Patient-Derived Xenograft Models for Endometrial Cancer Research

 , , ,
, , ,
Abstract
:
1. Introduction
1.1. Cancer Models
1.2. Endometrial Cancer
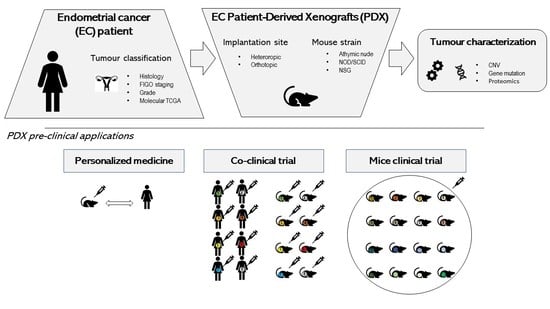
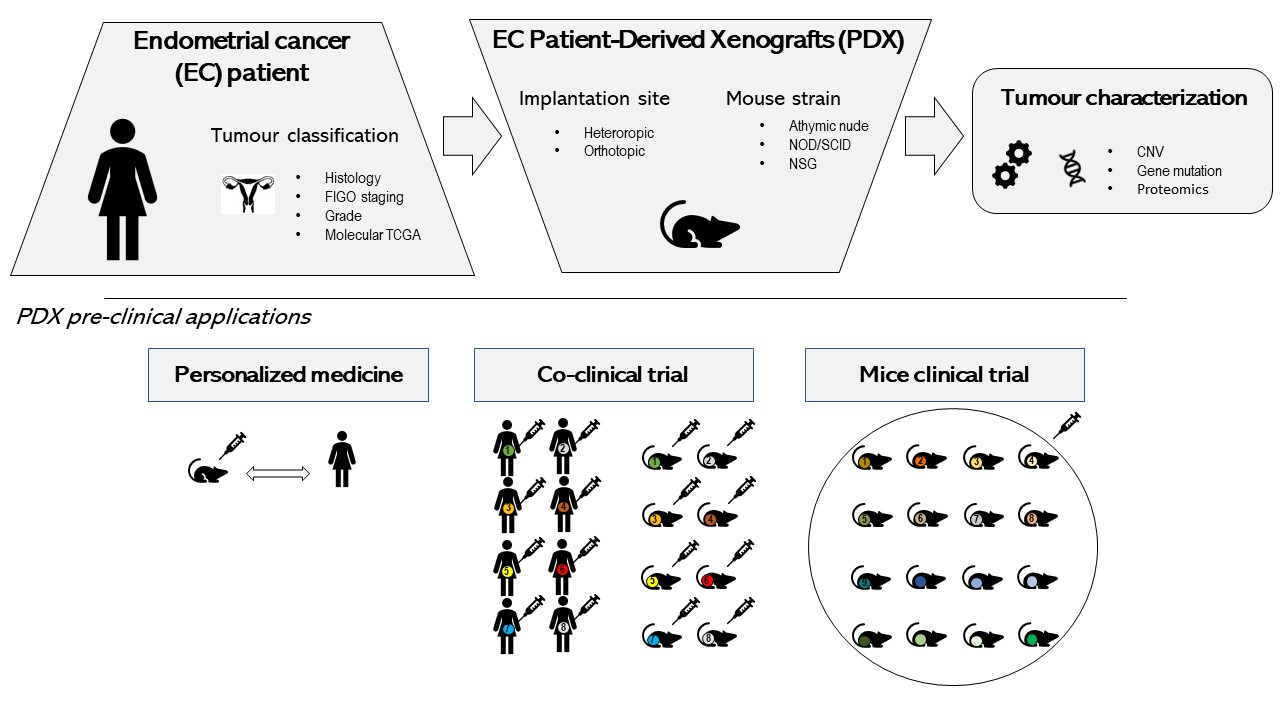
2. Endometrial Cancer PDX Models
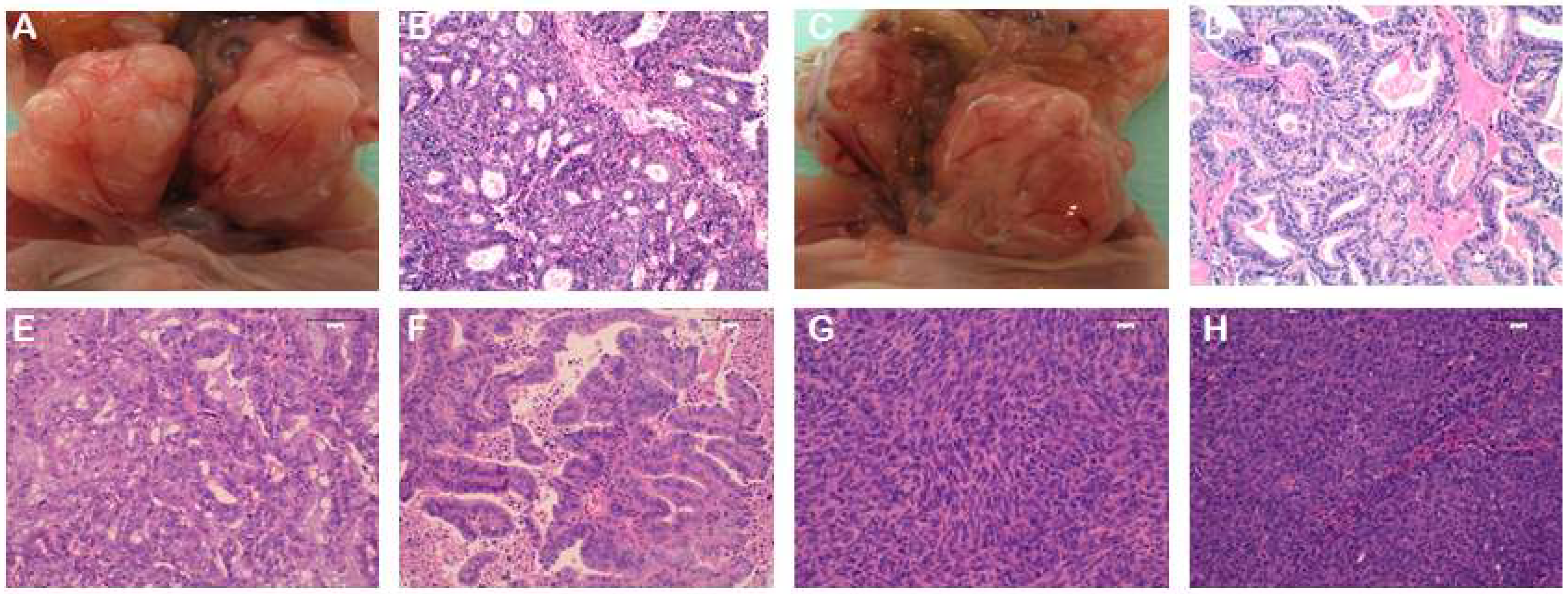
2.1. Strategies for EC PDX Model Development
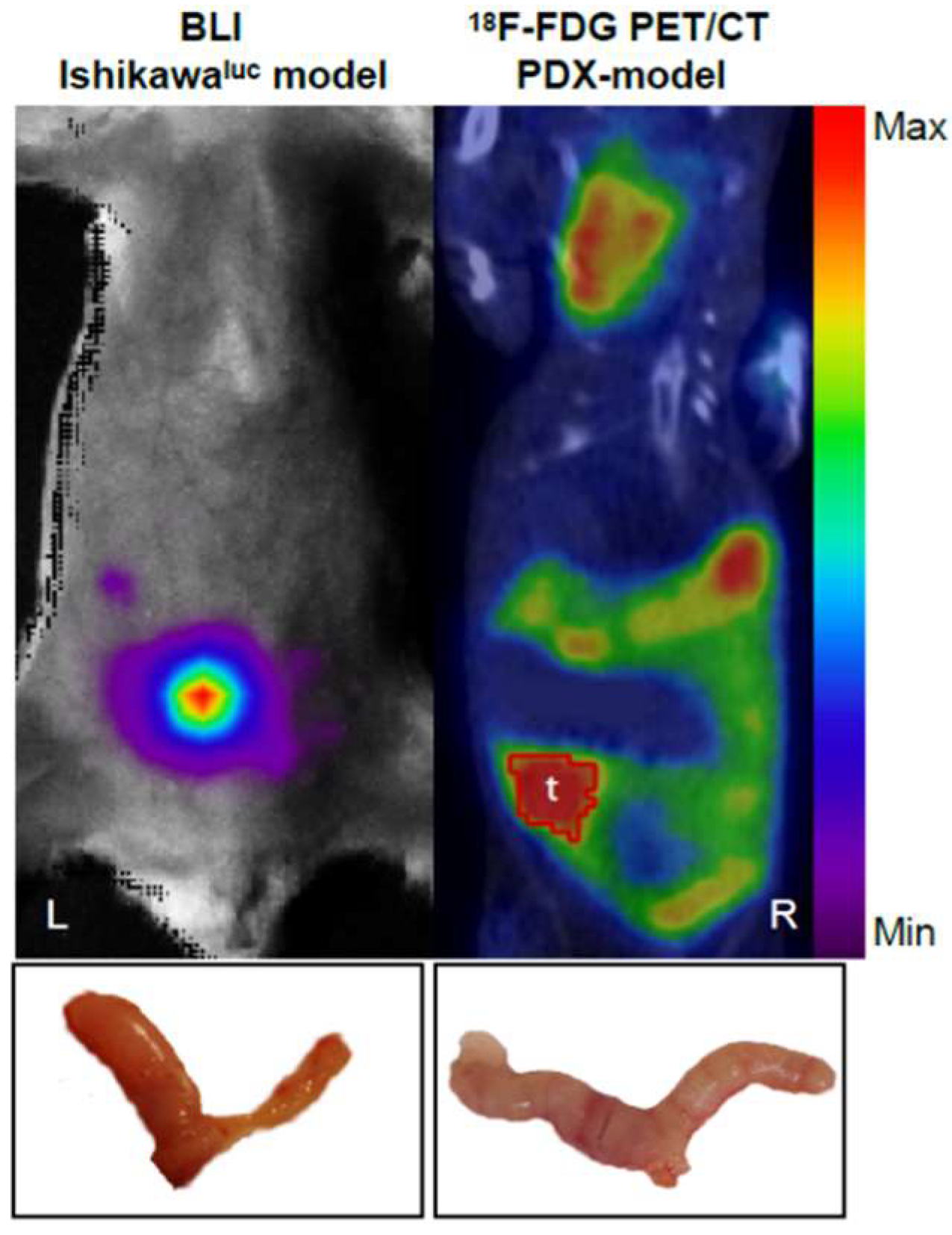
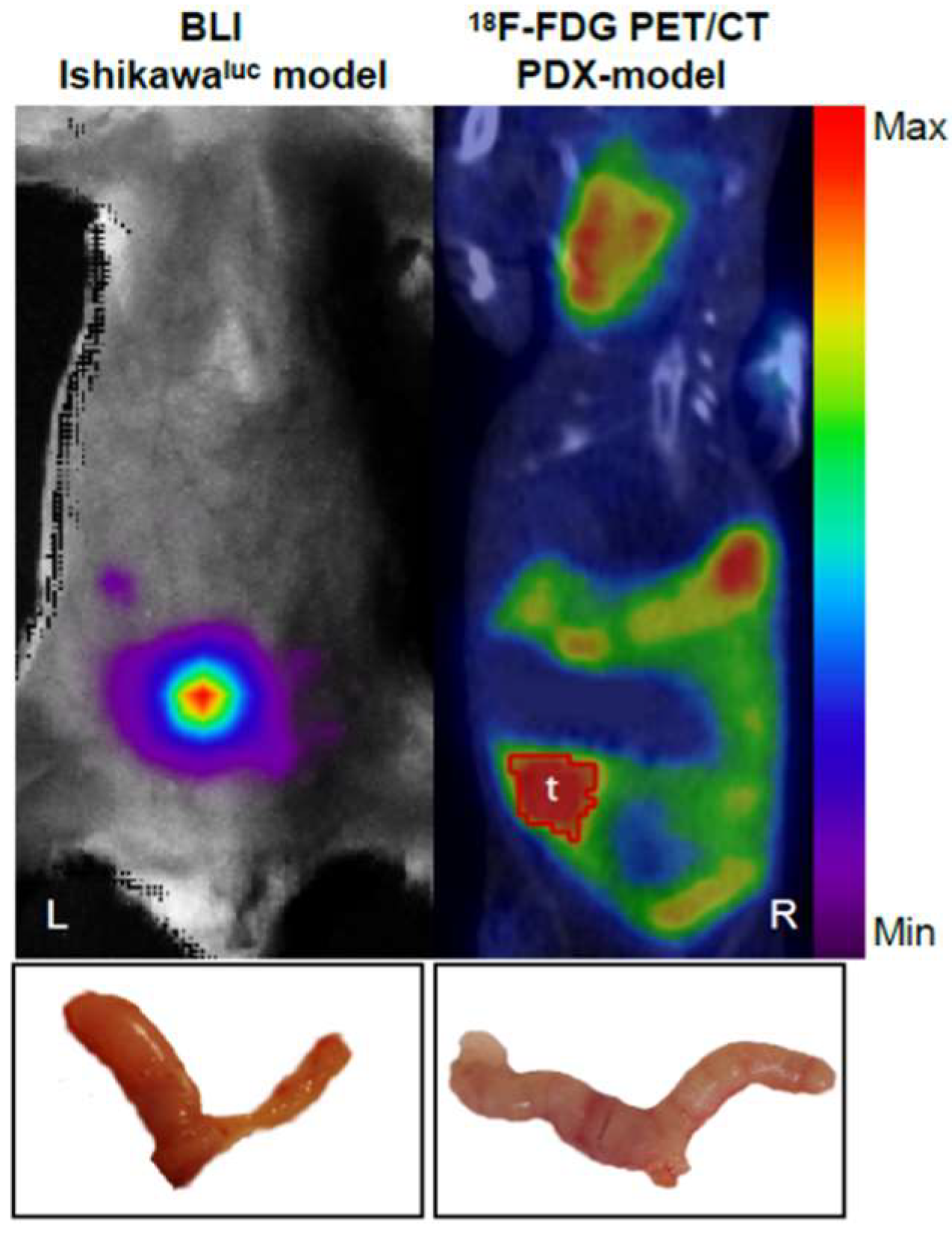
2.2. Strategies for EC PDX Model Monitoring
- Micro-computed tomography is a high image resolution technique with great potential for in vivo use since it could be coupled with other imaging modalities providing three-dimensional (3D) reconstruction of bone and soft tissue. It has been used to study bone metastases [37] in mice and also has been applied to monitor the progression of lung and liver tumours [38,39,40].
- Magnetic resonance imaging (MRI) also offers in-vivo, non-invasive, 3D, and high-resolution images. In recent years, MRI has gained great importance due to the absence of ionizing radiation and its soft tissue contrast. Hence, MRI is used not only for its ability to define lesions with great spatial resolution but also to recover quantitative features that might be able to predict cancer progression.
- Positron emission tomography (PET) is a highly sensitive and specific imaging technique used to visualize the distribution and concentration of radiolabelled molecules injected into murine models. It is a form of quantitative whole-body imaging used for the in vivo monitoring of biological processes, such as enzymatic reactions, cellular metabolism, and cell proliferation and migration [41], which makes it an ideal tool for the imaging of cancer [32,42].
- Single-Photon Emission Computed Tomography is a technique that detects gamma radiation directly emitted by a radionuclide during decay and provides 3D information by acquiring multiple two-dimensional (2D) images while rotating around the imaged object. This technique is frequently used in oncology to visualize neuroendocrine tumours [43] and thyroid cancer [44,45] and to perform bone scintigraphy.
- Ultrasound is an ideal technique for detecting tumour growth in mice since it produces high-resolution images of small structures. It is a non-ionizing radiation technique, portable, easy to use, and quickly generates relevant images.
- Intravital microscopy is an optical imaging technique that enables highly sensitive in vivo imaging of tissue structure and function at high spatial resolution (cellular and sub-cellular) and temporal resolution. However, a surgical procedure is required to access the tissue/organ of interest for microscopy, having therefore the consequence that immediately after image acquisition the animal must be sacrificed. Intravital microscopy has been reported to be used in studies involving a metastatic process [46,47] and the response of tumour blood vessels to vascular targeted therapy [48,49].
- Whole-body optical imaging is a sensitive technique based on fluorochromes excitation by an external light source (fluorescence) or by chemiluminescent enzymatic light emission reactions within the animal (bioluminescence). Despite the poor spatial resolution due to light scattering, this technique enables the integration of the light signal emitted to obtain a 2D planar image. Green fluorescence protein (GFP) has been widely used to measure in vivo tumour growth as well as the effect of metastatic spread and drug treatments on different types of cancer in mouse models [50,51].
2.3. Collaborative EC PDX Cohort
2.4. Use of EC PDX Models in Preclinical Studies
3. New Perspectives on the Use of EC PDX Models
4. Collaborative PDX Networks
5. PDX-Related Challenges
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GEM | Genetically engineered mouse |
| PDX | Patient-derived xenograft |
| SCID | Severe combined immunodeficient mice |
| NOD | Non-obese diabetic |
| NSG | NOD/SCID gamma mice |
| FIGO | International Federation of Gynaecology and Obstetrics |
| EC | Endometrial cancer |
| TCGA | The Cancer Genome Atlas |
| SCNA | Somatic copy number alterations |
| POLE | DNA polymerase epsilon |
| ENITEC | European Network of Individual Treatment in Endometrial Cancer |
| PDTO | Patient-derived tumour organoids |
| CT | Computed tomography |
| MRI | Magnetic resonance imaging |
| PET | Positron emission tomography |
| GFP | Green fluorescence protein |
| BLI | Bioluminescence imaging |
| IDIBELL-ICO | Institute of biomedical research from Bellvitge–institute Catalan of Oncology |
| VHIR | Vall d’Hebron Institute of research |
| H&E | Hematoxylin and eosin |
| AKT | Alpha serine threonine kinase |
| FGFR | Fibroblast growth factor receptor |
| CDK4 | Cyclin dependent kinase 4 |
| CDK6 | Cyclin dependent kinase 4 |
| PTEN | Phosphatase and Tensin Homolog |
| NCI | National Cancer Institute |
| PPTC | Pediatric Preclinical Testing Consortium |
| COG | Children’s Oncology Group |
| PRoXe | Public Repository of Xenografts |
| NIBR PDXE | Novartis Institutes for Biomedical Research PDX Encyclopedia |
References
- Cheon, D.-J.; Orsulic, S. Mouse models of cancer. Annu. Rev. Pathol. 2011, 6, 95–119. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Singh, R.K.; Fidler, I.J.; Raz, A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am. J. Pathol. 2007, 170, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Haber, D.A.; Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 2010, 10, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Suggitt, M.; Bibby, M.C. 50 years of preclinical anticancer drug screening: Empirical to target-driven approaches. Clin. Cancer Res. 2005, 11, 971–981. [Google Scholar] [PubMed]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Li, A.; Walling, J.; Kotliarov, Y.; Center, A.; Steed, M.E.; Ahn, S.J.; Rosenblum, M.; Mikkelsen, T.; Zenklusen, J.C.; Fine, H.A. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol. Cancer Res. MCR 2008, 6, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, M.; McGovern, J.A.; Friedl, P.; Hutmacher, D.W. Rational Design of Mouse Models for Cancer Research. Trends Biotechnol. 2018, 36, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.; Jodrell, D.I.; Tuveson, D.A. Predictive in vivo animal models and translation to clinical trials. Drug Discov. Today 2012, 17, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, C.L.; Houghton, P.J. Establishment of human tumor xenografts in immunodeficient mice. Nat. Protoc. 2007, 2, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.L.; Mackay, H.J.; Haluska, P. Patient-derived xenograft models in gynecologic malignancies. Am. Soc. Clin. Oncol. 2014, e258–e266. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Teng, L.; Shen, Y.; He, K.; Xu, Z.; Li, G. Patient-derived human tumour tissue xenografts in immunodeficient mice: A systematic review. Clin. Transl. Oncol. 2010, 12, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Seol, H.S.; Chang, S. The Generation and Application of Patient-Derived Xenograft Model for Cancer Research. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2018, 50, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J. Human tumor xenograft models for preclinical assessment of anticancer drug development. Toxicol. Res. 2014, 30, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A. Abnormal genital tract bleeding. Clin. Cornerstone. 2000, 3, 25–35. [Google Scholar] [CrossRef]
- Albers, J.R.; Hull, S.K.; Wesley, R.M. Abnormal uterine bleeding. Am. Fam. Physician. 2004, 69, 1915–1926. [Google Scholar] [PubMed]
- Bakkum-Gamez, J.N.; Gonzalez-Bosquet, J.; Laack, N.N.; Mariani, A.; Dowdy, S.C. Current issues in the management of endometrial cancer. Mayo Clin. Proc. 2008, 83, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Yeramian, A.; Moreno-Bueno, G.; Dolcet, X.; Catasus, L.; Abal, M.; Colas, E.; Reventos, J.; Palacios, J.; Prat, J.; Matias-Guiu, X. Endometrial carcinoma: Molecular alterations involved in tumor development and progression. Oncogene 2013, 32, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Amant, F.; Moerman, P.; Neven, P.; Timmerman, D.; Van Limbergen, E.; Vergote, I. Endometrial cancer. Lancet 2005, 366, 491–505. [Google Scholar] [CrossRef]
- Mendivil, A.; Schuler, K.M.; Gehrig, P.A. Non-endometrioid adenocarcinoma of the uterine corpus: A review of selected histological subtypes. Cancer Control J. 2009, 16, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Colombo, N.; Creutzberg, C.; Amant, F.; Bosse, T.; González-Martín, A.; Ledermann, J.; Marth, C.; Nout, R.; Querleu, D.; Mirza, M.R.; et al. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: Diagnosis, Treatment and Follow-up. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2016, 26, 2–30. [Google Scholar] [CrossRef] [PubMed]
- Llobet, D.; Pallares, J.; Yeramian, A.; Santacana, M.; Eritja, N.; Velasco, A.; Dolcet, X.; Matias-Guiu, X. Molecular pathology of endometrial carcinoma: Practical aspects from the diagnostic and therapeutic viewpoints. J. Clin. Pathol. 2009, 62, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [PubMed] [Green Version]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational implications of tumor heterogeneity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, S.; Llauradó, M.; Castellví, J.; Fernandez, Y.; Alameda, F.; Colás, E.; Ruiz, A.; Doll, A.; Schwartz, S., Jr.; Carreras, R.; et al. Generation and characterization of orthotopic murine models for endometrial cancer. Clin. Exp. Metast. 2012, 29, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Haldorsen, I.S.; Popa, M.; Fonnes, T.; Brekke, N.; Kopperud, R.; Visser, N.C.; Rygh, C.B.; Pavlin, T.; Salvesen, H.B.; McCormack, E.; et al. Multimodal Imaging of Orthotopic Mouse Model of Endometrial Carcinoma. PLoS ONE 2015, 10, e0135220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depreeuw, J.; Hermans, E.; Schrauwen, S.; Annibali, D.; Coenegrachts, L.; Thomas, D.; Luyckx, M.; Gutierrez-Roelens, I.; Debruyne, D.; Konings, K.; et al. Characterization of patient-derived tumor xenograft models of endometrial cancer for preclinical evaluation of targeted therapies. Gynecol. Oncol. 2015, 139, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unno, K.; Ono, M.; Winder, A.D.; Maniar, K.P.; Paintal, A.S.; Yu, Y.; Wie, J.J.; Lurain, J.R.; Kim, J.J. Establishment of human patient-derived endometrial cancer xenografts in NOD scid gamma mice for the study of invasion and metastasis. PLoS ONE 2014, 9, e116064. [Google Scholar] [CrossRef] [PubMed]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall’Ara, E.; Boudiffa, M.; Taylor, C.; Schug, D.; Fiegle, E.; Kennerley, A.J.; Damianou, C.; Tozer, G.M.; Kiessling, F.; Müller, R. Longitudinal imaging of the ageing mouse. Mech. Ageing Dev. 2016, 160, 93–116. [Google Scholar] [CrossRef] [PubMed]
- Ravoori, M.; Czaplinska, A.J.; Sikes, C.; Han, L.; Johnson, E.M.; Qiao, W.; Ng, C.; Cody, D.D.; Murphy, W.A.; Do, K.A.; et al. Quantification of mineralized bone response to prostate cancer by noninvasive in vivo microCT and non-destructive ex vivo microCT and DXA in a mouse model. PLoS ONE 2010, 5, e9854. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, M.; Xiao, Y.; Yang, L.; Zhang, J.; Ray, E.; Yang, X. Computed tomography fluoroscopy-guided percutaneous 125I seed implantation for safe, effective and real-time monitoring radiotherapy of inoperable stage T1-3N0M0 non-small-cell lung cancer. Mol. Clin. Oncol. 2013, 1, 1019–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodt, T.; von Falck, C.; Halter, R.; Ringe, K.; Shin, H.-O.; Galanski, M.; Borlak, J. In vivo microCT quantification of lung tumor growth in SPC-raf transgenic mice. Front. Biosci. 2009, 14, 1939–1944. [Google Scholar] [CrossRef]
- Ohta, S.; Lai, E.W.; Morris, J.C.; Bakan, D.A.; Klaunberg, B.; Cleary, S.; Powers, J.F.; Tischler, A.S.; Abu-Asab, M.; Schimel, S.; et al. MicroCT for high-resolution imaging of ectopic pheochromocytoma tumors in the liver of nude mice. Int. J. Cancer 2006, 119, 2236–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelps, M.E. Positron emission tomography provides molecular imaging of biological processes. Proc. Natl. Acad. Sci. USA 2000, 97, 9226–9233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambhir, S.S. Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2002, 2, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Bison, S.M.; Haeck, J.C.; Bol, K.; Koelewijn, S.J.; Groen, H.C.; Melis, M.; Veenland, J.F.; Bernsen, M.R.; de Jong, M. Optimization of combined temozolomide and peptide receptor radionuclide therapy (PRRT) in mice after multimodality molecular imaging studies. EJNMMI Res. 2015, 5, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-Y.; Brandt, M.P.; Shen, D.H.; Kloos, R.T.; Zhang, X.; Jhiang, S.M. Single photon emission computed tomography imaging for temporal dynamics of thyroidal and salivary radionuclide accumulation in 17-allyamino-17-demothoxygeldanamycin-treated thyroid cancer mouse model. Endocr. Relat. Cancer 2011, 18, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Van der Have, F.; Ivashchenko, O.; Goorden, M.C.; Ramakers, R.M.; Beekman, F.J. High-resolution clustered pinhole (131)Iodine SPECT imaging in mice. Nucl. Med. Biol. 2016, 43, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Beerling, E.; Ritsma, L.; Vrisekoop, N.; Derksen, P.W.B.; van Rheenen, J. Intravital microscopy: New insights into metastasis of tumors. J. Cell Sci. 2011, 124, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Segall, J.E. Intravital imaging of cell movement in tumours. Nat. Rev. Cancer 2003, 3, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Strieth, S.; Eichhorn, M.E.; Sutter, A.; Jonczyk, A.; Berghaus, A.; Dellian, M. Antiangiogenic combination tumor therapy blocking alpha(v)-integrins and VEGF-receptor-2 increases therapeutic effects in vivo. Int. J. Cancer 2006, 119, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Akerman, S.; Fisher, M.; Daniel, R.A.; Lefley, D.; Reyes-Aldasoro, C.C.; Lunt, S.J.; Harris, S.; Bjorndahl, M.; Williams, L.J.; Evans, H.; et al. Influence of soluble or matrix-bound isoforms of vascular endothelial growth factor-A on tumor response to vascular-targeted strategies. Int. J. Cancer 2013, 133, 2563–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, R.M. Visualization of GFP-expressing tumors and metastasis in vivo. BioTechniques 2001, 30, 1016–1022. [Google Scholar] [PubMed]
- Willmann, J.K.; van Bruggen, N.; Dinkelborg, L.M.; Gambhir, S.S. Molecular imaging in drug development. Nat. Rev. Drug Discov. 2008, 7, 591–607. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.C.; Sundar, R.; Lim, J.S.J.; Yap, T.A. Towards precision medicine in the clinic: From biomarker discovery to novel therapeutics. Trends Pharmacol. Sci. 2017, 38, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.D.; Hartley, L.; Cook, M.; Heong, V.; Boehm, E.; McShane, L.; Pyman, J.; McNally, O.; Ananda, S.; Harrell, M.; et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol. Oncol. 2014, 8, 656–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weroha, S.J.; Becker, M.A.; Enderica-Gonzalez, S.; Harrington, S.C.; Oberg, A.L.; Maurer, M.J.; Perkins, S.E.; AlHilli, M.; Butler, K.A.; McKinstry, S.; et al. Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Lodhia, K.A.; Hadley, A.M.; Haluska, P.; Scott, C.L. Prioritizing therapeutic targets using patient-derived xenograft models. Biochim. Biophys. Acta 2015, 1855, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winder, A.; Unno, K.; Yu, Y.; Lurain, J.; Kim, J.J. The allosteric AKT inhibitor, MK2206, decreases tumor growth and invasion in patient derived xenografts of endometrial cancer. Cancer Biol. Ther. 2017, 18, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hall, T.; Eathiraj, S.; Wick, M.J.; Schwartz, B.; Abbadessa, G. In-vitro and in-vivo combined effect of ARQ 092, an AKT inhibitor, with ARQ 087, a FGFR inhibitor. Anticancer Drugs 2017, 28, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, S. Palbociclib: First Global Approval. Drugs 2015, 75, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Dosil, M.A.; Mirantes, C.; Eritja, N.; Felip, I.; Navaridas, R.; Gatius, S.; Santacana, M.; Colàs, E.; Moiola, C.; Schoenenberger, J.A.; et al. Palbociclib has antitumour effects on Pten-deficient endometrial neoplasias: Inhibition of cyclin D-CDK4/6 axis in Pten-deficient neoplasias. J. Pathol. 2017, 242, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Eritja, N.; Chen, B.-J.; Rodríguez-Barrueco, R.; Santacana, M.; Gatius, S.; Vidal, A.; Martí, M.D.; Ponce, J.; Bergadà, L.; Yeramian, A.; et al. Autophagy orchestrates adaptive responses to targeted therapy in endometrial cancer. Autophagy 2017, 13, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, J.W.; Hernandez, S.F.; Byron, V.F.; DiGloria, C.M.; Lopez, H.; Scialabba, V.; Kim, M.; Zhang, L.; Borger, D.R.; Tambouret, R.; et al. Dual HER2 targeting impedes growth of HER2 gene-amplified uterine serous carcinoma xenografts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6517–6528. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, J.W.; Hall, T.R.; Zhang, L.; Kim, M.; Byron, V.F.; Tambouret, R.; Sathayanrayanan, S.; Foster, R.; Rueda, B.R.; Growdon, W.B. Inhibition of gamma-secretase activity impedes uterine serous carcinoma growth in a human xenograft model. Gynecol. Oncol. 2014, 133, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Siolas, D.; Hannon, G.J. Patient-Derived Tumor Xenografts: Transforming Clinical Samples into Mouse Models. Cancer Res. 2013, 73, 5315–5319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliardi, G.; Sassi, F.; Torti, D.; Galimi, F.; Zanella, E.R.; Buscarino, M.; Ribero, D.; Muratore, A.; Massuco, P.; Pisacane, A.; et al. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2515–2525. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Terada, N.; Kobayashi, T.; Ogawa, O. Patient-derived xenografts as in vivo models for research in urological malignancies. Nat. Rev. Urol. 2017, 14, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felix, A.S.; Weissfeld, J.; Edwards, R.; Linkov, F. Future directions in the field of endometrial cancer research: The need to investigate the tumor microenvironment. Eur. J. Gynaecol. Oncol. 2010, 31, 139–144. [Google Scholar] [PubMed]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruna, A.; Rueda, O.M.; Greenwood, W.; Batra, A.S.; Callari, M.; Batra, R.N.; Pogrebniak, K.; Sandoval, J.; Cassidy, J.W.; Tufegdzic-Vidakovic, A.; et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell 2016, 167, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.J.; Bird, G.; Refaeli, Y.; Jimeno, A. Humanized Mouse Xenograft Models: Narrowing the Tumor-Microenvironment Gap. Cancer Res. 2016, 76, 6153–6158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Research Centre | Type of Sample | Type of PDX | Tissue | Engraftment Rate & Time | Mouse Strain | Number Models | Type of Models | Preclinical Drug Tested | |
|---|---|---|---|---|---|---|---|---|---|
| IDIBELL-ICO | Primary tumor, metastases | orthotopic | small tissue fragment | 75–90% | 1–5 months | Athymic nude | 64 | 60%EEC; 10%PS; 20%CS; 3%CC; 7%other types | Sorafenib, Chloroquine (61) |
| VHIR | Primary tumor, metastases, recurrences | heterotopic (s.c) | 5–10 mm3 tissue fragment | 60–80% | 2–3 months | Athymic nude | 40 | 43%EEC; 32%PS; 10%CS; 2.5%CC; 5%undifferentiated 7.5%other types | Carboplatin Paclitaxel, Palbociclib (60) |
| KUL | Primary tumor, metastases, recurrences | heterotopic (s.c) | 8–10 mm3 tissue fragment | 100% | 3–5 months | Athymic nude | 15 | 46%EEC; 13%PS; 13%CS; 7%undifferentiated 21%other types | Carboplatin, NVP-BEZ235, AZD 6244 (33) |
| HUHB | Primary tumor, metastases | orthotopic | Cell suspension | 25–100% | 3–13 months | NSG | 5 | 60%EEC; 20%PS; 20%undifferentiated | |
| Endometrioid EC | Non-Endometrioid EC | ||||
|---|---|---|---|---|---|
| FIGO stage | I | 39 | 62% | 18 | 32% |
| II | 7 | 11% | 4 | 7% | |
| III | 13 | 21% | 27 | 48% | |
| IV | 2 | 3% | 5 | 9% | |
| Grade | 1 | 20 | 32% | ||
| 2 | 23 | 37% | |||
| 3 | 20 | 32% | 56 | 100% | |
| Histology | Serous carcinoma | 22 | 39% | ||
| Carcinosarcoma | 19 | 34% | |||
| Clear Cell carcinoma | 4 | 7% | |||
| Others | 11 | 20% | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moiola, C.P.; Lopez-Gil, C.; Cabrera, S.; Garcia, A.; Van Nyen, T.; Annibali, D.; Fonnes, T.; Vidal, A.; Villanueva, A.; Matias-Guiu, X.; et al. Patient-Derived Xenograft Models for Endometrial Cancer Research. Int. J. Mol. Sci. 2018, 19, 2431. https://doi.org/10.3390/ijms19082431
Moiola CP, Lopez-Gil C, Cabrera S, Garcia A, Van Nyen T, Annibali D, Fonnes T, Vidal A, Villanueva A, Matias-Guiu X, et al. Patient-Derived Xenograft Models for Endometrial Cancer Research. International Journal of Molecular Sciences. 2018; 19(8):2431. https://doi.org/10.3390/ijms19082431
Chicago/Turabian StyleMoiola, Cristian P., Carlos Lopez-Gil, Silvia Cabrera, Angel Garcia, Tom Van Nyen, Daniela Annibali, Tina Fonnes, August Vidal, Alberto Villanueva, Xavier Matias-Guiu, and et al. 2018. "Patient-Derived Xenograft Models for Endometrial Cancer Research" International Journal of Molecular Sciences 19, no. 8: 2431. https://doi.org/10.3390/ijms19082431