Emerging Roles of Sonic Hedgehog in Adult Neurological Diseases: Neurogenesis and Beyond


Abstract
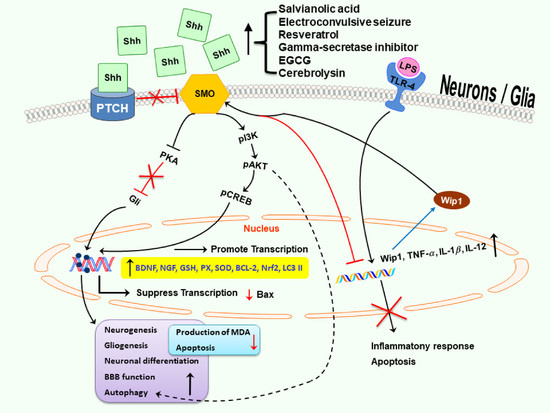
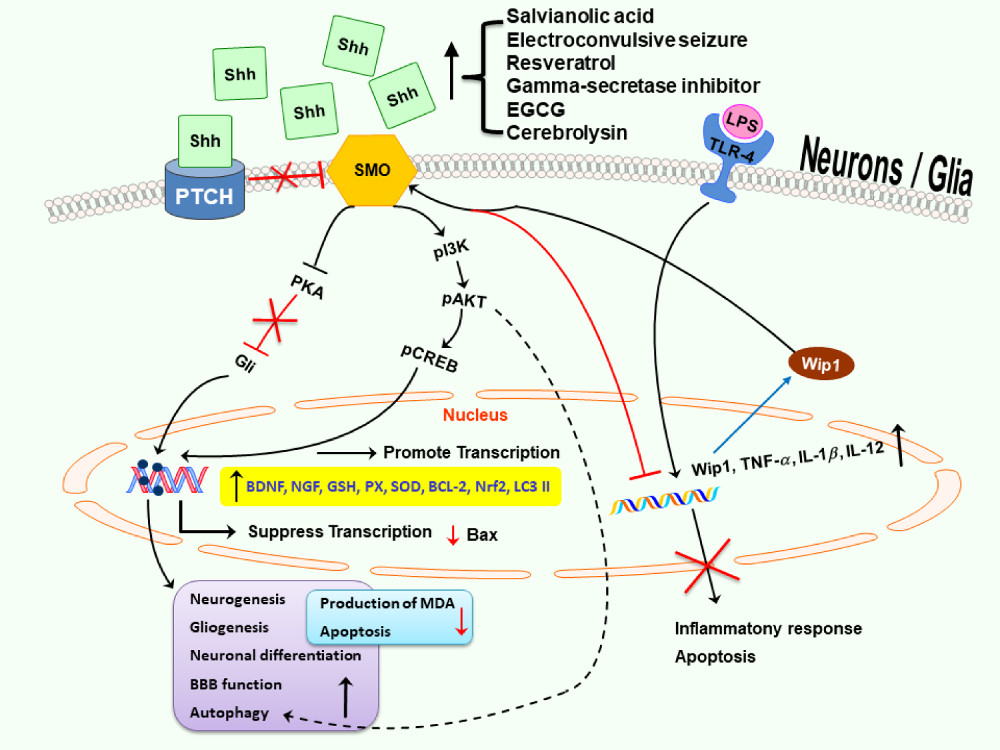
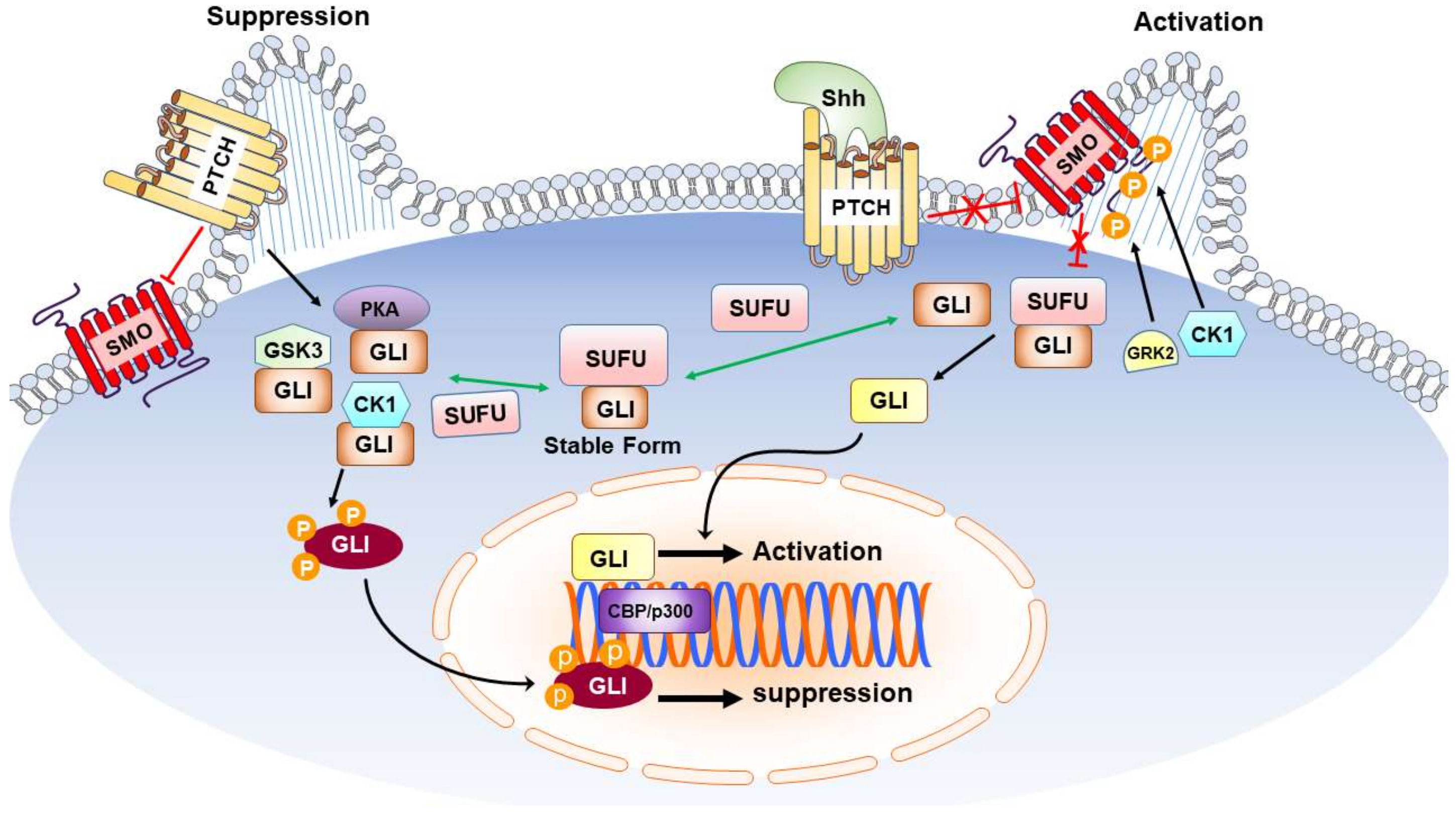
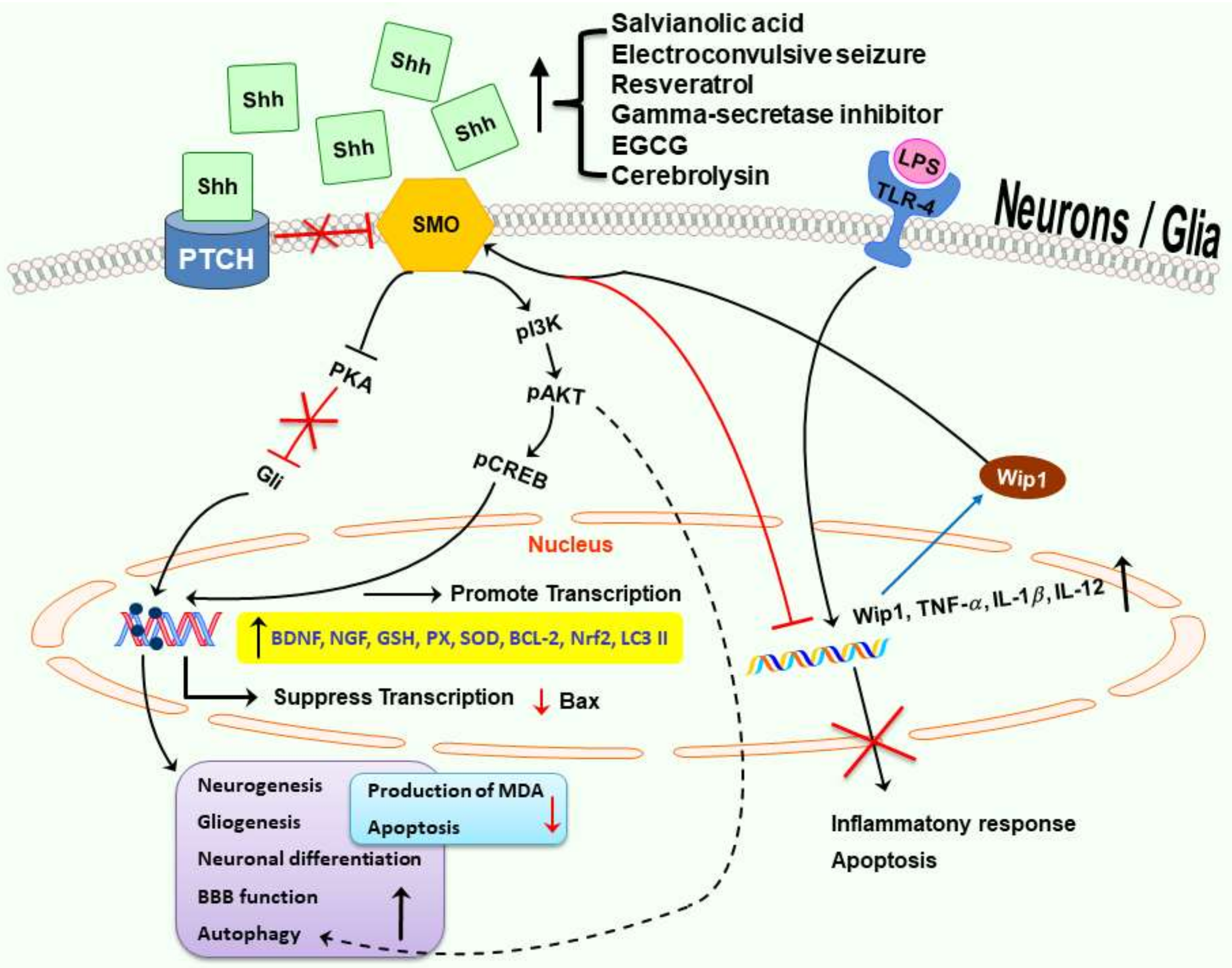
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Sonic Hedgehog and Neurogenesis in Neurological Diseases
3. Sonic Hedgehog and Antioxidation in Neurological Diseases
4. Sonic Hedgehog and Anti-Inflammation in Neurological Diseases
5. Sonic Hedgehog and Autophagy in Neurological Diseases
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3-NP | 3-nitropropionic acid |
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| APP | amyloid precursor protein |
| ASD | autism spectrum disorders |
| BBB | blood-brain barrier |
| BDNF | brain-derived neurotrophic factor |
| BMP | bone morphogenetic protein |
| BrdU | 5-bromo-2′-deoxyuridine |
| CK1 | casein kinase 1 |
| CNS | central nervous system |
| DG | dentate gyrus |
| DS | Down syndrome |
| ECS | electroconvulsive seizure |
| EGCG | epigallocatechin-3-gallate |
| EPO | erythropoietin |
| FU | Fused |
| Gas1 | growth arrest-specific gene 1 |
| Gli | glioma-associated oncogene homolog |
| GSK3 | glycogen synthase kinase-3 |
| GSH-PX | glutathione peroxidase |
| H2O2 | hydrogen peroxide |
| HAND | human immunodeficiency virus type-1 (HIV)-associated neurocognitive disorder |
| HD | Huntington’s disease |
| Hh | hedgehog gene |
| HI | hypoxia ischemia |
| IL-1β | interleukin-1β |
| KA | kainic acid |
| LC-3 | microtubule-associated protein 1 light chain-3 |
| LPS | lipopolysaccharide |
| MBMECs | murine brain microvascular endothelial cells |
| MCAO | middle cerebral artery occlusion |
| MDA | malondialdehyde |
| MMP-9 | matrix metalloproteinase-9 |
| MS | multiple sclerosis |
| Mtb | mycobacterium tuberculosis |
| NO | nitric oxide |
| NPCs | neural progenitor cells |
| NSCs | neural stem cells |
| OGD | oxygen glucose deprivation |
| PD | Parkinson’s disease |
| ONOO− | peroxynitrite anion |
| oxLDL | oxidized low-density lipoprotein |
| PD | Parkinson’s disease |
| PKA | protein kinase A |
| Ptch | Patched |
| PUR | purmorphamine |
| RMS | rostral migratory stream |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SAG | smoothened agonist |
| SAH | subarachnoid hemorrhage |
| SGZ | subgranular zone |
| Shh | sonic hedgehog |
| Shh-N | N-terminal fragment of Shh |
| SMCs | smooth muscle cells |
| Smo | smoothened |
| SODs | superoxide dismutases |
| SUFU | suppressor of FU |
| SVZ | subventricular zone |
| TBI | traumatic brain injury |
| TJP | tight junction protein |
| TNF-α | tumor necrosis factor-α |
| UCBMC | umbilical cord blood mononuclear cells |
| VEGF | vascular endothelial growth factor |
References
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Ruiz i Altaba, A.; Palma, V.; Dahmane, N. Hedgehog-Gli signalling and the growth of the brain. Nat. Rev. Neurosci. 2002, 3, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Wechsler-Reya, R.J.; Scott, M.P. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999, 22, 103–114. [Google Scholar] [CrossRef]
- Hynes, M.; Porter, J.A.; Chiang, C.; Chang, D.; Tessier-Lavigne, M.; Beachy, P.A.; Rosenthal, A. Induction of midbrain dopaminergic neurons by Sonic hedgehog. Neuron 1995, 15, 35–44. [Google Scholar] [CrossRef]
- Ericson, J.; Muhr, J.; Placzek, M.; Lints, T.; Jessell, T.M.; Edlund, T. Sonic hedgehog induces the differentiation of ventral forebrain neurons: A common signal for ventral patterning within the neural tube. Cell 1995, 81, 747–756. [Google Scholar] [CrossRef]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Fuccillo, M.; Joyner, A.L.; Fishell, G. Morphogen to mitogen: The multiple roles of hedgehog signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Xavier, G.M.; Seppala, M.; Barrell, W.; Birjandi, A.A.; Geoghegan, F.; Cobourne, M.T. Hedgehog receptor function during craniofacial development. Dev. Biol. 2016, 415, 198–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlgren, S.C.; Bronner-Fraser, M. Inhibition of sonic hedgehog signaling in vivo results in craniofacial neural crest cell death. Curr. Biol. 1999, 9, 1304–1314. [Google Scholar] [CrossRef]
- Petryk, A.; Graf, D.; Marcucio, R. Holoprosencephaly: Signaling interactions between the brain and the face, the environment and the genes, and the phenotypic variability in animal models and humans. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Marcucio, R.S.; Young, N.M.; Hu, D.; Hallgrimsson, B. Mechanisms that underlie co-variation of the brain and face. Genesis 2011, 49, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Tabler, J.M.; Bolger, T.G.; Wallingford, J.; Liu, K.J. Hedgehog activity controls opening of the primary mouth. Dev. Biol. 2014, 396, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, C.; Yamagishi, H.; Maeda, J.; Tsuchihashi, T.; Ivey, K.; Hu, T.; Srivastava, D. Sonic hedgehog is essential for first pharyngeal arch development. Pediatr. Res. 2006, 59, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Washington Smoak, I.; Byrd, N.A.; Abu-Issa, R.; Goddeeris, M.M.; Anderson, R.; Morris, J.; Yamamura, K.; Klingensmith, J.; Meyers, E.N. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev. Biol. 2005, 283, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Yin, V.T.; Esmaeli, B. Targeting the Hedgehog pathway for locally advanced and metastatic basal cell carcinoma. Curr. Pharm. Des. 2017, 23, 655–659. [Google Scholar] [PubMed]
- Samkari, A.; White, J.; Packer, R. SHH inhibitors for the treatment of medulloblastoma. Expert Rev. Neurother. 2015, 15, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Laukkanen, M.O.; Castellone, M.D. Hijacking the Hedgehog pathway in cancer therapy. Anticancer Agents Med. Chem. 2016, 16, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.J.; Robinson, G.W. Medulloblastoma-translating discoveries from the bench to the bedside. Nat. Rev. Clin. Oncol. 2014, 11, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, J. Decoding the phosphorylation code in Hedgehog signal transduction. Cell Res. 2013, 23, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic hedgehog dependent phosphorylation by CK1α and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Kenney, A.M.; Cole, M.D.; Rowitch, D.H. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 2003, 130, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayuso, J.; Ulloa, F.; Cox, B.; Briscoe, J.; Marti, E. The Sonic hedgehog pathway independently controls the patterning, proliferation and survival of neuroepithelial cells by regulating Gli activity. Development 2006, 133, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regl, G.; Kasper, M.; Schnidar, H.; Eichberger, T.; Neill, G.W.; Philpott, M.P.; Esterbauer, H.; Hauser-Kronberger, C.; Frischauf, A.M.; Aberger, F. Activation of the BCL2 promoter in response to Hedgehog/GLI signal transduction is predominantly mediated by GLI2. Cancer Res. 2004, 64, 7724–7731. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Lingbeek, M.; Shakhova, O.; Liu, J.; Tanger, E.; Saremaslani, P.; Van Lohuizen, M.; Marino, S. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 2004, 428, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Buylla, A.; Ihrie, R.A. Sonic hedgehog signaling in the postnatal brain. Semin. Cell Dev. Biol. 2014, 33, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, K.; Kaspar, B.K.; Gage, F.H.; Schaffer, D.V. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat. Neurosci. 2003, 6, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.J.; Petralia, R.S.; Mattson, M.P. Sonic hedgehog signaling and hippocampal neuroplasticity. Trends Neurosci. 2016, 39, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Dashti, M.; Peppelenbosch, M.P.; Rezaee, F. Hedgehog signalling as an antagonist of ageing and its associated diseases. Bioessays 2012, 34, 849–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.G.; Spassky, N.; Romaguera-Ros, M.; Garcia-Verdugo, J.M.; Aguilar, A.; Schneider-Maunoury, S.; Alvarez-Buylla, A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat. Neurosci. 2008, 11, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Traiffort, E.; Charytoniuk, D.A.; Faure, H.; Ruat, M. Regional distribution of Sonic Hedgehog, patched, and smoothened mRNA in the adult rat brain. J. Neurochem. 1998, 70, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Miao, J.; Zhang, X.; Du, Y.; Liu, H.; Li, S.; Li, L. Inhibition of sonic hedgehog signaling aggravates brain damage associated with the down-regulation of Gli1, Ptch1 and SOD1 expression in acute ischemic stroke. Neurosci. Lett. 2012, 506, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Amankulor, N.M.; Hambardzumyan, D.; Pyonteck, S.M.; Becher, O.J.; Joyce, J.A.; Holland, E.C. Sonic hedgehog pathway activation is induced by acute brain injury and regulated by injury-related inflammation. J. Neurosci. 2009, 29, 10299–10308. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.H.; Chang, S.H.; Huang, C.T.; Yin, J.H.; Hwang, C.S.; Yang, L.Y.; Yang, D.I. Inhibitor of differentiation-1 and hypoxia-inducible factor-1 mediate sonic hedgehog induction by amyloid β-peptide in rat cortical neurons. Mol. Neurobiol. 2016, 53, 793–809. [Google Scholar] [CrossRef] [PubMed]
- Pitter, K.L.; Tamagno, I.; Feng, X.; Ghosal, K.; Amankulor, N.; Holland, E.C.; Hambardzumyan, D. The SHH/Gli pathway is reactivated in reactive glia and drives proliferation in response to neurodegeneration-induced lesions. Glia 2014, 62, 1595–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.P.; Dai, R.L.; Li, Y.N.; Mao, L.; Xue, Y.M.; He, Q.W.; Huang, M.; Huang, Y.; Mei, Y.W.; Hu, B. The protective effect of sonic hedgehog is mediated by the phosphoinositide 3-kinase/AKT/Bcl-2 pathway in cultured rat astrocytes under oxidative stress. Neuroscience 2012, 209, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Hambardzumyan, D.; Fomchenko, E.I.; Momota, H.; Mainwaring, L.; Bleau, A.M.; Katz, A.M.; Edgar, M.; Kenney, A.M.; Cordon-Cardo, C. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008, 68, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Wu, C.L.; Hwang, W.C.; Yang, D.I. More insight into BDNF against neurodegeneration: Anti-apoptosis, anti-oxidation, and suppression of autophagy. Int. J. Mol. Sci. 2017, 18, 545. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, L.; Daley, B.F.; Davidson, B.L.; Boudreau, R.L.; Lipton, J.W.; Cole-Strauss, A.; Steece-Collier, K.; Collier, T.J. Sonic hedgehog controls the phenotypic fate and therapeutic efficacy of grafted neural precursor cells in a model of nigrostriatal neurodegeneration. PLoS ONE 2015, 10, e0137136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, W.; Guo, S.; Zhao, S.; He, S.; Zhang, L.; Tang, Y.; Wang, H. Lentivirus-mediated delivery of sonic hedgehog into the striatum stimulates neuroregeneration in a rat model of Parkinson disease. Neurol. Sci. 2014, 35, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.S.; Cheng, H.; Tang, C.M.; Nien, M.W.; Huang, Y.S.; Lee, I.H.; Yin, J.H.; Kuo, T.B.; Yang, C.C.; Tsai, S.K.; et al. Anti-oxidative, anti-apoptotic, and pro-angiogenic effects mediate functional improvement by sonic hedgehog against focal cerebral ischemia in rats. Exp. Neurol. 2013, 247, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Chen, S.D.; Yin, J.H.; Hwang, C.S.; Yang, D.I. Erythropoietin and sonic hedgehog mediate the neuroprotective effects of brain-derived neurotrophic factor against mitochondrial inhibition. Neurobiol. Dis. 2010, 40, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Chen, S.D.; Hwang, C.S.; Yang, D.I. Sonic hedgehog mediates BDNF-induced neuroprotection against mitochondrial inhibitor 3-nitropropionic acid. Biochem. Biophys. Res. Commun. 2009, 385, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Suwelack, D.; Hurtado-Lorenzo, A.; Millan, E.; Gonzalez-Nicolini, V.; Wawrowsky, K.; Lowenstein, P.R.; Castro, M.G. Neuronal expression of the transcription factor Gli1 using the Tα1 α-tubulin promoter is neuroprotective in an experimental model of Parkinson’s disease. Gene Ther. 2004, 11, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Hwang, C.S.; Chen, S.D.; Yin, J.H.; Yang, D.I. Neuroprotective mechanisms of brain-derived neurotrophic factor against 3-nitropropionic acid toxicity: Therapeutic implications for Huntington’s disease. Ann. N. Y. Acad. Sci. 2010, 1201, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chopp, M.; Meier, D.H.; Winter, S.; Wang, L.; Szalad, A.; Lu, M.; Wei, M.; Cui, Y.; Zhang, Z.G.; et al. Sonic hedgehog signaling pathway mediates cerebrolysin-improved neurological function after stroke. Stroke 2013, 44, 1965–1972. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Li, Y.; Liu, Z.; Zhang, J.; Cui, Y.; Chen, X.; Chopp, M. The sonic hedgehog pathway mediates brain plasticity and subsequent functional recovery after bone marrow stromal cell treatment of stroke in mice. J. Cereb. Blood Flow Metab. 2013, 33, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.R.; Lee, S.W.; Topalkara, K.; Qiu, J.; Xu, J.; Zhou, Z.; Moskowitz, M.A. Sonic hedgehog regulates ischemia/hypoxia-induced neural progenitor proliferation. Stroke 2009, 40, 3618–3626. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.J.; Manor, U.; Petralia, R.S.; Brose, R.D.; Wu, R.T.; Ott, C.; Wang, Y.X.; Charnoff, A.; Lippincott-Schwartz, J.; Mattson, M.P. Sonic hedgehog pathway activation increases mitochondrial abundance and activity in hippocampal neurons. Mol. Biol. Cell 2017, 28, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauth, M. Sonic the Hedgehog: A game about aging? Emerging evidence for anti-geriatric effects of Hedgehog signaling. Bioessays 2014, 36, 1128. [Google Scholar] [PubMed]
- Renault, M.A.; Robbesyn, F.; Chapouly, C.; Yao, Q.; Vandierdonck, S.; Reynaud, A.; Belloc, I.; Traiffort, E.; Ruat, M.; Desgranges, C.; et al. Hedgehog-dependent regulation of angiogenesis and myogenesis is impaired in aged mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Bishop, C.L.; Bergin, A.M.; Fessart, D.; Borgdorff, V.; Hatzimasoura, E.; Garbe, J.C.; Stampfer, M.R.; Koh, J.; Beach, D.H. Primary cilium-dependent and -independent Hedgehog signaling inhibits p16(INK4A). Mol. Cell 2010, 40, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Teperino, R.; Amann, S.; Bayer, M.; McGee, S.L.; Loipetzberger, A.; Connor, T.; Jaeger, C.; Kammerer, B.; Winter, L.; Wiche, G.; et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell 2012, 151, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Song, H.; Gage, F.H. Neurogenesis in the adult hippocampus. Cold Spring Harb. Perspect. Biol. 2015, 7, a018812. [Google Scholar] [CrossRef] [PubMed]
- Sakalem, M.E.; Seidenbecher, T.; Zhang, M.; Saffari, R.; Kravchenko, M.; Wordemann, S.; Diederich, K.; Schwamborn, J.C.; Zhang, W.; Ambrée, O. Environmental enrichment and physical exercise revert behavioral and electrophysiological impairments caused by reduced adult neurogenesis. Hippocampus 2017, 27, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Winkler, J. Adult neurogenesis in neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2015, 7, a021287. [Google Scholar] [CrossRef] [PubMed]
- Seib, D.R.; Martin-Villalba, A. Neurogenesis in the normal ageing hippocampus: A mini-review. Gerontology 2015, 61, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Hidaka, R.; Fujimaki, S.; Asashima, M.; Kuwabara, T. MicroRNAs and epigenetics in adult neurogenesis. Adv. Genet. 2014, 86, 27–44. [Google Scholar] [PubMed]
- Jessberger, S.; Parent, J.M. Epilepsy and adult neurogenesis. Cold Spring Harb. Perspect. Biol. 2015, 7, a020677. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Murao, N.; Katano, Y.; Juliandi, B.; Kohyama, J.; Akira, S.; Kawai, T.; Nakashima, K. TLR9 signalling in microglia attenuates seizure-induced aberrant neurogenesis in the adult hippocampus. Nat. Commun. 2015, 6, 6514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokaia, M. Seizure-induced neurogenesis in the adult brain. Eur. J. Neurosci. 2011, 33, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, O.; Kokaia, Z. Neurogenesis following stroke affecting the adult brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a019034. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Cai, J.; Nathan, C.; Wei, X.; Schleidt, S.; Rosenwasser, R.; Iacovitti, L. Neurogenesis is enhanced by stroke in multiple new stem cell niches along the ventricular system at sites of high BBB permeability. Neurobiol. Dis. 2015, 74, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.L.; Chopp, M.; Roberts, C.; Liu, X.; Wei, M.; Nejad-Davarani, S.P.; Wang, X.; Zhang, Z.G. Stroke increases neural stem cells and angiogenesis in the neurogenic niche of the adult mouse. PLoS ONE 2014, 9, e113972. [Google Scholar] [CrossRef] [PubMed]
- Charron, F.; Tessier-Lavigne, M. The Hedgehog, TGF-β/BMP and Wnt families of morphogens in axon guidance. Adv. Exp. Med. Biol. 2007, 621, 116–133. [Google Scholar] [PubMed]
- Yam, P.T.; Charron, F. Signaling mechanisms of non-conventional axon guidance cues: The Shh, BMP and Wnt morphogens. Curr. Opin. Neurobiol. 2013, 23, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Choy, S.W.; Cheng, S.H. Hedgehog signaling. Vitam. Horm. 2012, 88, 1–23. [Google Scholar] [PubMed]
- Zhang, Y.; Zhang, X.; Cui, L.; Chen, R.; Zhang, C.; Li, Y.; He, T.; Zhu, X.; Shen, Z.; Dong, L.; et al. Salvianolic Acids for Injection (SAFI) promotes functional recovery and neurogenesis via sonic hedgehog pathway after stroke in mice. Neurochem. Int. 2017, 110, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.S.; Dempsey, R.J.; Vemuganti, R. Resveratrol neuroprotection in stroke and traumatic CNS injury. Neurochem. Int. 2015, 89, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastianetto, S.; Menard, C.; Quirion, R. Neuroprotective action of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Agrawal, M.; Dore, S. Neuroprotective properties and mechanisms of resveratrol in in vitro and in vivo experimental cerebral stroke models. ACS Chem. Neurosci. 2013, 4, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Wang, L.; Tang, F.; Zeng, L.; Zhou, L.; Song, X.; Jia, W.; Chen, J.; Yang, Q. Resveratrol pretreatment decreases ischemic injury and improves neurological function via Sonic hedgehog signaling after stroke in rats. Mol. Neurobiol. 2017, 54, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Segre, T. Green tea: Potential health benefits. Am. Fam. Phys. 2009, 79, 591–594. [Google Scholar]
- Kuriyama, S.; Shimazu, T.; Ohmori, K.; Kikuchi, N.; Nakaya, N.; Nishino, Y.; Tsubono, Y.; Tsuji, I. Green tea consumption and mortality due to cardiovascular disease, cancer, and all causes in Japan: The Ohsaki study. JAMA 2006, 296, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Xu, X.; Song, M.; Tao, H.; Bai, Y. Green tea epigallocatechin-3-gallate (EGCG) promotes neural progenitor cell proliferation and sonic hedgehog pathway activation during adult hippocampal neurogenesis. Mol. Nutr. Food Res. 2012, 56, 1292–1303. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank-Kamenetsky, M.; Zhang, X.M.; Bottega, S.; Guicherit, O.; Wichterle, H.; Dudek, H.; Bumcrot, D.; Wang, F.Y.; Jones, S.; Shulok, J.; et al. Small-molecule modulators of Hedgehog signaling: Identification and characterization of Smoothened agonists and antagonists. J. Biol. 2002, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Barnett, A.; Zhang, Y.; Yu, X.; Luo, Y. Poststroke Sonic hedgehog agonist treatment improves functional recovery by enhancing neurogenesis and angiogenesis. Stroke 2017, 48, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Dierssen, M. Down syndrome: The brain in trisomic mode. Nat. Rev. Neurosci. 2012, 13, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, R.; Guidi, S.; Ciani, E. Is it possible to improve neurodevelopmental abnormalities in Down syndrome? Rev. Neurosci. 2011, 22, 419–455. [Google Scholar] [CrossRef] [PubMed]
- Trazzi, S.; Fuchs, C.; Valli, E.; Perini, G.; Bartesaghi, R.; Ciani, E. The amyloid precursor protein (APP) triplicated gene impairs neuronal precursor differentiation and neurite development through two different domains in the Ts65Dn mouse model for Down syndrome. J. Biol. Chem. 2013, 288, 20817–20829. [Google Scholar] [CrossRef] [PubMed]
- Trazzi, S.; Mitrugno, V.M.; Valli, E.; Fuchs, C.; Rizzi, S.; Guidi, S.; Perini, G.; Bartesaghi, R.; Ciani, E. APP-dependent up-regulation of Ptch1 underlies proliferation impairment of neural precursors in Down syndrome. Hum. Mol. Genet. 2011, 20, 1560–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomini, A.; Stagni, F.; Trazzi, S.; Guidi, S.; Emili, M.; Brigham, E.; Ciani, E.; Bartesaghi, R. Inhibition of APP gamma-secretase restores Sonic Hedgehog signaling and neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 2015, 82, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Stagni, F.; Raspanti, A.; Giacomini, A.; Guidi, S.; Emili, M.; Ciani, E.; Giuliani, A.; Bighinati, A.; Calzà, L.; Magistretti, J.; et al. Long-term effect of neonatal inhibition of APP gamma-secretase on hippocampal development in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 2017, 103, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Das, I.; Park, J.M.; Shin, J.H.; Jeon, S.K.; Lorenzi, H.; Linden, D.J.; Worley, P.F.; Reeves, R.H. Hedgehog agonist therapy corrects structural and cognitive deficits in a Down syndrome mouse model. Sci. Transl. Med. 2013, 5, 201ra120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chopp, M.; Cui, Y.; Wang, L.; Zhang, R.; Zhang, L.; Lu, M.; Szalad, A.; Doppler, E.; Hitzl, M. Cerebrolysin enhances neurogenesis in the ischemic brain and improves functional outcome after stroke. J. Neurosci. Res. 2010, 88, 3275–3281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, X.A.; Cacabelos, R.; Sampedro, C.; Aleixandre, M.; Linares, C.; Granizo, E.; Doppler, E.; Moessler, H. Efficacy and safety of Cerebrolysin in moderate to moderately severe Alzheimer’s disease: Results of a randomized, double-blind, controlled trial investigating three dosages of Cerebrolysin. Eur. J. Neurol. 2011, 18, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Bambakidis, N.C.; Petrullis, M.; Kui, X.; Rothstein, B.; Karampelas, I.; Kuang, Y.; Selman, W.R.; LaManna, J.C.; Miller, R.H. Improvement of neurological recovery and stimulation of neural progenitor cell proliferation by intrathecal administration of Sonic hedgehog. J. Neurosurg. 2012, 116, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Lipsman, N.; Sankar, T.; Downar, J.; Kennedy, S.H.; Lozano, A.M.; Giacobbe, P. Neuromodulation for treatment-refractory major depressive disorder. CMAJ 2014, 186, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Madsen, T.M.; Treschow, A.; Bengzon, J.; Bolwig, T.G.; Lindvall, O.; Tingstrom, A. Increased neurogenesis in a model of electroconvulsive therapy. Biol. Psychiatry 2000, 47, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, V.A.; Siuciak, J.A.; Du, F.; Duman, R.S. Hippocampal mossy fiber sprouting induced by chronic electroconvulsive seizures. Neuroscience 1999, 89, 157–166. [Google Scholar] [CrossRef]
- Banerjee, S.B.; Rajendran, R.; Dias, B.G.; Ladiwala, U.; Tole, S.; Vaidya, V.A. Recruitment of the sonic hedgehog signalling cascade in electroconvulsive seizure-mediated regulation of adult rat hippocampal neurogenesis. Eur. J. Neurosci. 2005, 22, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.I.; Shtaya, A.B.; Zaben, M.J.; Owens, E.V.; Kiecker, C.; Gray, W.P. Endogenous GFAP-positive neural stem/progenitor cells in the postnatal mouse cortex are activated following traumatic brain injury. J. Neurotrauma 2012, 29, 828–842. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasmurthy, S.G.; Liu, J.Y.; Song, J.X.; Yang, C.B.; Malampati, S.; Wang, Z.Y.; Huang, Y.Y.; Li, M. Neurogenic traditional Chinese medicine as a promising strategy for the treatment of Alzheimer’s disease. Int. J. Mol. Sci. 2017, 18, 272. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Staufenbiel, M.; Li, R.; Shen, Y. Deficiency of patched 1-induced Gli1 signal transduction results in astrogenesis in Swedish mutated APP transgenic mice. Hum. Mol. Genet. 2014, 23, 6512–6527. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Zhen, Y.Y.; Lin, J.W.; Lin, T.K.; Huang, C.W.; Liou, C.W.; Chan, S.H.; Chuang, Y.C. Dynamin-related protein 1 promotes mitochondrial fission and contributes to the hippocampal neuronal cell death following experimental status epilepticus. CNS Neurosci. Ther. 2016, 22, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Pollard, H.; Charriaut-Marlangue, C.; Cantagrel, S.; Represa, A.; Robain, O.; Moreau, J.; Ben-Ari, Y. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience 1994, 63, 7–18. [Google Scholar] [CrossRef]
- Geissler, M.; Dinse, H.R.; Neuhoff, S.; Kreikemeier, K.; Meier, C. Human umbilical cord blood cells restore brain damage induced changes in rat somatosensory cortex. PLoS ONE 2011, 6, e20194. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Hong, X.; Chen, X.; Lan, F.; Zhang, G.; Liao, L. Intracerebral transplantation of mesenchymal stem cells derived from human umbilical cord blood alleviates hypoxic ischemic brain injury in rat neonates. J. Perinat. Med. 2010, 38, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Gornicka-Pawlak el, B.; Janowski, M.; Habich, A.; Jablonska, A.; Drela, K.; Kozlowska, H.; Lukomska, B.; Sypecka, J.; Domanska-Janik, K. Systemic treatment of focal brain injury in the rat by human umbilical cord blood cells being at different level of neural commitment. Acta Neurobiol. Exp. (Wars) 2011, 71, 46–64. [Google Scholar] [PubMed]
- Wang, X.; Zhao, Y.; Wang, X. Umbilical cord blood cells regulate the differentiation of endogenous neural stem cells in hypoxic ischemic neonatal rats via the hedgehog signaling pathway. Brain Res. 2014, 1560, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novo, E.; Parola, M. The role of redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogen. Tissue Repair 2012, 5, S4. [Google Scholar] [CrossRef] [PubMed]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta 2010, 1802, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayir, H.; Kagan, V.E. Bench-to-bedside review: Mitochondrial injury, oxidative stress and apoptosis--there is nothing more practical than a good theory. Crit. Care 2008, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Wu, H.Y.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Lin, T.K.; Liou, C.W.; Chuang, Y.C. Effects of rosiglitazone on global ischemia-induced hippocampal injury and expression of mitochondrial uncoupling protein 2. Biochem. Biophys. Res. Commun. 2006, 351, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Lin, T.K.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Protective effects of peroxisome proliferator-activated receptors gamma coactivator-1α against neuronal cell death in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res. 2010, 88, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. Mitochondrial dysfunction and oxidative stress: Cause and consequence of epileptic seizures. Free Radic. Biol. Med. 2004, 37, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.V.; Schapira, A.H. Mitochondrial respiratory chain disorders II: Neurodegenerative disorders and nuclear gene defects. Lancet 2000, 355, 389–394. [Google Scholar] [CrossRef]
- Dai, R.L.; Zhu, S.Y.; Xia, Y.P.; Mao, L.; Mei, Y.W.; Yao, Y.F.; Xue, Y.M.; Hu, B. Sonic hedgehog protects cortical neurons against oxidative stress. Neurochem. Res. 2011, 36, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Xia, Y.; Mao, L.; Mei, Y.; Xue, Y.; Hu, B. Involvement of PI3K/Akt pathway in the neuroprotective effect of Sonic hedgehog on cortical neurons under oxidative stress. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 856–860. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Cui, L.; Zhang, C.; Zhang, X.; He, J.; Xie, Y.; Chen, Y. Sonic hedgehog promotes neurite outgrowth of cortical neurons under oxidative stress: Involving of mitochondria and energy metabolism. Exp. Cell Res. 2017, 350, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, J.; Liu, R.Y.; Lian, Z.G.; Chen, X.L.; Ma, L.; Sun, H.M.; Zhao, Y.L. The role of the sonic hedgehog signaling pathway in early brain injury after experimental subarachnoid hemorrhage in rats. Neurosci. Lett. 2013, 552, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Li, T.; Wang, L.; Xie, Y.; Liu, S.; Bai, X.; Zhang, T.; Bo, S.; Xin, D.; Xue, H.; et al. Neuroprotective effects of a Smoothened receptor agonist against early brain injury after experimental subarachnoid hemorrhage in rats. Front. Cell. Neurosci. 2016, 10, 306. [Google Scholar] [CrossRef] [PubMed]
- Volkmar, F.R.; Lord, C.; Bailey, A.; Schultz, R.T.; Klin, A. Autism and pervasive developmental disorders. J. Child Psychol. Psychiatry 2004, 45, 135–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, A.; Chauhan, V. Oxidative stress in autism. Pathophysiology 2006, 13, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Al-Ayadhi, L.Y. Relationship between sonic hedgehog protein, brain-derived neurotrophic factor and oxidative stress in autism spectrum disorders. Neurochem. Res. 2012, 37, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mehta, J.L. Antisense to LOX-1 inhibits oxidized LDL-mediated upregulation of monocyte chemoattractant protein-1 and monocyte adhesion to human coronary artery endothelial cells. Circulation 2000, 101, 2889–2895. [Google Scholar] [CrossRef] [PubMed]
- Schreurs, M.P.; Cipolla, M.J. Cerebrovascular dysfunction and blood-brain barrier permeability induced by oxidized LDL are prevented by apocynin and magnesium sulfate in female rats. J. Cardiovasc. Pharmacol. 2014, 63, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.L.; Chen, T.; Zhang, X. Activation of sonic hedgehog signaling attenuates oxidized low-density lipoprotein-stimulated brain microvascular endothelial cells dysfunction in vitro. Int. J. Clin. Exp. Pathol. 2015, 8, 12820–12828. [Google Scholar] [PubMed]
- Chen, K.Y.; Chiu, C.H.; Wang, L.C. Anti-apoptotic effects of Sonic hedgehog signalling through oxidative stress reduction in astrocytes co-cultured with excretory-secretory products of larval Angiostrongylus cantonensis. Sci. Rep. 2017, 7, 41574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, R.; Turnbull, J. Sonic hedgehog is cytoprotective against oxidative challenge in a cellular model of amyotrophic lateral sclerosis. J. Mol. Neurosci. 2012, 47, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.C.; Neher, J.J. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol. Neurobiol. 2010, 41, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Brilha, S.; Ong, C.W.M.; Weksler, B.; Romero, N.; Couraud, P.O.; Friedland, J.S. Matrix metalloproteinase-9 activity and a downregulated Hedgehog pathway impair blood-brain barrier function in an in vitro model of CNS tuberculosis. Sci. Rep. 2017, 7, 16031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, H.; Zhao, L.; Ling, Z.; Kuo, L.; Xue, X.; Feng, J. Wip1 regulates blood-brain barrier function and neuro-inflammation induced by lipopolysaccharide via the sonic hedgehog signaling signaling pathway. Mol. Immunol. 2018, 93, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.B.; Singh, M.V.; Piekna-Przybylska, D.; Gorantla, S.; Poluektova, L.Y.; Maggirwar, S.B. Sonic Hedgehog mimetic prevents leukocyte infiltration into the CNS during acute HIV infection. Sci. Rep. 2017, 7, 9578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.B.; Singh, M.V.; Gorantla, S.; Poluektova, L.Y.; Maggirwar, S.B. Smoothened agonist reduces human immunodeficiency virus type-1-induced blood-brain barrier breakdown in humanized mice. Sci. Rep. 2016, 6, 26876. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.F.; Zhao, Y.; Jiang, J.P.; Guan, W.X.; Du, J.F. Phosphatase Wip1 in immunity: An overview and update. Front. Immunol. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Cui, L.; Xu, F.; Chen, L.; Jiang, L.; Huang, H.; Xu, J.; Zhao, X.; Li, L.; Zeng, S.; et al. Up-regulation of Wip1 involves in neuroinflammation of retinal astrocytes after optic nerve crush via NF-κB signaling pathway. Inflamm. Res. 2016, 65, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.; Cha, H.; Lee, M.O.; Mazur, S.J.; Appella, E.; Fornace, A.J., Jr. Regulation of the Wip1 phosphatase and its effects on the stress response. Front. Biosci. 2012, 17, 1480–1498. [Google Scholar] [CrossRef]
- Macurek, L.; Lindqvist, A.; Voets, O.; Kool, J.; Vos, H.R.; Medema, R.H. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene 2010, 29, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Nguyen, T.A.; Moon, S.H.; Darlington, Y.; Sommer, M.; Donehower, L.A. The type 2C phosphatase Wip1: An oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metast. Rev. 2008, 27, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.; Lee, J.; Malhotra, A.; Nahta, R.; Arnold, A.R.; Buss, M.C.; Brown, B.D.; Maier, C.; Kenney, A.M.; Remke, M.; et al. WIP1 modulates responsiveness to Sonic Hedgehog signaling in neuronal precursor cells and medulloblastoma. Oncogene 2016, 35, 5552–5564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, G.A.; Yang, Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg. Focus 2007, 22, E4. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Paul-Satyaseela, M.; Lamichhane, G.; Kim, K.S.; Bishai, W.R. Mycobacterium tuberculosis invasion and traversal across an in vitro human blood-brain barrier as a pathogenic mechanism for central nervous system tuberculosis. J. Infect. Dis. 2006, 193, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Namer, I.J.; Steibel, J.; Poulet, P.; Mauss, Y.; Mohr, M.; Chambron, J. The role of Mycobacterium tuberculosis in experimental allergic encephalomyelitis. Eur. Neurol. 1994, 34, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Antinori, A.; Arendt, G.; Becker, J.T.; Brew, B.J.; Byrd, D.A.; Cherner, M.; Clifford, D.B.; Cinque, P.; Epstein, L.G.; Goodkin, K.; et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007, 69, 1789–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Lopez, N.; Athonvarangkul, D.; Singh, R. Autophagy and aging. Adv. Exp. Med. Biol. 2015, 847, 73–87. [Google Scholar] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Nah, J.; Yuan, J.; Jung, Y.K. Autophagy in neurodegenerative diseases: From mechanism to therapeutic approach. Mol. Cells 2015, 38, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, A.; Yue, Z. Autophagy and its normal and pathogenic states in the brain. Annu. Rev. Neurosci. 2014, 37, 55–78. [Google Scholar] [CrossRef] [PubMed]
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta 2008, 1782, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radad, K.; Moldzio, R.; Al-Shraim, M.; Kranner, B.; Krewenka, C.; Rausch, W.D. Recent advances in autophagy-based neuroprotection. Expert Rev. Neurother. 2015, 15, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.C.; Yu, J.T.; Jiang, T.; Tan, L. Autophagy modulation for Alzheimer’s disease therapy. Mol. Neurobiol. 2013, 48, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Chen, Y.; Sullivan, M.L.; Kochanek, P.M.; Clark, R.S. Autophagy in acute brain injury: Feast, famine, or folly? Neurobiol. Dis. 2011, 43, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinby, G.E. Feasibility of prophylaxis by oral pralidoxime. Cholinesterase inactivation by organophosphorus pesticides. Arch. Environ. Health 1968, 16, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Omata, Y.; Lim, Y.M.; Akao, Y.; Tsuda, L. Age-induced reduction of autophagy-related gene expression is associated with onset of Alzheimer’s disease. Am. J. Neurodegen. Dis. 2014, 3, 134–142. [Google Scholar]
- Baldo, B.; Soylu, R.; Petersen, A. Maintenance of basal levels of autophagy in Huntington’s disease mouse models displaying metabolic dysfunction. PLoS ONE 2013, 8, e83050. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhami, F.; Schloemer, A.; Kuan, C.Y. The roles of autophagy in cerebral ischemia. Autophagy 2007, 3, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, X.; Xu, F.; Bahr, B.A.; Shibata, M.; Uchiyama, Y.; Hagberg, H.; Blomgren, K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005, 12, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rami, A.; Langhagen, A.; Steiger, S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol. Dis. 2008, 29, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Vosler, P.S.; Graham, S.H.; Wechsler, L.R.; Chen, J. Mitochondrial targets for stroke: Focusing basic science research toward development of clinically translatable therapeutics. Stroke 2009, 40, 3149–3155. [Google Scholar] [CrossRef] [PubMed]
- Petralia, R.S.; Schwartz, C.M.; Wang, Y.X.; Kawamoto, E.M.; Mattson, M.P.; Yao, P.J. Sonic hedgehog promotes autophagy in hippocampal neurons. Biol. Open 2013, 2, 499–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Sanchez, M.; Menzies, F.M.; Chang, Y.Y.; Simecek, N.; Neufeld, T.P.; Rubinsztein, D.C. The Hedgehog signalling pathway regulates autophagy. Nat. Commun. 2012, 3, 1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.Y.; He, Y.J.; Yang, Q.H.; Huang, W.; Liu, Z.H.; Ye, G.R.; Tang, S.H.; Shu, J.C. Induction of autophagy and apoptosis by miR-148a through the sonic hedgehog signaling pathway in hepatic stellate cells. Am. J. Cancer Res. 2015, 5, 2569–2589. [Google Scholar] [PubMed]
- Xiao, Q.; Yang, Y.; Qin, Y.; He, Y.H.; Chen, K.X.; Zhu, J.W.; Zhang, G.P.; Luo, J.D. AMP-activated protein kinase-dependent autophagy mediated the protective effect of sonic hedgehog pathway on oxygen glucose deprivation-induced injury of cardiomyocytes. Biochem. Biophys. Res. Commun. 2015, 457, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Gagne-Sansfacon, J.; Allaire, J.M.; Jones, C.; Boudreau, F.; Perreault, N. Loss of Sonic hedgehog leads to alterations in intestinal secretory cell maturation and autophagy. PLoS ONE 2014, 9, e98751. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, J.; Li, Y.; Singh, P.; Cao, L.; Xu, L.J.; Li, D.; Wang, Y.; Xie, Z.; Gui, Y. Sonic hedgehog promotes autophagy of vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1319–H1331. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.; Fauconnier, J.; Cazorla, O.; Thireau, J.; Soleti, R.; Vidal, B.; Ouille, A.; Bartholome, M.; Bideaux, P.; Roubille, F.; et al. Activation of Sonic hedgehog signaling in ventricular cardiomyocytes exerts cardioprotection against ischemia reperfusion injuries. Sci. Rep. 2015, 5, 7983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, K.; Takano, H.; Niitsuma, Y.; Hasegawa, H.; Uchiyama, R.; Oka, T.; Miyazaki, M.; Nakaya, H.; Komuro, I. Sonic hedgehog is a critical mediator of erythropoietin-induced cardiac protection in mice. J. Clin. Investig. 2010, 120, 2016–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Cheng, G.; Agrawal, D.K. Autophagy of vascular smooth muscle cells in atherosclerotic lesions. Autophagy 2007, 3, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Cheng, G.; Gangahar, D.M.; Agrawal, D.K. Insulin-like growth factor-1 and TNF-α regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol. Cell Biol. 2006, 84, 448–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-D.; Yang, J.-L.; Hwang, W.-C.; Yang, D.-I. Emerging Roles of Sonic Hedgehog in Adult Neurological Diseases: Neurogenesis and Beyond. Int. J. Mol. Sci. 2018, 19, 2423. https://doi.org/10.3390/ijms19082423
Chen S-D, Yang J-L, Hwang W-C, Yang D-I. Emerging Roles of Sonic Hedgehog in Adult Neurological Diseases: Neurogenesis and Beyond. International Journal of Molecular Sciences. 2018; 19(8):2423. https://doi.org/10.3390/ijms19082423
Chicago/Turabian StyleChen, Shang-Der, Jenq-Lin Yang, Wei-Chao Hwang, and Ding-I Yang. 2018. "Emerging Roles of Sonic Hedgehog in Adult Neurological Diseases: Neurogenesis and Beyond" International Journal of Molecular Sciences 19, no. 8: 2423. https://doi.org/10.3390/ijms19082423
APA StyleChen, S.-D., Yang, J.-L., Hwang, W.-C., & Yang, D.-I. (2018). Emerging Roles of Sonic Hedgehog in Adult Neurological Diseases: Neurogenesis and Beyond. International Journal of Molecular Sciences, 19(8), 2423. https://doi.org/10.3390/ijms19082423