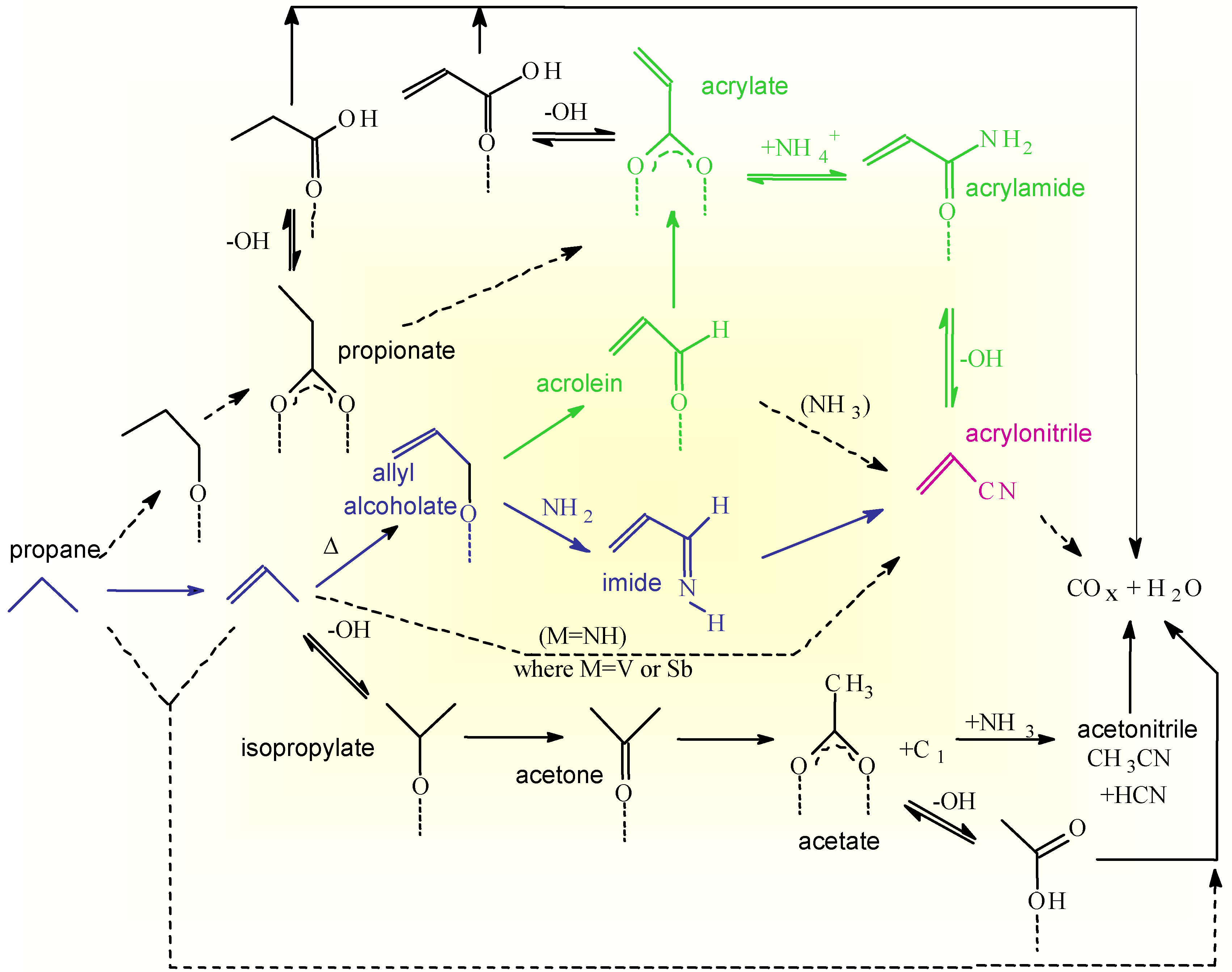
The main route of propane conversion is through the formation of propene as an intermediate, but a side reaction of acrylate formation via a propionate intermediate is possible. The relative rate of this side reaction is higher in the case of oxidation than in ammoxidation, because chemisorption of ammonia inhibits the activity of the Lewis acid sites responsible for the alkane activation according to this side route. Direct conversion of propane to carbon oxides is also indicated, but this pathway is less important in stationary conditions especially in the presence of ammonia.
Propene may be oxidized according to two routes, the first reversible and favored at low temperatures with the formation of an isopropylate species by reaction with Brönsted sites, and the second with the formation of an allyl alcoholate by abstraction of allylic hydrogen and nucleophilic lattice oxygen attack. The first adspecies easily transforms to acetone which may undergo oxidative cleavage to an acetate species and a C1 fragment. The reaction of Brönsted acid sites with ammonia to give ammonium ions slows down the rate of the first unselective side reaction. The second adspecies (allyl alcoholate) transforms easily to acrolein which further converts to an acrylate species. The acrylate species strongly interacts with the surface and is thermally stable up to relatively high temperatures, but reacts faster with ammonium ions to give an acrylamide intermediate. At higher temperatures the amide transforms to acrylonitrile, but when Brönsted sites are present, the amide is hydrolized to reform ammonia and free, weakly bonded, acrylic acid. The latter product decarboxylates easily forming carbon oxides. In the presence of strong Brönsted acid sites acrylonitrile also can be hydrolized back to free acrylic acid and ammonium ions, and thus then transformed to carbon oxides. A different activated ammonia adspecies (NH2 type species formed by heterolytic dissociation of ammonia coordinated to Lewis acid sites) reacts with the allyl alcoholate or acrolein intermediate to form the acrylimine intermediate which transforms faster to acrylonitrile than the acrylamide intermediate.
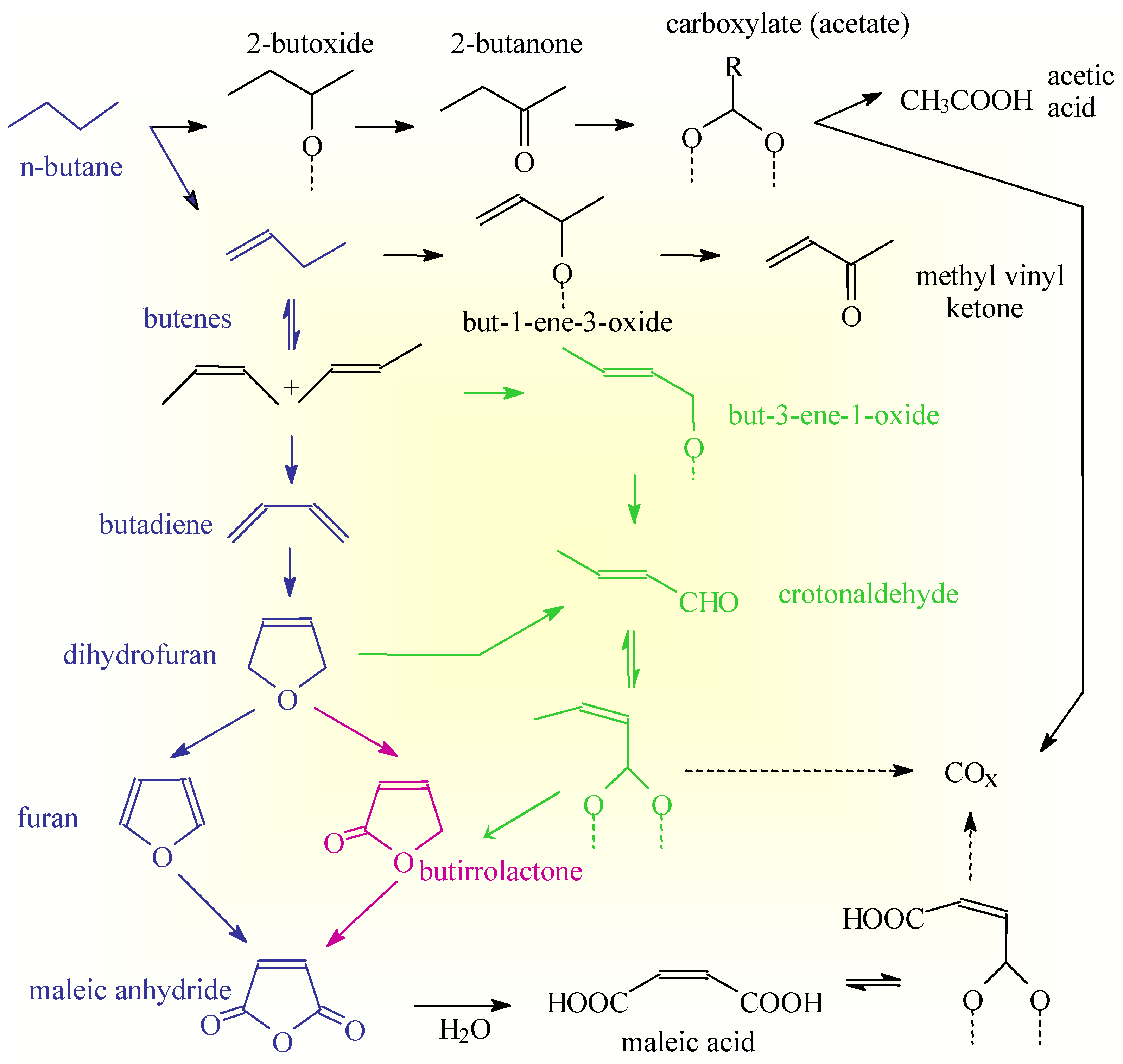
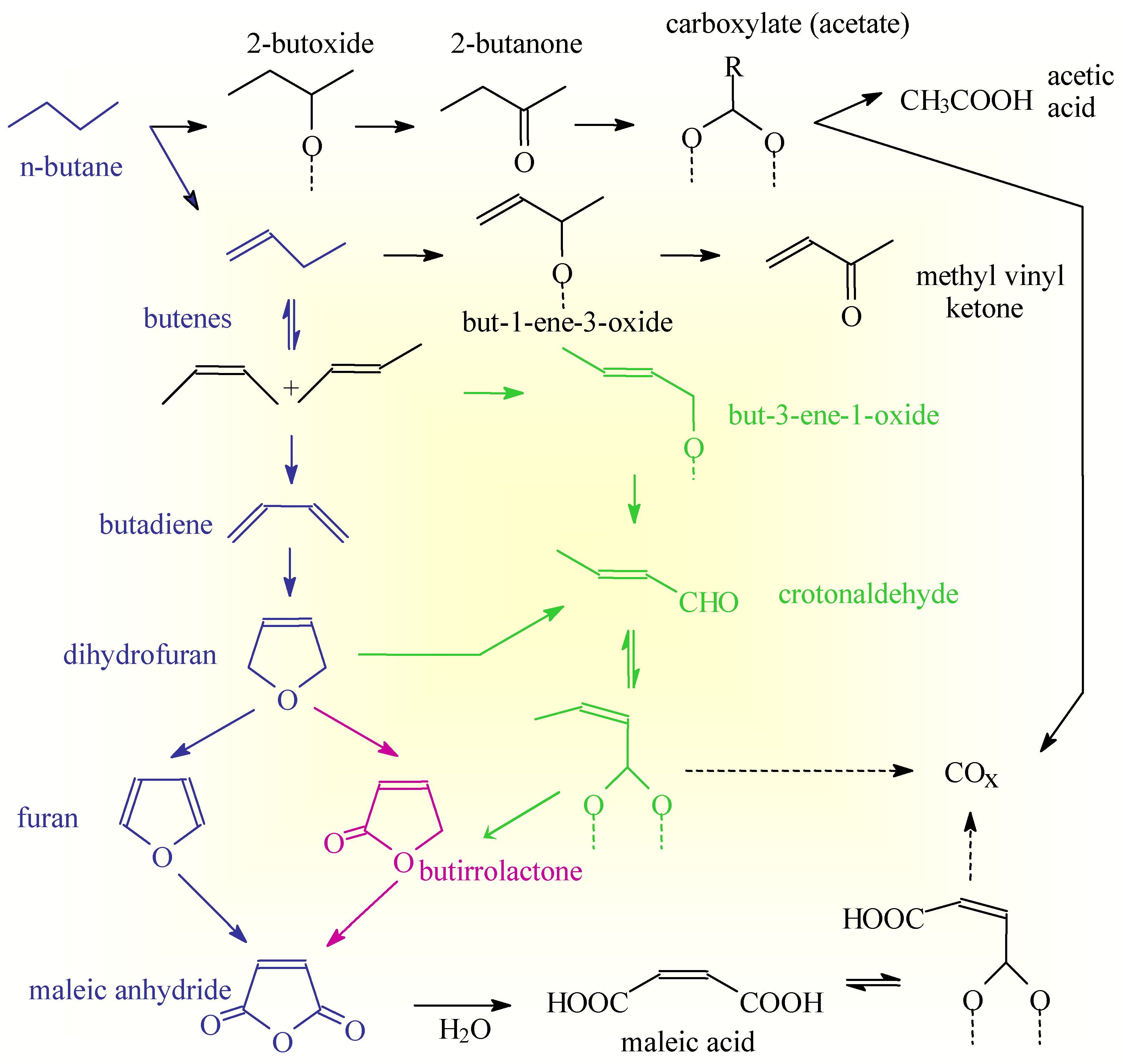
The reaction scheme reported in Scheme 2 thus shows that different pathways exist on the surface of VSbO4+Sb2O4 to form acrylonitrile and that the intrinsic selectivity of each pathway is different due to the different possibilities of having side reactions. The overall selectivity to acrylonitrile depends on the relative rates of these competitive pathways which, in turn, depend on the reaction conditions and nature of the catalyst [14]. The presence of multiple pathways of reaction is not a specific feature of the reaction pattern of acrylonitrile synthesis on VSbO4+Sb2O4, but common characteristics of the reaction patterns in complex and multisteps reactions of selective oxidation on oxides. As a further demonstration of this concept, reported in the Scheme 3 is the reaction pattern in n-butane selective oxidation on (VO)2P2O7 [3] which show several common features with that discussed above (Scheme 2).
Scheme 3.
Reaction network in n-butane oxidation on (VO)2P2O7.
Conclusions
The understanding of the catalytic chemistry on metal-oxide surface is a challenge for fundamental research in catalysis and surface science, but also a necessity to develop new reactions for a sustainable (green) chemistry. We have discussed here some aspects of the reaction mechanisms over oxide surfaces with specific reference to multistep selective oxidation reactions. In particular, two cases were discussed in detail: propane (amm)oxidation on VSbO4+Sb2O4 catalyst and n-butane oxidation on (VO)2P2O7 catalyst. Emphasis was given to the concepts of (i) modification of the surface reactivity due to chemisorption of reactants, intermediate and products and (ii) multiple pathways of reaction even for the same final reaction product. The relative rates of these multiple pathways depend on the reaction conditions (temperature, feed composition). These two concepts are needed to understand the surface catalytic chemistry and kinetic behavior of the cited catalysts, and to explain the differences in the behavior when using alkenes or alkanes as feedstock. At the same time, these concepts represent a challenge for surface science and theory to describe the catalytic chemistry and kinetic of oxide-type catalysts in complex, multistep reactions.
It was also evidenced that it is not possible to discuss of a unique reaction mechanism, but the reaction mechanism is a function of the reaction conditions. In other words, the complexity issue in bridging the gap between surface science & theory and applied catalysis is the key challenge that must be addressed to bridge this gap. Therefore, it is necessary to try to develop new models of the surface reactivity which include new evidence on aspects such as (i) the role of chemisorbed species on the surface reactivity, (ii) the presence of multiple pathways of reaction, (iii) the dynamics of catalyst reconstruction, (iv) the mobility of surface adspecies, etc [1]. The consideration of all these possible effects in analyzing the surface reactivity will make possible the design of new catalysts as well as the understanding of surface reactivity at oxide surfaces.
References
- Centi, G.; Cavani, F.; Trifirò, F. Selective Oxidation by Heterogeneous Catalysis; Kluwer Academic/Plenum Pub.: New York, 2000. [Google Scholar]
- Vision 2020 Catalysis Report. http://www.ccrhq.org/vision/index/roadmaps/catrep.html.
- Centi, G. (Ed.) Vanadyl Pyrophosphate Catalyst. Catal. Today 1993, 16, 1.
- Centi, G.; Trifirò, F.; Ebner, J.R.; Franchetti, V.M. Chem. Rev. 1988, 88, 55. [CrossRef]
- Centi, G.; Trifirò, F. Chem. Eng. Science 1990, 45, 2589. [CrossRef]
- Tundo, P.; Anastas, P.T. Green Chemistry : Challenging Perspectives; Oxford Univ. Press: New NY, 1999. [Google Scholar]
- Freund, H.-J.; Bäumer, M.; Kuhlenbeck, H. Adv. Catal. 2000, 45, 333.
- Idriss, H.; Barteau, M.A. Adv. Catal. 2000, 45, 261.
- Schlögl, R.; Knop-Gericke, A.; Havecker, M.; Wild, U.; Frickel, D.; Ressler, T.; Jentoft, R.E.; Wienold, J.; Mestl, G.; Blume, A.; Timpe, O.; Uchida, I. Topics in Catal. 2001, 15, 219.
- Topsφe, H. Studies in Surface Science and Catal. 2000, 130, 1.
- Centi, G.; Perathoner, S. Catal. Rev.-Sci. Eng. 1998, 40, 175. [CrossRef]
- Centi, G.; Perathoner, S. Catal. Today 1998, 41, 457. [CrossRef]
- Centi, G.; Perathoner, S.; Trifirò, F. Appl. Catal. A: General 1997, 157, 143. [CrossRef]
- Centi, G.; Perathoner, S. CHEMTECH 1998, 28, 13.
- Busca, G.; Centi, G.; Trifirò, F. J. Am. Chem. Soc. 1985, 107, 7757.
- Centi, G.; Marchi, F.; Perathoner, S. Appl. Catal. A 1997, 149, 225. [CrossRef]
- Centi, G.; Marchi, F.; Perathoner. J. Chem. Soc. Faraday 1996, 92, 5151. [CrossRef]
© 2001 by MDPI (http://www.mdpi.org).
Share and Cite
MDPI and ACS Style
Centi, G.; Perathoner, S. Reaction Mechanism and Control of Selectivity in Catalysis by Oxides: Some Challenges and Open Questions. Int. J. Mol. Sci. 2001, 2, 183-196. https://doi.org/10.3390/i2050183
AMA Style
Centi G, Perathoner S. Reaction Mechanism and Control of Selectivity in Catalysis by Oxides: Some Challenges and Open Questions. International Journal of Molecular Sciences. 2001; 2(5):183-196. https://doi.org/10.3390/i2050183
Chicago/Turabian StyleCenti, Gabriele, and Siglinda Perathoner. 2001. "Reaction Mechanism and Control of Selectivity in Catalysis by Oxides: Some Challenges and Open Questions" International Journal of Molecular Sciences 2, no. 5: 183-196. https://doi.org/10.3390/i2050183