The Na/K-ATPase Signaling: From Specific Ligands to General Reactive Oxygen Species


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Na/K-ATPase Signaling and Intracellular Ionic Concentration
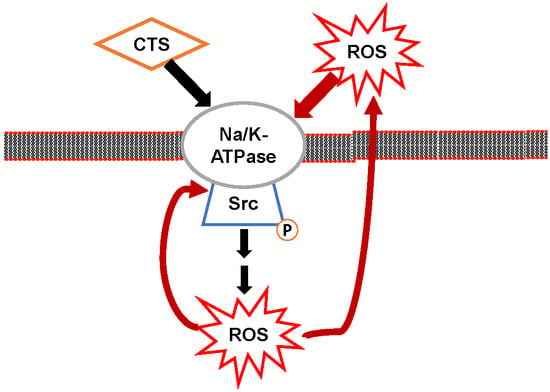
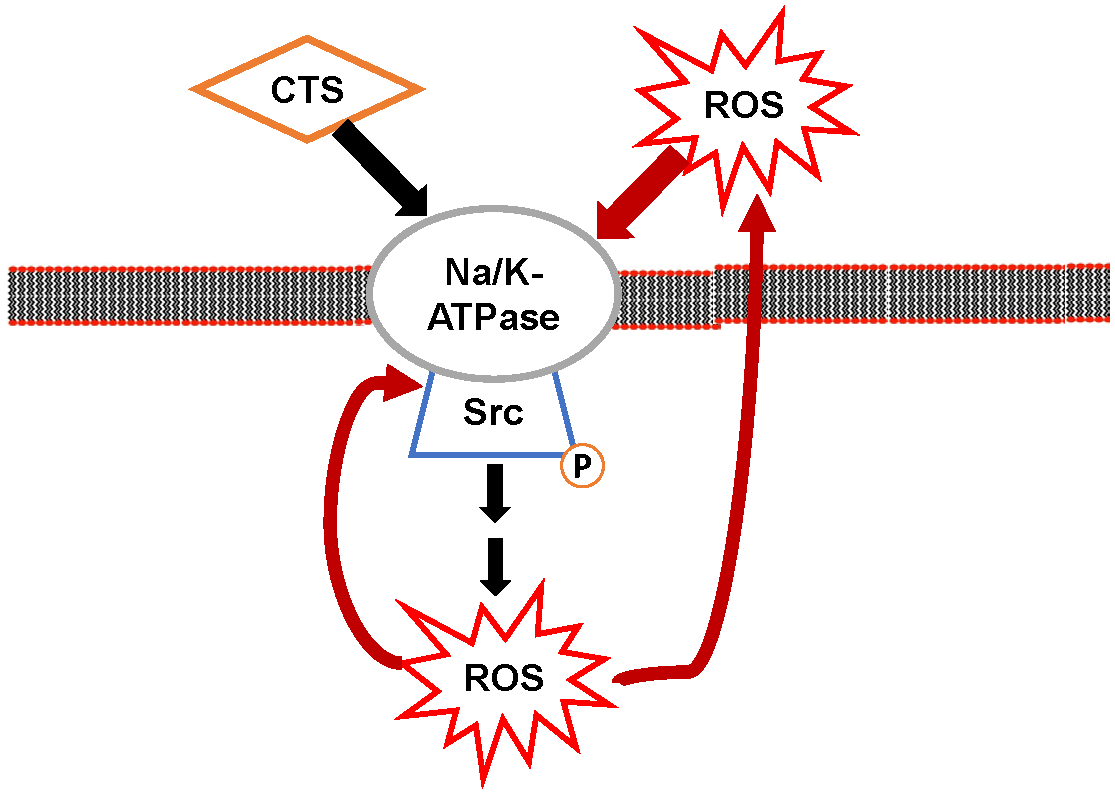
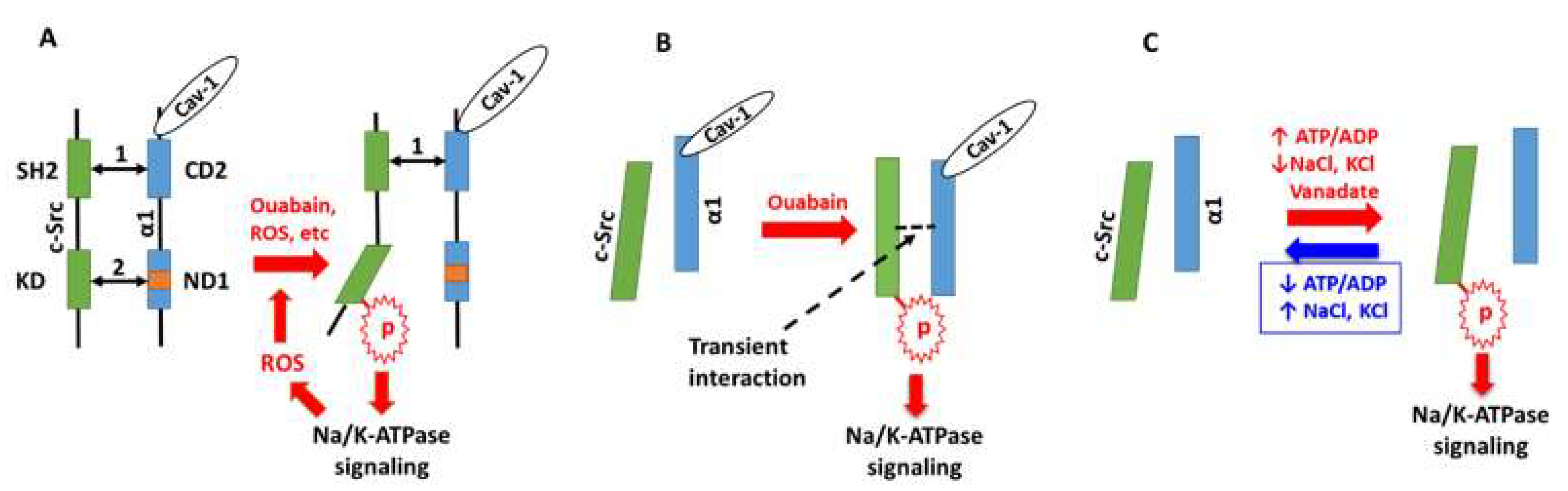
3. Na/K-ATPase Signaling and Reactive Oxygen Species (ROS): The Positive Oxidant Amplification Loop
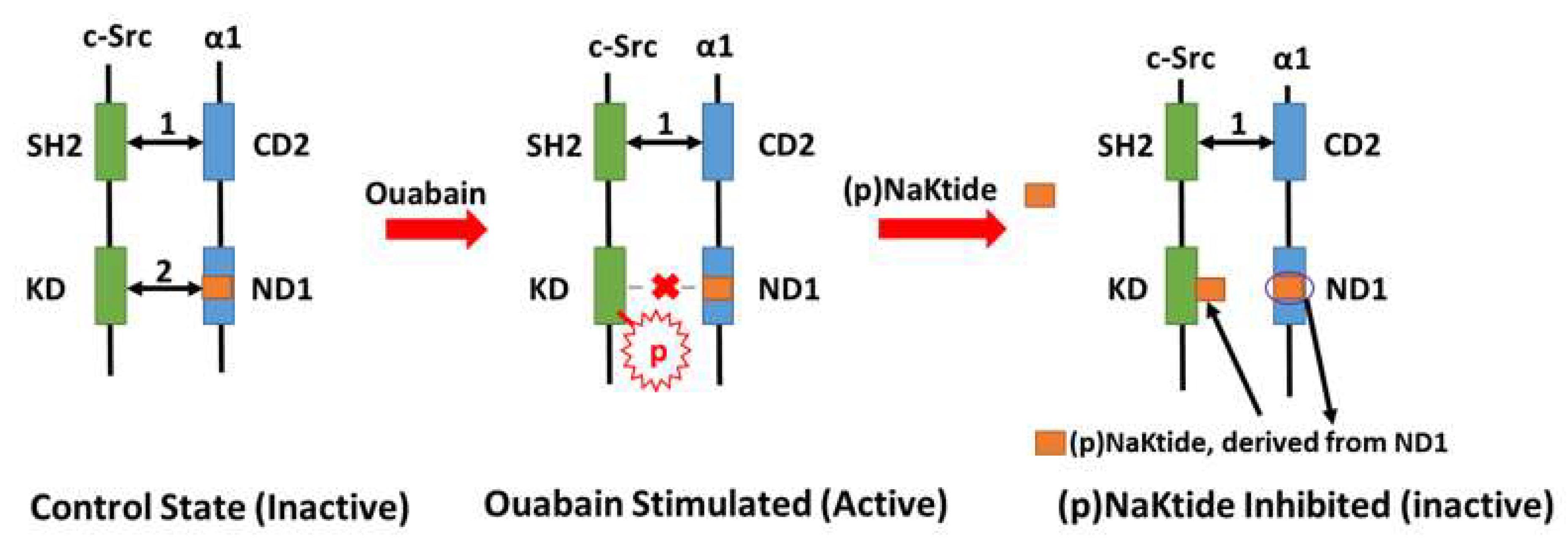
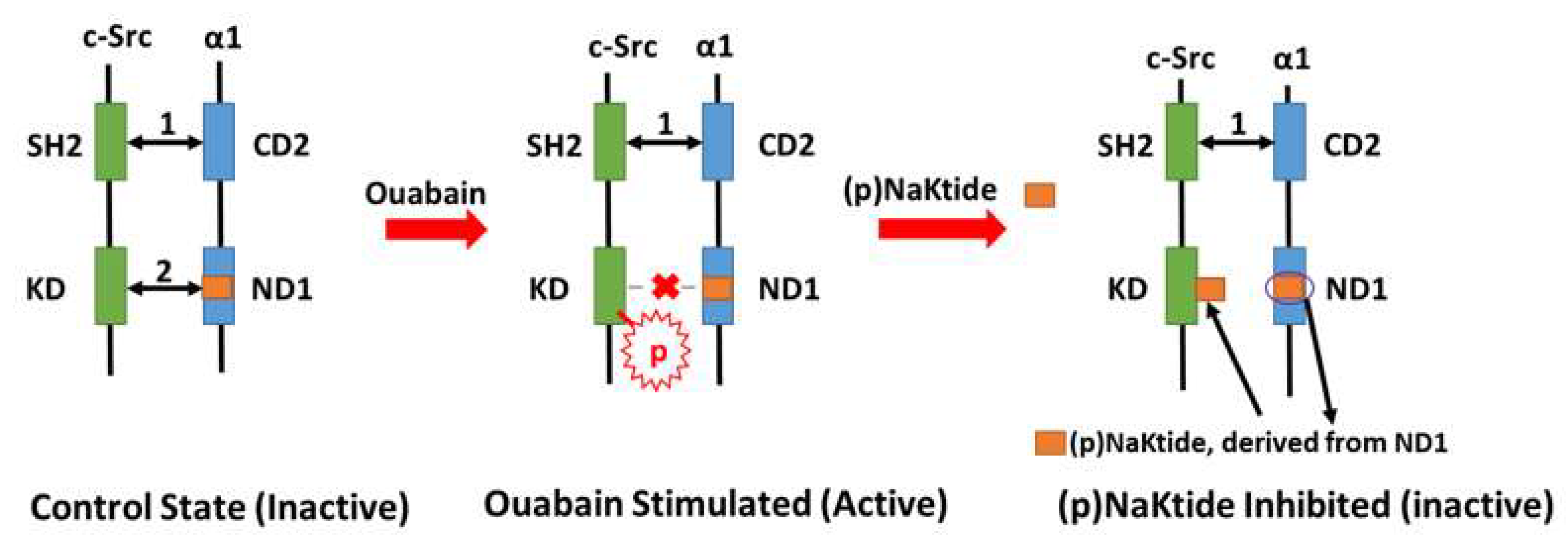
4. Na/K-ATPase Signaling and pNaKtide: A Specific Antagonist of c-Src Kinase that Breaks the Oxidant Amplification Loop
5. Na/K-ATPase Signaling-Mediated Transporter Endocytosis and Renal Sodium Handling
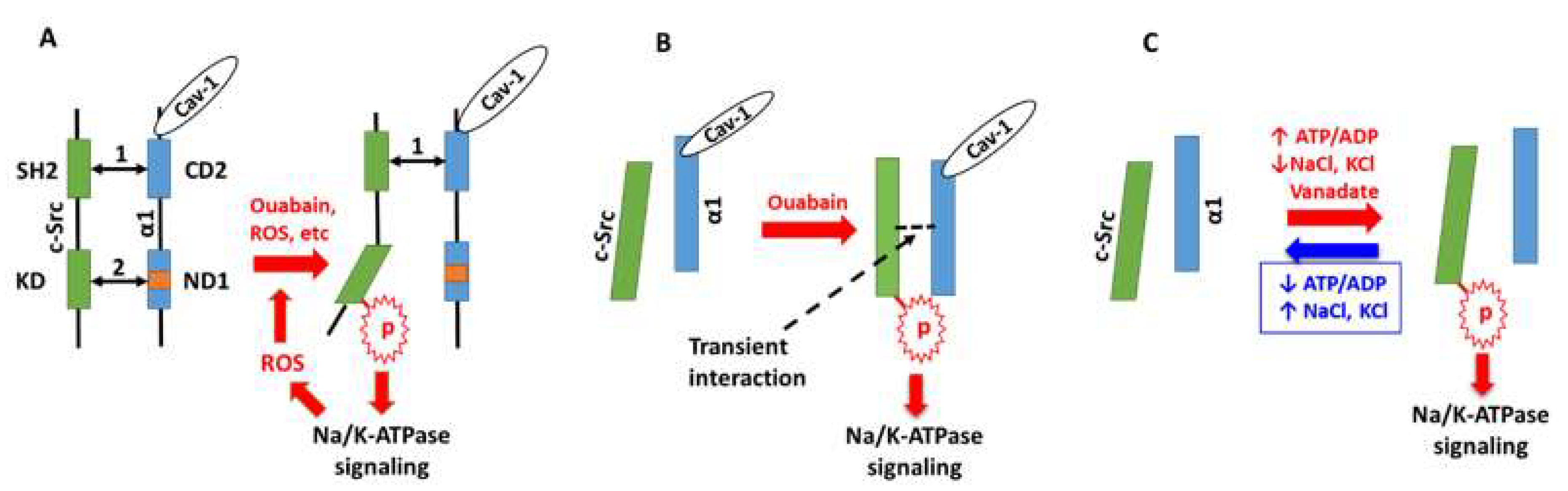
6. Perspectives: The Working Models of Na/K-ATPase Signaling
Funding
Conflicts of Interest
References
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar] [CrossRef]
- Peng, M.; Huang, L.; Xie, Z.; Huang, W.H.; Askari, A. Partial inhibition of Na+/K+-ATPase by ouabain induces the Ca2+-dependent expressions of early-response genes in cardiac myocytes. J. Biol. Chem. 1996, 271, 10372–10378. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, H.; Xie, Z. Ouabain-induced hypertrophy in cultured cardiac myocytes is accompanied by changes in expression of several late response genes. J. Mol. Cell. Cardiol. 1997, 29, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Kometiani, P.; Xie, Z. Differential regulation of Na/K-ATPase alpha-subunit isoform gene expressions in cardiac myocytes by ouabain and other hypertrophic stimuli. J. Mol. Cell. Cardiol. 1997, 29, 3157–3167. [Google Scholar] [CrossRef] [PubMed]
- Kometiani, P.; Li, J.; Gnudi, L.; Kahn, B.B.; Askari, A.; Xie, Z. Multiple signal transduction pathways link Na+/K+-ATPase to growth-related genes in cardiac myocytes. The roles of Ras and mitogen-activated protein kinases. J. Biol. Chem. 1998, 273, 15249–15256. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Kometiani, P.; Liu, J.; Li, J.; Shapiro, J.I.; Askari, A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J. Biol. Chem. 1999, 274, 19323–19328. [Google Scholar] [CrossRef] [PubMed]
- Sugden, P.H. Signaling in myocardial hypertrophy: Life after calcineurin? Circ. Res. 1999, 84, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Askari, A. Na/K-ATPase as a signal transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Barry, W.H.; Hasin, Y.; Smith, T.W. Sodium pump inhibition, enhanced calcium influx via sodium-calcium exchange, and positive inotropic response in cultured heart cells. Circ. Res. 1985, 56, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Reuter, H.; Reuter, H.; Pott, C.; Goldhaber, J.I.; Henderson, S.A.; Philipson, K.D.; Schwinger, R.H. Na+–Ca2+ exchange in the regulation of cardiac excitation–contraction coupling. Cardiovasc. Res. 2005, 67, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar] [PubMed]
- Tian, J.; Gong, X.; Xie, Z. Signal-transducing function of Na+-K+-ATPase is essential for ouabain’s effect on [Ca2+]i in rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1899–H1907. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Tian, J.; Liu, J.; Garlid, K.D.; Shapiro, J.I.; Xie, Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial KATP channels. Mol. Cell. Biochem. 2003, 242, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Aizman, O.; Uhlén, P.; Lal, M.; Brismar, H.; Aperia, A. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc. Natl. Acad. Sci. USA 2001, 98, 13420–13424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyakawa-Naito, A.; Uhlén, P.; Lal, M.; Aizman, O.; Mikoshiba, K.; Brismar, H.; Zelenin, S.; Aperia, A. Cell signaling microdomain with Na,K-ATPase and inositol 1,4,5-trisphosphate receptor generates calcium oscillations. J. Biol. Chem. 2003, 278, 50355–50361. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Cai, T.; Tian, J.; Ivanov, A.V.; Giovannucci, D.R.; Xie, Z. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol. Biol. Cell 2005, 16, 4034–4045. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cai, T.; Yang, C.; Turner, D.A.; Giovannucci, D.R.; Xie, Z. Regulation of inositol 1,4,5-trisphosphate receptor-mediated calcium release by the Na/K-ATPase in cultured renal epithelial cells. J. Biol. Chem. 2008, 283, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Malmersjo, S.; Li, J.; Ando, H.; Aizman, O.; Uhlén, P.; Mikoshiba, K.; Aperia, A. Distinct role of the N-terminal tail of the Na,K-ATPase catalytic subunit as a signal transducer. J. Biol. Chem. 2006, 281, 21954–21962. [Google Scholar] [CrossRef] [PubMed]
- Marban, E.; Tsien, R.W. Enhancement of calcium current during digitalis inotropy in mammalian heart: Positive feed-back regulation by intracellular calcium? J. Physiol. 1982, 329, 589–614. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, E.M. Regulation of voltage-dependent calcium channels in rat sensory neurones involves a Ras-mitogen-activated protein kinase pathway. J. Physiol. 2000, 527, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydemir-Koksoy, A.; Abramowitz, J.; Allen, J.C. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J. Biol. Chem. 2001, 276, 46605–46611. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Wang, H.; Tian, J.; Xie, Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J. Biol. Chem. 2002, 277, 18694–18702. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Askari, A.; Xie, Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J. Biol. Chem. 2000, 275, 27832–27837. [Google Scholar] [CrossRef] [PubMed]
- Pressley, T.A. Ionic regulation of Na+, K+-ATPase expression. Semin. Nephrol. 1992, 12, 67–71. [Google Scholar] [PubMed]
- Kuroki, D.W.; Minden, A.; Sánchez, I.; Wattenberg, E.V. Regulation of a c-Jun amino-terminal kinase/stress-activated protein kinase cascade by a sodium-dependent signal transduction pathway. J. Biol. Chem. 1997, 272, 23905–23911. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wattenberg, E.V. Differential activation of mitogen-activated protein kinases by palytoxin and ouabain, two ligands for the Na+, K+-ATPase. Toxicol. Appl. Pharmacol. 1998, 151, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Orlov, S.N.; Thorin-Trescases, N.; Pchejetski, D.; Taurin, S.; Farhat, N.; Tremblay, J.; Thorin, E.; Hamet, P. Na+/K+ pump and endothelial cell survival: [Na+]i/[K+]i-independent necrosis triggered by ouabain, and protection against apoptosis mediated by elevation of [Na+]i. Pflugers Arch. 2004, 448, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Taurin, S.; Dulin, N.O.; Pchejetski, D.; Grygorczyk, R.; Tremblay, J.; Hamet, P.; Orlov, S.N. c-Fos expression in ouabain-treated vascular smooth muscle cells from rat aorta: Evidence for an intracellular-sodium-mediated, calcium-independent mechanism. J. Physiol. 2002, 543, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.Y.; Wei, L.; Xia, S.; Rothman, S.; Yu, S.P. Ionic mechanism of ouabain-induced concurrent apoptosis and necrosis in individual cultured cortical neurons. J. Neurosci. 2002, 22, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Golovina, V.A.; Song, H.; James, P.F.; Lingrel, J.B.; Blaustein, M.P. Na+ pump alpha 2-subunit expression modulates Ca2+ signaling. Am. J. Physiol. Cell Physiol. 2003, 284, C475–C486. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Blaustein, M.P. Na+ pump low and high ouabain affinity alpha subunit isoforms are differently distributed in cells. Proc. Natl. Acad. Sci. USA 1997, 94, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Blaustein, M.P. Distinct distribution of different Na+ pump alpha subunit isoforms in plasmalemma. Ann. N. Y. Acad. Sci. 1997, 834, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.D.; Etter, E.F.; Philipson, K.D.; Carrington, W.A.; Fogarty, K.E.; Lifshitz, L.M.; Fay, F.S. Coupling of the Na+/Ca2+ exchanger, Na+/K+ pump and sarcoplasmic reticulum in smooth muscle. Nature 1993, 365, 657–660. [Google Scholar] [CrossRef] [PubMed]
- Arnon, A.; Hamlyn, J.M.; Blaustein, M.P. Ouabain augments Ca2+ transients in arterial smooth muscle without raising cytosolic Na+. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H679–H691. [Google Scholar] [CrossRef] [PubMed]
- Arnon, A.; Hamlyn, J.M.; Blaustein, M.P. Na+ entry via store-operated channels modulates Ca2+ signaling in arterial myocytes. Am. J. Physiol. Cell Physiol. 2000, 278, C163–C173. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Song, H.; Nakai, J.; Ohkura, M.; Kotlikoff, M.I.; Kinsey, S.P.; Golovina, V.A.; Blaustein, M.P. Local subplasma membrane Ca2+ signals detected by a tethered Ca2+ sensor. Proc. Natl. Acad. Sci. USA 2006, 103, 13232–13237. [Google Scholar] [CrossRef] [PubMed]
- Contreras, R.G.; Shoshani, L.; Flores-Maldonado, C.; Lazaro, A.; Cereijido, M. Relationship between Na(+),K(+)-ATPase and cell attachment. J. Cell Sci. 1999, 112, 4223–4232. [Google Scholar] [PubMed]
- Huang, W.H.; Wang, Y.; Askari, A. (Na+ + K+)-ATPase: Inactivation and degradation induced by oxygen radicals. Int. J. Biochem. 1992, 24, 621–626. [Google Scholar] [PubMed]
- Huang, W.H.; Wang, Y.; Askari, A.; Zolotarjova, N.; Ganjeizadeh, M. Different sensitivities of the Na+/K+-ATPase isoforms to oxidants. Biochim. Biophys. Acta 1994, 1190, 108–114. [Google Scholar] [CrossRef]
- Figtree, G.A.; Keyvan, K.G.; Liu, C.C.; Rasmussen, H.H. Oxidative regulation of the Na(+)-K(+) pump in the cardiovascular system. Free Radic. Biol. Med. 2012, 53, 2263–2268. [Google Scholar] [CrossRef] [PubMed]
- Figtree, G.A.; Liu, C.C.; Bibert, S.; Hamilton, E.J.; Garcia, A.; White, C.N.; Chia, K.K.M.; Cornelius, F.; Geering, K.; Rasmussen, H.H. Reversible oxidative modification: A key mechanism of Na+-K+ pump regulation. Circ. Res. 2009, 105, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V.A.; Kamanina, Y.V.; Ziganshin, R.H.; Meng, X.; Anashkina, A.A.; Makhro, A.; Lopina, O.D.; Gassmann, M.; et al. S-glutathionylation of the Na,K-ATPase catalytic alpha subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef] [PubMed]
- ThÉvenod, F.; Friedmann, J.M. Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+/K+-ATPase through proteasomal and endo-/lysosomal proteolytic pathways. FASEB J. 1999, 13, 1751–1761. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Wang, Y.; Askari, A.; Huang, W.H.; Klaunig, J.E.; Askari, A. Studies on the specificity of the effects of oxygen metabolites on cardiac sodium pump. J. Mol. Cell. Cardiol. 1990, 22, 911–920. [Google Scholar] [CrossRef]
- Yan, Y.; Shapiro, A.P.; Haller, S.; Katragadda, V.; Liu, L.; Tian, J.; Basrur, V.; Malhotra, D.; Xie, Z.; Abraham, N.G.; et al. The Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J. Biol. Chem. 2013, 288, 34249–34258. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shapiro, A.P.; Mopidevi, B.R.; Chaudhry, M.A.; Maxwell, K.; Haller, S.T.; Drummond, C.A.; Kennedy, D.J.; Tian, J.; Malhotra, D.; et al. Protein carbonylation of an amino acid residue of the Na/K-ATPase alpha1 subunit determines Na/K-ATPase signaling and sodium transport in renal proximal tubular cells. J. Am. Heart Assoc. 2016, 5, e003675. [Google Scholar] [CrossRef] [PubMed]
- Bibert, S.; Liu, C.C.; Figtree, G.A.; Garcia, A.; Hamilton, E.J.; Marassi, F.M.; Sweadner, K.J.; Cornelius, F.; Geering, K.; Rasmussen, H.H. FXYD proteins reverse inhibition of the Na+-K+ pump mediated by glutathionylation of its β1 subunit. J. Biol. Chem. 2011, 286, 18562–18572. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, A.; Petrushanko, I.Y.; Hernansanz-Agustín, P.; Martínez-Ruiz, A. “Oxygen Sensing” by Na,K-ATPase: These miraculous thiols. Front. Physiol. 2016, 7, 314. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Chen, Y.; Huang, W.; Viterna, J.; Liu, J.; Westfall, K.; Tian, J.; Bartlett, D.J.; Tang, W.H.W.; Xie, Z.; et al. CD36 and Na/K-ATPase-alpha1 form a proinflammatory signaling loop in kidney. Hypertension 2013, 61, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, W.; Yang, M.; Xin, G.; Cui, W.; Xie, Z.; Silverstein, R.L. Cardiotonic steroids stimulate macrophage inflammatory responses through a pathway involving CD36, TLR4, and Na/K-ATPase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kennedy, D.J.; Ramakrishnan, D.P.; Yang, M.; Huang, W.; Li, Z.; Xie, Z.; Chadwick, A.C.; Sahoo, D.; Silverstein, R.L. Oxidized LDL–bound CD36 recruits an Na+/K+-ATPase–Lyn complex in macrophages that promotes atherosclerosis. Sci. Signal. 2015, 8, ra91. [Google Scholar] [CrossRef] [PubMed]
- Pasdois, P.; Quinlan, C.L.; Rissa, A.; Tariosse, L.; Vinassa, B.; Costa, A.D.; Pierre, S.V.; Santos, P.D.; Garlid, K.D. Ouabain protects rat hearts against ischemia-reperfusion injury via pathway involving src kinase, mitoKATP, and ROS. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1470–H1478. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, J.; Liu, J.; Yuan, Z.; Pierre, S.V.; Qu, W.; Zhao, X.; Xie, Z. Involvement of Na+/K+-ATPase in hydrogen peroxide-induced hypertrophy in cardiac myocytes. Free Radic. Biol. Med. 2006, 41, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ye, Q.; Liu, C.; Xie, J.X.; Yan, Y.; Lai, F.; Duan, Q.; Li, X.; Tian, J.; Xie, Z. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic. Biol. Med. 2014, 71, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.; Xie, Z. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Z.; Xie, J.X.; Li, X.; Tian, J.; Cai, T.; Cui, H.; Ding, H.; Shapiro, J.I.; Xie, Z. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J. Biol. Chem. 2011, 286, 32394–32403. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Chaudhry, M.; Maxwell, K.; Yan, Y.; Wang, X.; Shah, P.T.; Khawaja, A.A.; Martin, R.; Robinette, T.J.; et al. Attenuation of Na/K-ATPase mediated oxidant amplification with pNaKtide ameliorates experimental uremic cardiomyopathy. Sci. Rep. 2016, 6, 34592. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Maxwell, K.; Yan, Y.; Liu, J.; Chaudhry, M.A.; Getty, M.; Xie, Z.; Abraham, N.G.; Shapiro, J.I. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci. Adv. 2015, 1, e1500781. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Srikanthan, K.; Goguet-Rubio, P.; Nichols, A.; Mallick, A.; Nawab, A.; Martin, R.; Shah, P.T.; Chaudhry, M.; Sigdel, S.; et al. pNaKtide Attenuates steatohepatitis and atherosclerosis by blocking Na/K-ATPase/ROS amplification in C57Bl6 and ApoE knockout mice fed a western diet. Sci. Rep. 2017, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Nichols, A.; Mallick, A.; Klug, R.L.; Liu, J.; Wang, X.; Srikanthan, K.; Goguet-Rubio, P.; Nawab, A.; Pratt, R.; et al. The Na/K-ATPase oxidant amplification loop regulates aging. Sci. Rep. 2018, 8, 9721. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Song, Y.; Wang, Y. pNaKtide ameliorates renal interstitial fibrosis through inhibition of sodium-potassium adenosine triphosphatase-mediated signaling pathways in unilateral ureteral obstruction mice. Nephrol. Dial. Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xie, Z.J. The sodium pump and cardiotonic steroids-induced signal transduction protein kinases and calcium-signaling microdomain in regulation of transporter trafficking. Biochim. Biophys. Acta 2010, 1802, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- McDonough, A.A. Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am. J. Physiol. 2010, 298, R851–R861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonough, A.A.; Leong, P.K.; Yang, L.E. Mechanisms of pressure natriuresis: How blood pressure regulates renal sodium transport. Ann. N. Y. Acad. Sci. 2003, 986, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Fan, L.; Crowder, L.A.; Karim-Jimenez, Z.; Murer, H.; Moe, O.W. Dopamine acutely stimulates Na+/H+ exchanger (NHE3) endocytosis via clathrin-coated vesicles: Dependence on protein kinase A-mediated NHE3 phosphorylation. J. Biol. Chem. 2001, 276, 26906–26915. [Google Scholar] [CrossRef] [PubMed]
- Chibalin, A.V.; Katz, A.I.; Berggren, P.O.; Bertorello, A.M. Receptor-mediated inhibition of renal Na(+)-K(+)-ATPase is associated with endocytosis of its α- and β-subunits. Am. J. Physiol. 1997, 273, C1458–C1465. [Google Scholar] [CrossRef] [PubMed]
- Chibalin, A.V.; Ogimoto, G.; Pedemonte, C.H.; Pressley, T.A.; Katz, A.I.; Féraille, E.; Berggren, P.-O.; Bertorello, A.M. Dopamine-induced endocytosis of Na+,K+-ATPase is initiated by phosphorylation of Ser-18 in the rat alpha subunit and is responsible for the decreased activity in epithelial cells. J. Biol. Chem. 1999, 274, 1920–1927. [Google Scholar] [CrossRef] [PubMed]
- Bacic, D.; Kaissling, B.; McLeroy, P.; Zou, L.; Baum, M.; Moe, O.W. Dopamine acutely decreases apical membrane Na/H exchanger NHE3 protein in mouse renal proximal tubule. Kidney Int. 2003, 64, 2133–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J. Ouabain-induced endocytosis and signal transduction of the Na/K-ATPase. Front. Biosci. 2005, 10, 2056–2063. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kesiry, R.; Periyasamy, S.M.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney Int. 2004, 66, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liang, M.; Liu, L.; Malhotra, D.; Xie, Z.; Shapiro, J. IOuabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int. 2005, 67, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Periyasamy, S.M.; Gunning, W.; Fedorova, O.V.; Bagrov, A.Y.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Effects of cardiac glycosides on sodium pump expression and function in LLC-PK1 and MDCK cells. Kidney Int. 2002, 62, 2118–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Shapiro, J.I. Regulation of sodium pump endocytosis by cardiotonic steroids: Molecular mechanisms and physiological implications. Pathophysiology 2007, 14, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periyasamy, S.M.; Liu, J.; Tanta, F.; Kabak, B.; Wakefield, B.; Malhotra, D.; Kennedy, D.J.; Nadoor, A.; Fedorova, O.V.; Gunning, W.; et al. Salt loading induces redistribution of the plasmalemmal Na/K-ATPase in proximal tubule cells. Kidney Int. 2005, 67, 1868–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Haller, S.; Shapiro, A.; Malhotra, N.; Tian, J.; Xie, Z.; Malhotra, D.; Shapiro, J.I.; Liu, J. Ouabain-stimulated trafficking regulation of the Na/K-ATPase and NHE3 in renal proximal tubule cells. Mol. Cell. Biochem. 2012, 367, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oweis, S.; Wu, L.; Kiela, P.R.; Zhao, H.; Malhotra, D.; Ghishan, F.K.; Xie, Z.; Shapiro, J.I.; Liu, J. Cardiac glycoside downregulates NHE3 activity and expression in LLC-PK1 cells. Am. J. Physiol. Renal Physiol. 2006, 290, F997–F1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Wu, L.; Qu, W.; Malhotra, D.; Xie, Z.; Shapiro, J.I.; Liu, J. Regulation of apical NHE3 trafficking by ouabain-induced activation of basolateral Na/K-ATPase receptor complex. Am. J. Physiol. Cell Physiol. 2008, 294, C555–C563. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yan, Y.; Liu, L.; Xie, Z.; Malhotra, D.; Joe, B.; Shapiro, J.I. Impairment of Na/K-ATPase signaling in renal proximal tubule contributes to Dahl salt-sensitive hypertension. J. Biol. Chem. 2011, 286, 22806–22813. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.T.; Martin, R.; Yan, Y.; Shapiro, J.I.; Liu, J. Carbonylation modification regulates Na/K-ATPase signaling and salt sensitivity: A review and a hypothesis. Front. Physiol. 2016, 7, 256. [Google Scholar] [CrossRef] [PubMed]
- McPherson, P.S.; Kay, B.K.; Hussain, N.K. Signaling on the endocytic pathway. Traffic 2001, 2, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, V.; Corti, M.; Gruenberg, J. Endocytosis and signaling cascades: A close encounter. FEBS Lett. 2001, 498, 190–196. [Google Scholar] [CrossRef]
- Di Guglielmo, G.M.; Baass, P.C.; Ou, W.J.; Posner, B.I.; Bergeron, J.J. Compartmentalization of SHC, GRB2 and mSOS, and hyperphosphorylation of Raf-1 by EGF but not insulin in liver parenchyma. EMBO J. 1994, 13, 4269–4277. [Google Scholar] [PubMed]
- Roy, S.; Wyse, B.; Hancock, J.F. H-Ras signaling and K-Ras signaling are differentially dependent on endocytosis. Mol. Cell. Biol. 2002, 22, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.; Beattie, E.C.; Lem, L.; Riethof, D.A.; Liu, S.H.; Mobley, W.C.; Soriano, P.; Brodsky, F.M. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell 1999, 96, 677–687. [Google Scholar] [CrossRef]
- Ware, M.F.; Tice, D.A.; Parsons, S.J.; Lauffenburger, D.A. Overexpression of cellular Src in fibroblasts enhances endocytic internalization of epidermal growth factor receptor. J. Biol. Chem. 1997, 272, 30185–30190. [Google Scholar] [CrossRef] [PubMed]
- Wiley, H.S.; Burke, P.M. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic 2001, 2, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, S.K.; Lund, K.A.; Li, X.F.; Cliften, P.; Amsler, K.; Opresko, L.K.; Wiley, H.S. Differential signaling and regulation of apical vs. basolateral EGFR in polarized epithelial cells. Am. J. Physiol. 1998, 275, C1419–C1428. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lilly, M.N.; Shapiro, J.I. Targeting Na/K-ATPase signaling: A new approach to control oxidative stress. Curr. Pharm. Des. 2018, 24, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xie, Z. The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflugers Arch. 2009, 457, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.V.; Xie, Z. The Na,K-ATPase receptor complex: Its organization and membership. Cell Biochem. Biophys. 2006, 46, 303–316. [Google Scholar] [CrossRef]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Weigand, K.M.; Swarts, H.G.; Fedosova, N.U.; Russel, F.G.; Koenderink, J.B. Na,K-ATPase activity modulates Src activation: A role for ATP/ADP ratio. Biochim. Biophys. Acta 2012, 1818, 1269–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gable, M.E.; Abdallah, S.L.; Najjar, S.M.; Liu, L.; Askari, A. Digitalis-induced cell signaling by the sodium pump: On the relation of Src to Na+/K+-ATPase. Biochem. Biophys. Res. Commun. 2014, 446, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Yosef, E.; Katz, A.; Peleg, Y.; Mehlman, T.; Karlish, S.J. Do Src kinase and Caveolin interact directly with Na,K-ATPase? J. Biol. Chem. 2016, 291, 11736–11750. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pratt, R.D.; Brickman, C.R.; Cottrill, C.L.; Shapiro, J.I.; Liu, J. The Na/K-ATPase Signaling: From Specific Ligands to General Reactive Oxygen Species. Int. J. Mol. Sci. 2018, 19, 2600. https://doi.org/10.3390/ijms19092600
Pratt RD, Brickman CR, Cottrill CL, Shapiro JI, Liu J. The Na/K-ATPase Signaling: From Specific Ligands to General Reactive Oxygen Species. International Journal of Molecular Sciences. 2018; 19(9):2600. https://doi.org/10.3390/ijms19092600
Chicago/Turabian StylePratt, Rebecca D., Cameron R. Brickman, Cameron L. Cottrill, Joseph I. Shapiro, and Jiang Liu. 2018. "The Na/K-ATPase Signaling: From Specific Ligands to General Reactive Oxygen Species" International Journal of Molecular Sciences 19, no. 9: 2600. https://doi.org/10.3390/ijms19092600