Characterizing Cavities in Model Inclusion Fullerenes: A Comparative Study

Abstract
:Introduction
Methods
Calculation results and discussion
Geometric descriptors

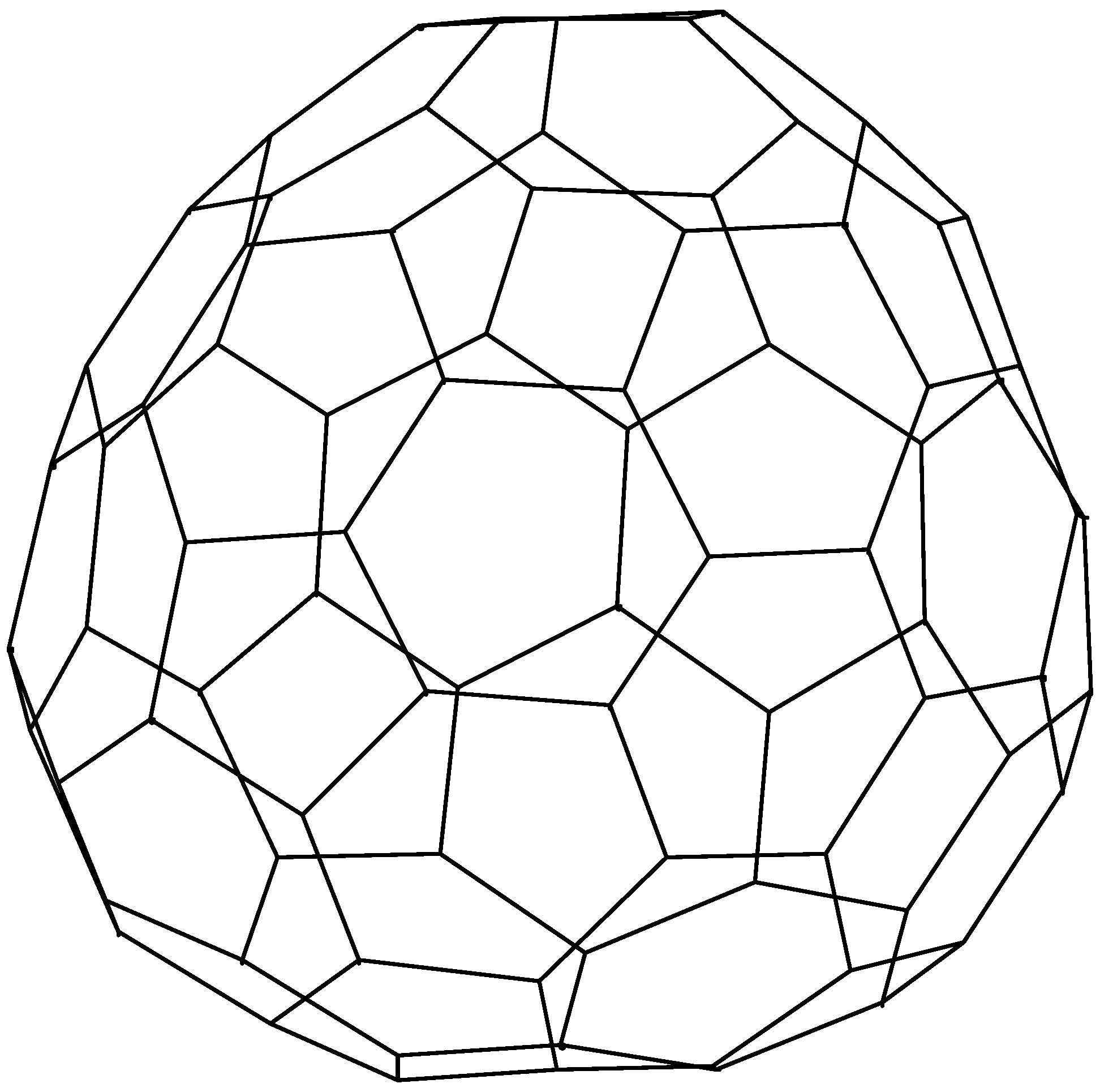
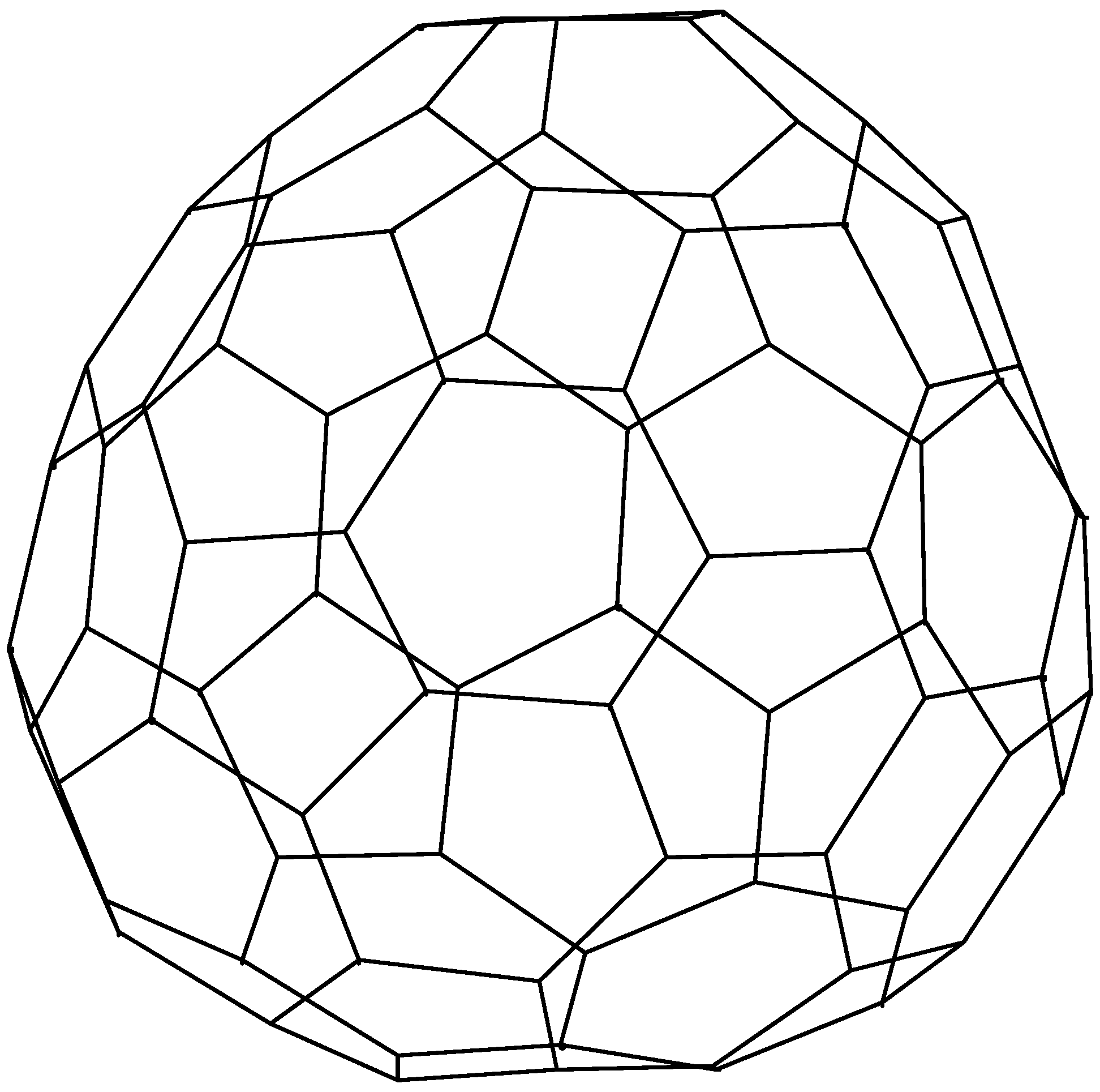{kind=link}
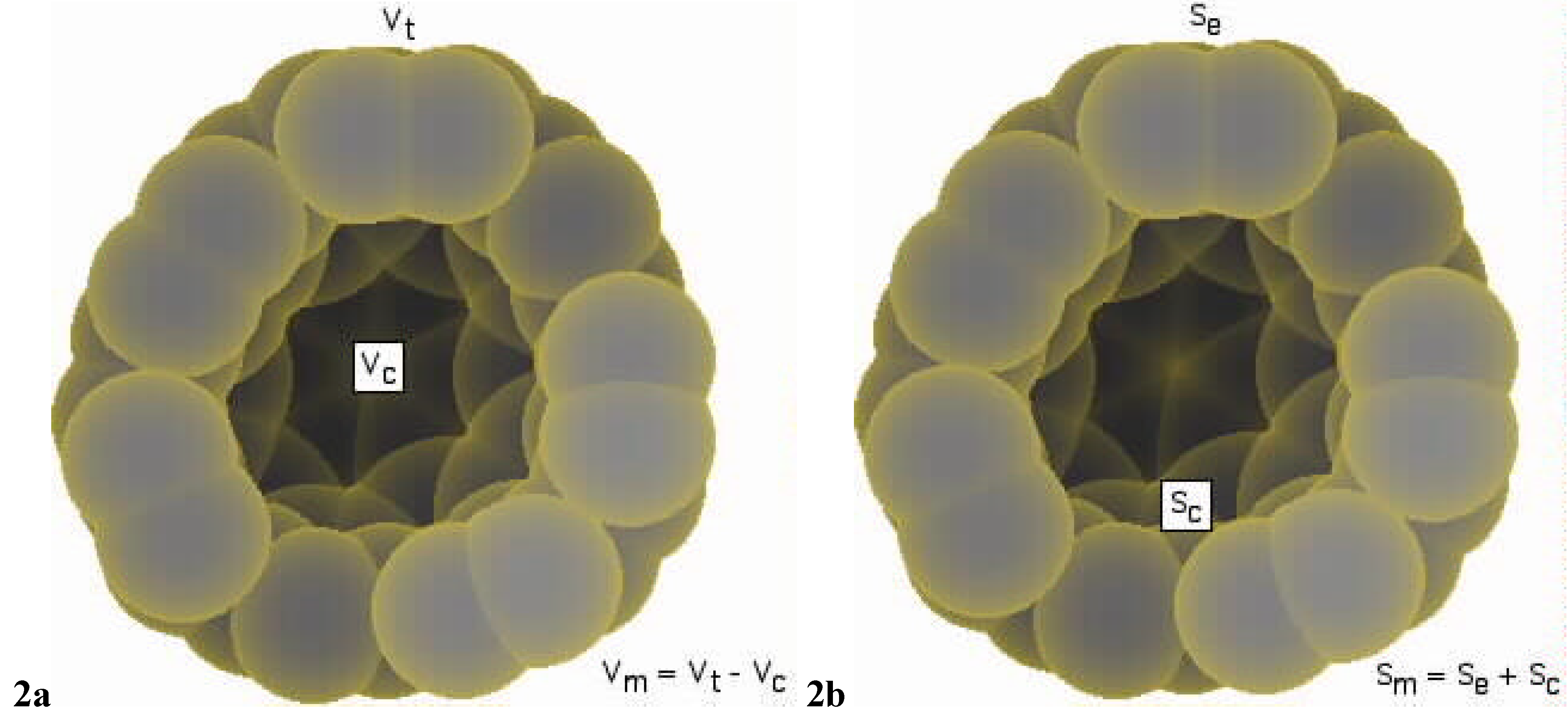
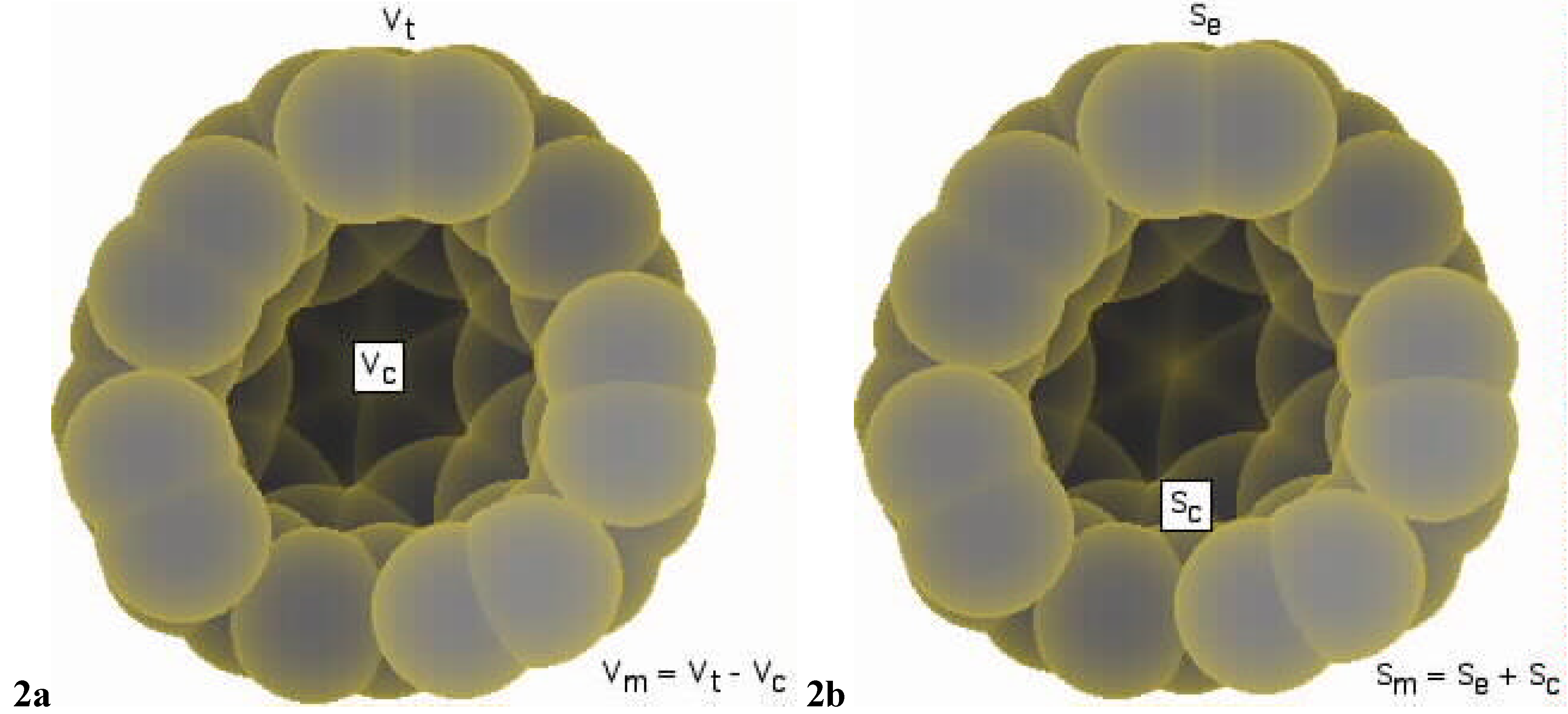{kind=link}
| Descriptor | Based on: | Limiting Parameter | Usual | This work |
| SURMO2 | Square/triangular tessellation | N1 and N2 | 12 | 72 |
| MS | Spherical tessellation | Density of surface pointsa | 10 | 100 |
| SCAP | Numerical integration | b | b | b |
| GEPOL | Triangular tessellation | NDIV | 5 | 5 |
| CSAM | Cubic lattice | Mesh gridc | 0.5 | 0.1 |
| TOPO | Cubic lattice | Mesh gridc | 0.5 | 0.1 |
| MCVS | Pseudorandom Monte Carlo | Number of observations | 104 | 107 |
| UMCVS | Uniform Monte Carlo | Number of observations | 104 | 107 |
| Property | Molecule+cavity | Cavity free molecule | ||||||
| SURMO-Ib | SURMO-IIc | SCAP | MS | GEPOL | CSAM | TOPO | Combinedd | |
| Ve | 730.6 | 730.6 | 678.5 | f | 686.4 | 683.8 | 678.8 | 678.8 |
| Sg | 391.3 | 429.4 | 503.6 | 504.9 | 505.9 | 500.3 | 477.6 | 505.9 |
| ASh | 598.0 | 617.0 | 633.8 | 627.8 | 628.8 | 627.6 | 616.5 | 628.8 |
| AS'i | 1061 | 1073 | 1078 | 1072.7 | 1072 | 1072 | 1063 | 1072 |
| Gj | 1.0025 | 0.9135 | 0.7415 | f | 0.7438 | 0.7502 | 0.7820 | 0.7383 |
| G'k | 0.5356 | 0.5878 | 0.7423 | f | 0.7371 | 0.7317 | 0.7036 | 0.7453 |
| Dl | 1.44 | 1.46 | 1.49 | 1.48 | 1.48 | 1.48 | 1.47 | 1.48 |
| Property | Cavity free molecule | |
| MCVS | UMCVS | |
| Ve | 681.4 (≥0.5) | 678.9 (0.5) |
| Sg | 508.4 (≥0.7) | 505.5 (0.7) |
| ASh | 632.5 (≥1.5) | 629.7 (1.5) |
| AS'i | 1071 (≥3) | 1074 (3) |
| Gj | 0.7365 (≥0.0014) | 0.7390 (0.0014) |
| G'k | 0.7461 (≥0.0016) | 0.7445 (0.0016) |
| Dl | 1.49 (≥0.04) | 1.48 (0.04) |
| Property | SCAP | MS | GEPOL | CSAM | TOPO | averagea | Combinedb | MCVS |
| Vc | -0.06 | - | 1 | 0.7 | -0.01 | 0.4 | -0.01 | 0.4 |
| Sd | -0.2 | 0.007 | -0.04 | -1 | -5 | 1 | -0.04 | 0.01 |
| ASe | -0.8 | -0.6 | -0.008 | -0.2 | -2 | 0.7 | -0.008 | 0.7 |
| AS'f | -0.7 | -0.4 | -0.1 | -0.1 | -1 | 0.5 | -0.1 | 0.6 |
| Gg | 0.2 | - | 0.7 | 1 | 6 | 2 | 0.04 | 0.3 |
| G’h | -0.1 | - | -1 | -2 | -5 | 2 | -0.03 | -0.4 |
| Di | -0.1 | -0.3 | 0.07 | -0.09 | -0.8 | 0.3 | 0.07 | 0.06 |
| Cavity | Va | Sb | ASc | AS'd | Ge | G'f | Dg |
| Fullerene-60 | 26.6 | 48.0 | 3.2 | 0 | 0.8978 | 1.8045 | 2.75 |
| Fullerene-70 | 39.5 | 63.8 | 8.4 | 2 | 0.8791 | 1.6152 | 2.89 |
| Fullerene-82 | 51.7 | 76.1 | 12.7 | 1 | 0.8819 | 1.4720 | 5.21 |
Solvation descriptors
| Fullerene | ΔGs,wa | ΔGs,ob | ΔGs,chc | ΔGs,cfd | log Po (SCAP)e | log Po (CDHI)f | log Pch (SCAP)g | log Pchh | log Pcf (SCAP)i | log Pcfh |
| fullerene-60 | -15.60 | -128.7 | -78.48 | -103.2 | 19.9 | 13.8 | 11.1 | 11.6 | 15.4 | 21.0 |
| fullerene-70 | -18.09 | -148.4 | -91.20 | -119.9 | 22.9 | 15.8 | 12.8 | 13.6 | 17.9 | 24.4 |
| fullerene-82 | -20.86 | -172.4 | -106.2 | -139.2 | 26.6 | 17.6 | 15.0 | 16.1 | 20.8 | 28.6 |
Conclusions
- The use of SURMO2, which do not recognize the cavities, may not be convenient for intercalation compounds, since this method gives an estimate of the global fraction of occupied space within the volume, not allowing distinction among different niches [83].
- The programs that do recognize the cavities (SCAP, MS, GEPOL, CSAM and TOPO) never exceed 1% relative deviation in molecular volume and 5% in surface area, in spite of the sometimes relevant differences in method of calculation. This means that the choice should rely mainly on other possibilities offered by the different methods such as computational performance, possibility of fragment analysis, etc. GEPOL shows, in general, the best results and the outcome is remarkably good for the entire surface areas and surface-related properties. The combined (GEPOL/TOPO) method shows relative errors below 0.1% for all of the descriptors in C60, C70 and C82.
- The molecular volume and surface area were measured accurately with MCVS and UMCVS. The UMCVS measures the molecular volume and surface areas with high precision, so that the standard deviation is divided by 10 each time the number of points (and the CPU time) is multiplied by 100. This may be compared with the CSAM and TOPO methods, using a three-dimensional grid, for which a multiplication of the CPU time by about 1 000 is required to divide the error by 10.
- The URNG in UMCVS provides the fastest convergence for the algorithm and a better estimate of the standard deviations. The use of a pseudorandom generator in MCVS produces a bias in the calculated properties and underestimates their standard deviations and errors, so that it cannot be recommended for high-precision predictions or as a benchmark maker.
- In C60, C70 and C82 the effect of including the internal cavity surface in the calculation of the solvent-accessible surfaces is small but, evidently, if the cavity were much bigger (e.g. fullerene-240, -540, and -960) this effect would be much greater because the solvent molecule would then have room inside; at present, this effect can be corrected with an additional calculation carried out by use of a method that does not recognize the cavity.
Acknowledgments
References and Notes
- Gavezzotti, A. The calculation of molecular volumes and the use of volume analysis in the investigation of structured media and of solid-state organic reactivity. J. Am. Chem. Soc. 1983, 105, 5220–5225. [Google Scholar] [CrossRef]
- Rashin, A. A.; Iofin, M.; Honig, B. Internal cavities and buried waters in globular proteins. Biochemistry 1986, 25, 3619–3625. [Google Scholar] [CrossRef] [PubMed]
- Schoenborn, B. P. J. Mol. Biol. 1969, 45, 297. [CrossRef]
- Richards, F. M. J. Mol. Biol. 1974, 82, 1–14. [CrossRef]
- Tilton, R. F., Jr.; Kuntz, I. D., Jr.; Petsko, G. A. Cavities in proteins: Structure of a metmyoglobin-xenon complex solved to 1.9 angstrom. Biochemistry 1984, 23, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Lumry, R.; Rosenberg, A. Coll. Int. C.N.R.S. 1975, 246, 55–63.
- Richards, F. M. Packing defects, cavities, volume fluctuations and access to the interior of proteins, including some general comments on surface area and protein structure. Carlsberg Res. Commun. 1979, 44, 47–63. [Google Scholar] [CrossRef]
- Smith, J. L.; Hendrickson, W. A.; Honzatko, R. B.; Sheriff, S. Structural heterogeneity in protein crystals. Biochemistry 1986, 25, 5018–5027. [Google Scholar] [CrossRef] [PubMed]
- Dill, K. A. Dominant forces in protein folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef] [PubMed]
- Richards, F. M. Areas, volumes, packing, and protein structure. Annu. Rev. Biophys. Bioeng. 1977, 6, 151–176. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S. J.; Gross, K. H.; Argos, P. Intramolecular cavities in globular proteins. Protein Eng. 1994, 7, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Pesek, J. J.; Schneider, J. F. The detection of mercury, lead, and methylmercury binding sites on lysozyme by carbon-13 NMR chemical shifts of the carboxylate groups. J. Inorg. Biochem. 1988, 32, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Eisenman, G.; Oberhauser, A. Biophys. J. 1988, 53, A631.
- Hannon, R. A.; Ford, G. C. Analysis of lattice contacts in protein crystals. Biochem. Soc. Trans. 1988, 16, 961–962. [Google Scholar]
- Olah, G. A.; Huang, H. W.; Liu, W.; Wu, Y. Location of ion-binding sites in the gramicidin channel by X-ray diffraction. J. Mol. Biol. 1991, 218, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Leach, A. R.; Kuntz, I. D. Conformational analysis of flexible ligands in macromolecular receptor sites. J. Comput. Chem. 1992, 13, 730–748. [Google Scholar] [CrossRef]
- Meng, E. C.; Shoichet, B. K.; Kuntz, I. D. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992, 13, 505–524. [Google Scholar] [CrossRef]
- Shoichet, B. K.; Bodian, D. L.; Kuntz, I. D. Molecular docking using shape descriptors. J. Comput. Chem. 1992, 13, 380–397. [Google Scholar] [CrossRef]
- Lewis, R. A.; Roe, D. C.; Huang, C.; Ferrin, T. E.; Langridge, R.; Kuntz, I. D. Automated site-directed drug design using molecular lattices. J. Mol. Graphics 1992, 10, 66–78. [Google Scholar] [CrossRef]
- Kuntz, I. D. Structure-based strategies for drug design and discovery. Science 1992, 257, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K. D.; Scheraga, H. A. Exact calculation of the volume and surface area of fused hard-sphere molecules with unequal radii. Mol. Phys. 1987, 62, 1247–1265. [Google Scholar] [CrossRef]
- Arteca, G. A.; Mezey, P. G. Shape characterization of some molecular model surfaces. J. Comput. Chem. 1988, 9, 554–563. [Google Scholar] [CrossRef]
- Arteca, G. A.; Mezey, P. G. Shape group theory of van der Waals surfaces. J. Math. Chem. 1989, 3, 43–71. [Google Scholar] [CrossRef]
- Lee, B.; Richards, F. M. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 1971, 55, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Shrake, A.; Rupley, J. A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 1973, 79, 351–371. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M. L. Solvent-accessible surfaces of proteins and nucleic acids. Science 1983, 221, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M. L. Analytical molecular surface calculation. J. Appl. Crystallogr. 1983, 16, 548–558. [Google Scholar] [CrossRef]
- Richmond, T. J. Solvent accessible surface area and excluded volume in proteins: Analytical equations for overlapping spheres and implications for hydrophobic effect. J. Mol. Biol. 1984, 178, 63–90. [Google Scholar] [CrossRef] [PubMed]
- Wodak, S. J.; Janin, J. Analytical approximation to the accessible surface area of proteins. Proc. Natl. Acad. Sci. USA 1980, 77, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- Finney, J. L. Volume occupation, environment and accessibility in proteins. The problem of the protein surface. J. Mol. Biol. 1975, 96, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Alard, P.; Wodak, S. J. Detection of cavities in a set of interpenetrating spheres. J. Comput. Chem. 1991, 12, 918–922. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. Characterizing cavities in model inclusion molecules: A comparative study. J. Mol. Graphics Mod. 1998, 16, 57–71. [Google Scholar] [CrossRef]
- Terryn, B.; Barriol, J. On the evaluation of the usual quantities or coefficients related to the shape of a molecule approximated on the basis of the van der Waals radii. J. Chim. Phys. Phys.-Chim. Biol. 1981, 78, 207–212. [Google Scholar]
- Greer, J.; Bush, B. L. Macromolecular shape and surface maps by solvent exclusion. Proc. Natl. Acad. Sci. USA 1978, 75, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Hopfinger, A. J. Polymer-solvent interactions for homopolypeptides in aqueous solution. Macromolecules 1971, 4, 731–737. [Google Scholar] [CrossRef]
- Hopfinger, A. J.; Battershell, R. D. Application of SCAP to drug design: 1. Prediction of octanol-water partition coefficients using solvent-dependent conformational analyses. J. Med. Chem. 1976, 19, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K. D.; Scheraga, H. A. Minimization of polypeptide energy, I. Preliminary structures of bovine pancreatic ribonuclease S-peptide. Proc. Natl. Acad. Sci. USA 1967, 58, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Rekker, R. F. The Hydrophobic Fragmental Constant; Elsevier: Amsterdam, 1976. [Google Scholar]
- Pascal, P. Program SCAP, Université Henry Poincaré-Nancy I. 1991. [Google Scholar]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. A universal model for the calculation of all organic solvent/water partition coefficients. J. Chromatogr. A 1998, 827, 345–358. [Google Scholar] [CrossRef]
- Torrens, F. Universal organic solvent-water partition coefficient model. J. Chem. Inf. Comput. Sci. 2000, 40, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Torrens, F. Calculation of partition coefficient and hydrophobic moment of the secondary structure of lysozyme. J. Chromatogr. A 2001, 908, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Torrens, F. Free energy of solvation and partition coefficients in methanol—water binary mixtures. Chromatographia. in press.
- Torrens, F. A new tool for rational chiral-drug design. J. Pharm. Biomed. Anal. submitted for publication.
- Torrens, F. Application of SCAP to rational chiral-drug design. Chirality. submitted for publication.
- Huron, M. J.; Claverie, P. Calculation of the interaction energy of one molecule with its whole surrounding. I. Method and application to pure nonpolar compounds. J. Phys. Chem. 1972, 76, 2123–2133. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Franke, R.; Dove, S.; Kuhne, B. Eur. J. Med. Chem. 1977, 14, 363–374.
- Giesen, D. J.; Gu, M. Z.; Cramer, C. J.; Truhlar, D. G. A universal organic solvation model. J. Org. Chem. 1996, 61, 8720–8721. [Google Scholar] [CrossRef] [PubMed]
- Giesen, D. J.; Chambers, C. C.; Cramer, C. J.; Truhlar, D. G. Solvation model for chloroform based on class IV atomic charges. J. Phys. Chem. B 1997, 101, 2061–2069. [Google Scholar] [CrossRef]
- Leo, A.; Hansch, C.; Elkins, D. Partition coefficients and their uses. Chem. Rev. 1971, 71, 525–616. [Google Scholar] [CrossRef]
- Klopman, G.; Iroff, L. D. Calculation of partition coefficients by the charge density method. J. Comput. Chem. 1981, 2, 157–160. [Google Scholar] [CrossRef]
- Leo, A.; Jow, P. Y. C.; Silipo, C.; Hansch, C. J. Med. Chem. 1975, 18, 865–868. [CrossRef]
- Hansch, C.; Leo, A. J. Substituent Constants for Correlation Analysis in Chemistry and Biology; John Wiley and Sons: New York, 1979. [Google Scholar]
- Pascual-Ahuir, J. L.; Silla, E.; Tomasi, J.; Bonaccorsi, R. Electrostatic interaction of a solute with a continuum. Improved description of the cavity and of the surface cavity bound charge distribution. J. Comput. Chem. 1987, 8, 778–787. [Google Scholar] [CrossRef]
- Muller, J. J. Calculation of scattering curves for macromolecules in solution and comparison with results of methods using effective atomic scattering factors. J. Appl. Crystallogr. 1983, 16, 74–82. [Google Scholar] [CrossRef]
- Pavlov, M. Yu.; Fedorov, B. A. Improved technique for calculating X-ray scattering intensity of biopolymers in solution: evaluation of the form, volume, and surface of a particle. Biopolymers 1983, 22, 1507–1522. [Google Scholar] [CrossRef]
- Connolly, M. L. Computation of molecular volume. J. Am. Chem. Soc. 1985, 107, 1118–1124. [Google Scholar] [CrossRef]
- Higo, J.; Gõ, N. Algorithm for rapid calculation of excluded volume of large molecules. J. Comput. Chem. 1989, 10, 376–379. [Google Scholar] [CrossRef]
- Senn, P. Numerical computation of surface areas of molecules. J. Math. Chem. 1991, 6, 351–358. [Google Scholar]
- Torrens, F.; Ortí, E.; Sánchez-Marín, J. Vectorized TOPO program for the theoretical simulation of molecular shape. J. Chim. Phys. Phys.-Chim. Biol. 1991, 88, 2435–2441. [Google Scholar]
- Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Meyer, A. Y. Molecular mechanics and molecular shape. Part 1. Van der Waals descriptors of simple molecules. J. Chem. Soc., Perkin Trans. 2 1985, 1161–1169. [Google Scholar] [CrossRef]
- Meyer, A. Y. Molecular mechanics and molecular shape. V. On the computation of the bare surface area of molecules. J. Comput. Chem. 1988, 9, 18–24. [Google Scholar] [CrossRef]
- Hermann, R. B. Theory of hydrophobic bonding. II. The correlation of hydrocarbon solubility in water with solvent cavity surface area. J. Phys. Chem. 1972, 76, 2754–2759. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. New dimension indices for the characterization of the solvent-accessible surface. J. Comput. Chem. 2001, 22, 477–487. [Google Scholar] [CrossRef]
- Lewis, M.; Rees, D. C. Fractal surfaces of proteins. Science 1985, 230, 1163–1165. [Google Scholar] [CrossRef]
- Fraga, S. A semiempirical formulation for the study of molecular interactions. J. Comput. Chem. 1982, 3, 329–334. [Google Scholar] [CrossRef]
- Fraga, S. Molecular associations. Comput. Phys. Commun. 1983, 29, 351–359. [Google Scholar] [CrossRef]
- Torrens, F.; Ortí, E.; Sánchez-Marín, J. Pair-potential calculation of molecular associations: A vectorized version. Comput. Phys. Commun. 1991, 66, 341–362. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. AMYR 2: A new version of a computer program for pair potential calculation of molecular associations. Comput. Phys. Commun. 1998, 115, 87–89. [Google Scholar] [CrossRef]
- Hammersley, J. M.; Handscomb, D. C. Monte Carlo Methods; Chapman and Hall: London, 1983; Chap. 5. [Google Scholar]
- Petitjean, M. On the analytical calculation of van der Waals surfaces and volumes: Some numerical aspects. J. Comput. Chem. 1994, 15, 507–523. [Google Scholar] [CrossRef]
- Luscher, M. A portable high-quality random number generator for lattice field theory simulations. Comput. Phys. Comun. 1994, 79, 100–110. [Google Scholar] [CrossRef]
- James, F. RANLUX: A Fortran implementation of high-quality pseudorandom number generator of Luscher. Comput. Phys. Comun. 1994, 79, 111–114. [Google Scholar] [CrossRef]
- Allinger, N. L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar] [CrossRef]
- Deming, W. E. Statistical Adjustment of Data; Dover: New York, 1964. [Google Scholar]
- Kantola, A.; Villar, H. O.; Loew, G. H. Atom based parametrization for a conformationally dependent hydrophobic index. J. Comput. Chem. 1991, 12, 681–689. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity: A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Torrens, F.; Sánchez-Marín, J.; Nebot-Gil, I. Interacting induced dipoles polarization model for molecular polarizabilities. Reference molecules, amino acids and model peptides. J. Mol. Struct. (Theochem) 1999, 463, 27–39. [Google Scholar] [CrossRef]
- The CDHI program is available from the authors on request.
- Leo, A.; Hansch, C. Linear Free-Energy Relationships between Partitioning Solvent Systems. J. Org. Chem. 1971, 36, 1539–1544. [Google Scholar] [CrossRef]
- Torrens, F. Fractal dimension of different structural-type zeolites and of the active sites. Top. Catal. in press.
- Sample availability: Not available.
© 2001 by MDPI (http://www.mdpi.org).
Share and Cite
Torrens, F. Characterizing Cavities in Model Inclusion Fullerenes: A Comparative Study. Int. J. Mol. Sci. 2001, 2, 72-88. https://doi.org/10.3390/i2020072
Torrens F. Characterizing Cavities in Model Inclusion Fullerenes: A Comparative Study. International Journal of Molecular Sciences. 2001; 2(2):72-88. https://doi.org/10.3390/i2020072
Chicago/Turabian StyleTorrens, Francisco. 2001. "Characterizing Cavities in Model Inclusion Fullerenes: A Comparative Study" International Journal of Molecular Sciences 2, no. 2: 72-88. https://doi.org/10.3390/i2020072