Recent Advances by In Silico and In Vitro Studies of Amyloid-β 1-42 Fibril Depicted a S-Shape Conformation


Abstract
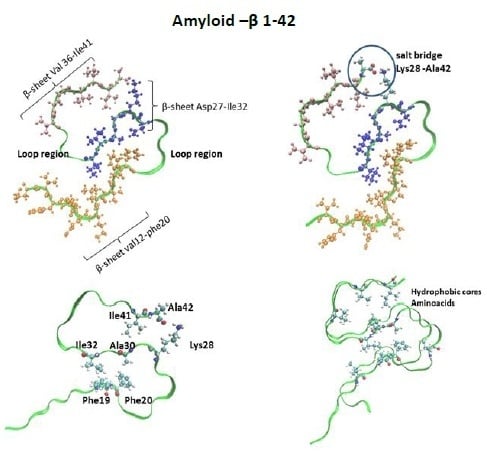
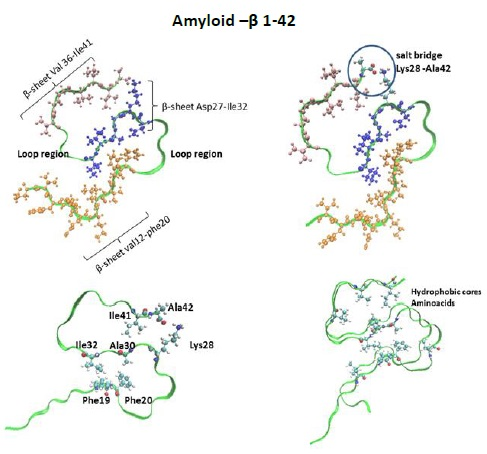
:
1. Introduction
1.1. Structural Properties of Aβ1-42 in Relation to Aβ1-40
- (a)
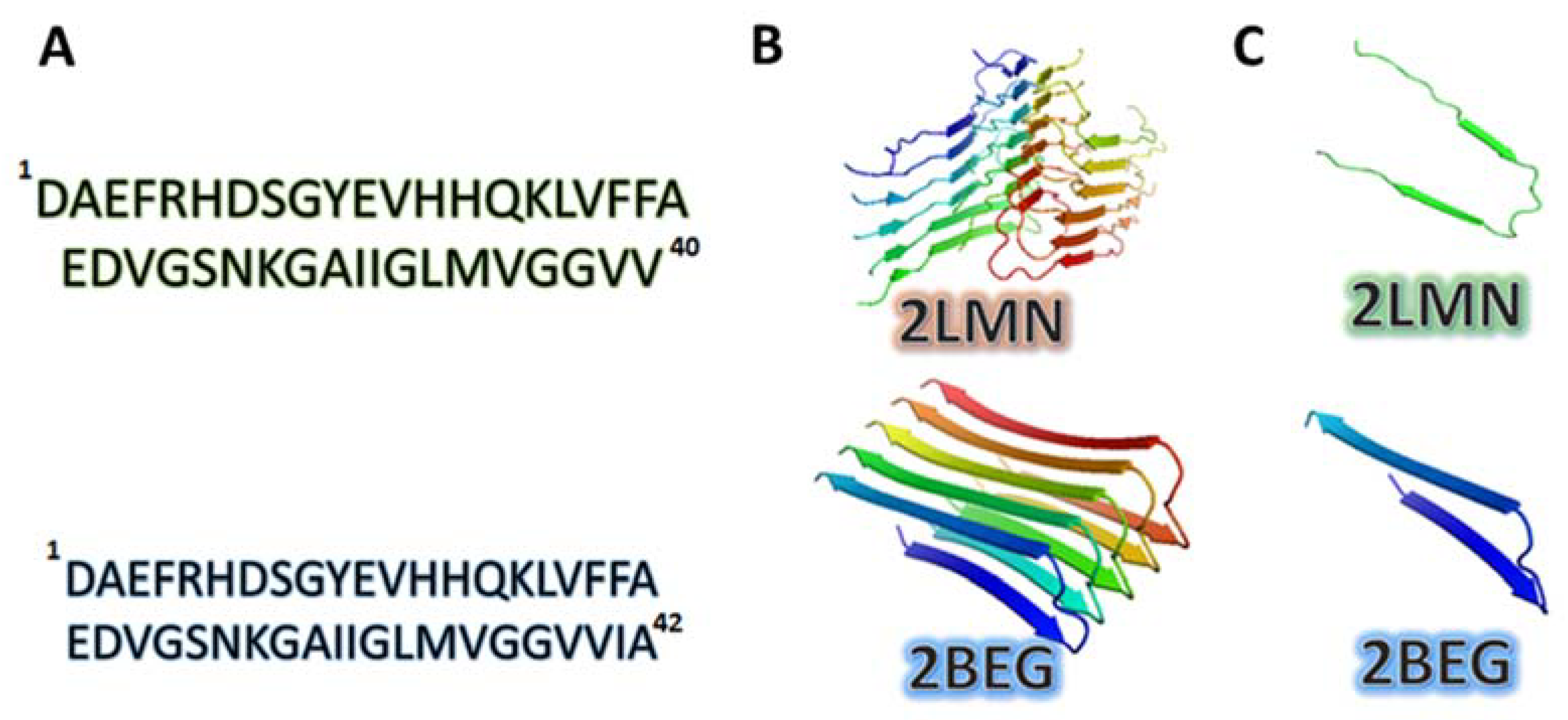
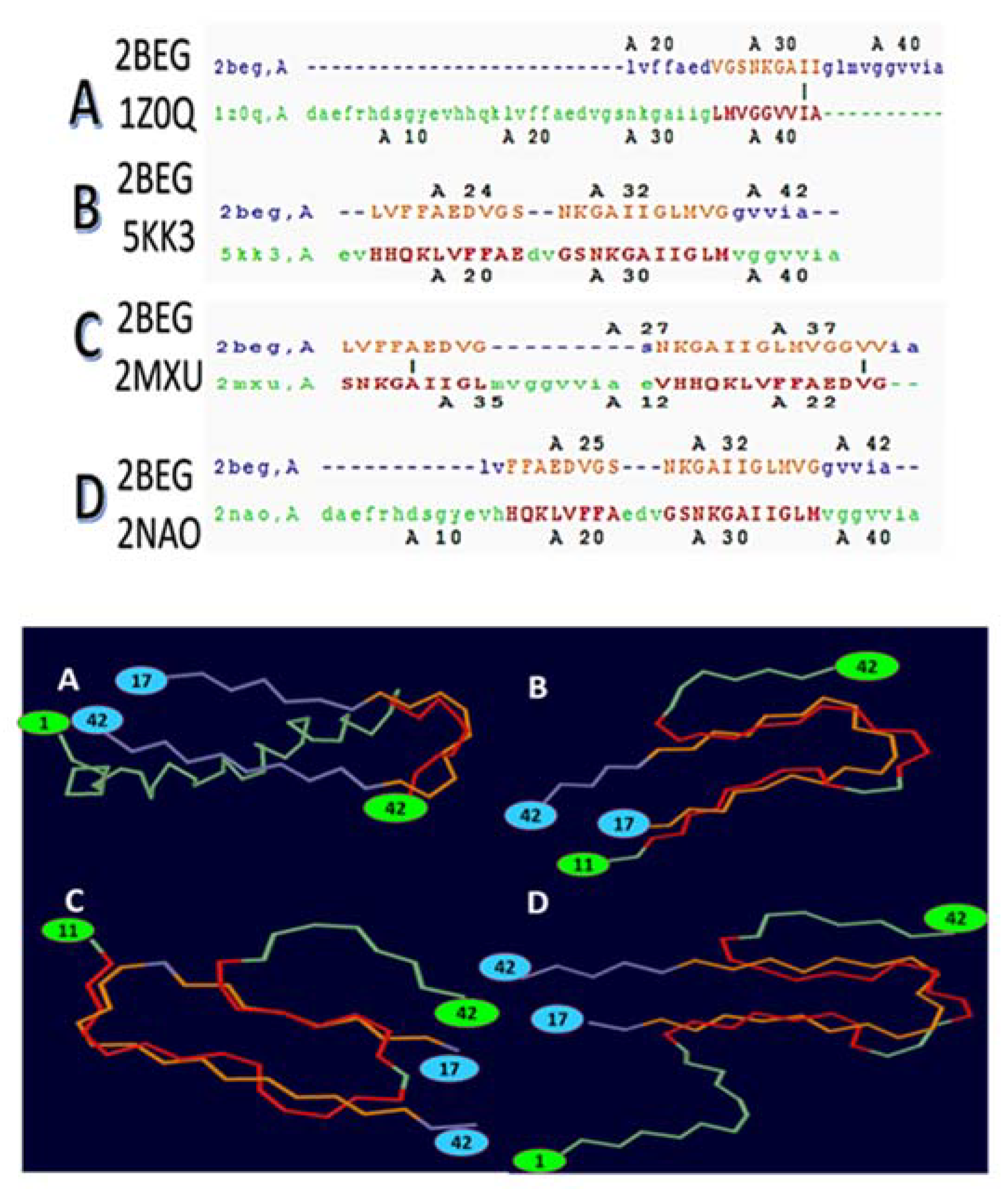
- Protofibril or a cross-β-subunit, Figure 1B);
- (b)
- Structure formed by two β-strands: β1 (residues 18–26) and β2 (residues 31–42);
- (c)
1.2. Aβ1-42 Fibril Preparation
1.3. Molecular Structure of S-Shape Aβ1-42 by Solid-State Nuclear Magnetic Resonance (ssNMR)
1.4. Molecular Structure of Aβ1-42 by Electron Microscopy (EM)
1.5. Biochemical Techniques to Determinate AB1-42 Aggregation
1.6. In Silico Studies Employing the S-Shape Aβ1-42 Structure
1.7. Structures Employed to Design Drugs
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ1-42 | β-amyloid 1-42 |
| APP | Amyloid precursor protein |
| AD | Alzheimer disease |
| Aβ | Amyloid beta |
| IR | Infrared spectroscopy |
| TEM | Transmision electron microscopy |
| AFM | Atomic force microscopy |
| ThT | Tioflavin T |
| SSNMR | Solid state nuclear magnetic resonance |
| FS-REDOR | Frequency selective rotational-echo, double resonance |
| MPL | Mass-per length |
| EM | Electron microscopy |
| STEM | Scanning transmission electron microscopy |
| HS-AFM | High-speed atomic force microscopy |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| CE | Capillary electrophoresis |
| ES-DMA | Electrospray differential mobility analysis |
| REMD | Replica Exchange Molecular Dynamics |
References
- Sipe, J.D.; Cohen, A.S. Review: History of the Amyloid Fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Fändrich, M. On the Structural Definition of Amyloid Fibrils and Other Polypeptide Aggregates. Cell. Mol. Life Sci. CMLS 2007, 64, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H. Alzheimer’s Disease and the Amyloid-Beta Peptide. J. Alzheimers Dis. JAD 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E.; Gold, G. Alzheimer Disease Therapy—Moving from Amyloid-β to Tau. Nat. Rev. Neurol. 2013, 9, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-W.; Chang, C.-F. Alzheimer’s Amyloid-β A2T Variant and Its N-Terminal Peptides Inhibit Amyloid-β Fibrillization and Rescue the Induced Cytotoxicity. PLoS ONE 2017, 12, e0174561. [Google Scholar] [CrossRef] [PubMed]
- Makin, O.S.; Atkins, E. Molecular Basis for Amyloid Fibril Formation and Stability. Proc. Natl. Acad. Sci. USA 2005, 102, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Dorostkar, M.M.; Zou, C.; Blazquez-Llorca, L.; Herms, J. Analyzing Dendritic Spine Pathology in Alzheimer’s Disease: Problems and Opportunities. Acta Neuropathol. 2015, 130, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Saftig, P. Deficiency of Presenilin-1 Inhibits the Normal Cleavage of Amyloid Precursor Protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP Processing and Synaptic Function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Kamal, A.; Stokin, G.B.; Yang, Z.; Xia, C.H.; Goldstein, L.S. Axonal Transport of Amyloid Precursor Protein Is Mediated by Direct Binding to the Kinesin Light Chain Subunit of Kinesin-I. Neuron 2000, 28, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Majd, S.; Power, J.H.; Grantham, H.J.M. Neuronal Response in Alzheimer’s and Parkinson’s Disease: The Effect of Toxic Proteins on Intracellular Pathways. BMC Neurosci. 2015, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Kagan, B.L.; Jang, H.; Capone, R.; Teran Arce, F.; Ramachandran, S.; Lal, R.; Nussinov, R. Antimicrobial Properties of Amyloid Peptides. Mol. Pharm. 2012, 9, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.V.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β Peptide Protects against Microbial Infection in Mouse and Worm Models of Alzheimer’s Disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef] [PubMed]
- Plant, L.D.; Boyle, J.P.; Smith, I.F.; Peers, C.; Pearson, H.A. The Production of Amyloid Beta Peptide Is a Critical Requirement for the Viability of Central Neurons. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 5531–5535. [Google Scholar] [CrossRef]
- Ow, S.-Y.; Dunstan, D.E. A Brief Overview of Amyloids and Alzheimer’s Disease. Protein Sci. Publ. Protein Soc. 2014, 23, 1315–1331. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-X.; Qiang, W. Molecular Structure of β-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.A.; Fox, N.C.; Sperling, R.A.; Klunk, W.E. Brain Imaging in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006213. [Google Scholar] [CrossRef] [PubMed]
- Omachi, Y.; Ito, K.; Arima, K.; Matsuda, H.; Nakata, Y.; Sakata, M.; Sato, N.; Nakagome, K.; Motohashi, N. Clinical Impact of (11)C-Pittsburgh Compound-B Positron Emission Tomography Carried out in Addition to Magnetic Resonance Imaging and Single-Photon Emission Computed Tomography on the Diagnosis of Alzheimer’s Disease in Patients with Dementia and Mild Cognitive Impairment. Psychiatry Clin. Neurosci. 2015, 69, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Vignaud, H.; Bobo, C.; Lascu, I.; Sörgjerd, K.M.; Zako, T.; Maeda, M.; Salin, B.; Lecomte, S.; Cullin, C. A Structure-Toxicity Study of Aß42 Reveals a New Anti-Parallel Aggregation Pathway. PLoS ONE 2013, 8, e80262. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Hoffmann, W.; Warnke, S.; Huang, X.; Gewinner, S.; Schöllkopf, W.; Bowers, M.T.; von Helden, G.; Pagel, K. An Infrared Spectroscopy Approach to Follow β-Sheet Formation in Peptide Amyloid Assemblies. Nat. Chem. 2017, 9, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Lomont, J.P.; Rich, K.L.; Maj, M.; Ho, J.-J.; Ostrander, J.S.; Zanni, M.T. Spectroscopic Signature for Stable β-Amyloid Fibrils versus β-Sheet-Rich Oligomers. J. Phys. Chem. B 2018, 122, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Gallardo, R.; Ungureanu, A.-A.; Castillo Cano, V.; Snellinx, A.; Ramakers, M.; Bartic, C.; Rousseau, F.; Schymkowitz, J.; De Strooper, B. The Alzheimer Disease Protective Mutation A2T Modulates Kinetic and Thermodynamic Properties of Amyloid-β (Aβ) Aggregation. J. Biol. Chem. 2014, 289, 30977–30989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krotee, P.; Rodriguez, J.A.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Hattne, J.; Nannenga, B.L.; Oskarsson, M.E.; Philipp, S.; et al. Atomic Structures of Fibrillar Segments of HIAPP Suggest Tightly Mated β-Sheets Are Important for Cytotoxicity. eLife 2017, 6, e19273. [Google Scholar] [CrossRef] [PubMed]
- Jahn, T.R.; Makin, O.S.; Morris, K.L.; Marshall, K.E.; Tian, P.; Sikorski, P.; Serpell, L.C. The Common Architecture of Cross-Beta Amyloid. J. Mol. Biol. 2010, 395, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Krotee, P.; Griner, S.L.; Sawaya, M.R.; Cascio, D.; Rodriguez, J.A.; Shi, D.; Philipp, S.; Murray, K.; Saelices, L.; Lee, J.; et al. Common Fibrillar Spines of Amyloid-β and Human Islet Amyloid Polypeptide Revealed by Microelectron Diffraction and Structure-Based Inhibitors. J. Biol. Chem. 2018, 293, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Cantu’, L.; Colombo, L.; Stoilova, T.; Demé, B.; Inouye, H.; Booth, R.; Rondelli, V.; Di Fede, G.; Tagliavini, F.; Del Favero, E.; et al. The A2V Mutation as a New Tool for Hindering Aβ Aggregation: A Neutron and X-ray Diffraction Study. Sci. Rep. 2017, 7, 5510. [Google Scholar] [CrossRef] [PubMed]
- Periole, X.; Huber, T.; Bonito-Oliva, A.; Aberg, K.C.; van der Wel, P.C.A.; Sakmar, T.P.; Marrink, S.J. Energetics Underlying Twist Polymorphisms in Amyloid Fibrils. J. Phys. Chem. B 2018, 122, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Yau, W.-M.; Luo, Y.; Mattson, M.P.; Tycko, R. Antiparallel β-Sheet Architecture in Iowa-Mutant β-Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2012, 109, 4443–4448. [Google Scholar] [CrossRef] [PubMed]
- Hubin, E.; Deroo, S.; Schierle, G.K.; Kaminski, C.; Serpell, L.; Subramaniam, V.; van Nuland, N.; Broersen, K.; Raussens, V.; Sarroukh, R. Two Distinct β-Sheet Structures in Italian-Mutant Amyloid-Beta Fibrils: A Potential Link to Different Clinical Phenotypes. Cell. Mol. Life Sci. CMLS 2015, 72, 4899–4913. [Google Scholar] [CrossRef] [PubMed]
- Norlin, N.; Hellberg, M. Aggregation and Fibril Morphology of the Arctic Mutation of Alzheimer’s Aβ Peptide by CD, TEM, STEM and in Situ AFM. J. Struct. Biol. 2012, 180, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Nakayama, T.; Ono, K.; Itami, M.; Takahashi, R.; Teplow, D.B.; Yamada, M. High-Speed Atomic Force Microscopy Reveals Structural Dynamics of Amyloid β1–42 Aggregates. Proc. Natl. Acad. Sci. USA 2016, 113, 5835–5840. [Google Scholar] [CrossRef] [PubMed]
- Murvai, Ü.; Somkuti, J.; Smeller, L.; Penke, B.; Kellermayer, M.S.Z. Structural and Nanomechanical Comparison of Epitaxially and Solution-Grown Amyloid Β25–35 Fibrils. Biochim. Biophys. Acta BBA Proteins Proteom. 2015, 1854, 327–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canale, C.; Relini, A.; Gliozzi, A. Atomic Force Microscopy of Ex Vivo Amyloid Fibrils. Methods Mol. Biol. 2011, 736, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Drolle, E.; Hane, F. Atomic Force Microscopy to Study Molecular Mechanisms of Amyloid Fibril Formation and Toxicity in Alzheimer’s Disease. Drug Metab. Rev. 2014, 46, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Canale, C.; Seghezza, S.; Vilasi, S.; Carrotta, R.; Bulone, D.; Diaspro, A.; San Biagio, P.L.; Dante, S. Different Effects of Alzheimer’s Peptide Aβ(1-40) Oligomers and Fibrils on Supported Lipid Membranes. Biophys. Chem. 2013, 182, 23–29. [Google Scholar] [CrossRef] [PubMed]
- LeVine, H. [18] Quantification of β-Sheet Amyloid Fibril Structures with Thioflavin T. In Methods in Enzymology; Amyloid, Prions, and Other Protein Aggregates; Academic Press: New York, NY, USA, 1999; Volume 309, pp. 274–284. [Google Scholar]
- Choi, S.R.; Schneider, J.A.; Bennett, D.A.; Beach, T.G.; Bedell, B.J.; Zehntner, S.P.; Krautkramer, M.J.; Kung, H.F.; Skovronsky, D.M.; Hefti, F.; et al. Correlation of Amyloid PET Ligand Florbetapir F 18 Binding with Aβ Aggregation and Neuritic Plaque Deposition in Postmortem Brain Tissue. Alzheimer Dis. Assoc. Disord. 2012, 26, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Tsuhara, S.; Tamura, A.; Chatani, E. A Specific Form of Prefibrillar Aggregates That Functions as a Precursor of Amyloid Nucleation. Sci. Rep. 2018, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.; Zhang-Haagen, B.; Decker, C.; Barz, B.; Schneider, M.; Biehl, R.; Radulescu, A.; Strodel, B.; Willbold, D.; Nagel-Steger, L. Aβ42 Pentamers/Hexamers Are the Smallest Detectable Oligomers in Solution. Sci. Rep. 2017, 7, 2493. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; So, M.; Sakurai, K.; Kardos, J.; Goto, Y. Supersaturation-Limited and Unlimited Phase Transitions Compete to Produce the Pathway Complexity in Amyloid Fibrillation. J. Biol. Chem. 2015, 290, 18134–18145. [Google Scholar] [CrossRef] [PubMed]
- Shigemitsu, Y.; Iwaya, N.; Goda, N.; Matsuzaki, M.; Tenno, T.; Narita, A.; Hoshi, M.; Hiroaki, H. Nuclear Magnetic Resonance Evidence for the Dimer Formation of Beta Amyloid Peptide 1-42 in 1,1,1,3,3,3-Hexafluoro-2-Propanol. Anal. Biochem. 2016, 498, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Pujol-Pina, R.; Vilaprinyó-Pascual, S.; Mazzucato, R.; Arcella, A.; Vilaseca, M.; Orozco, M.; Carulla, N. SDS-PAGE Analysis of Aβ Oligomers Is Disserving Research into Alzheimer´s Disease: Appealing for ESI-IM-MS. Sci. Rep. 2015, 5, 14809. [Google Scholar] [CrossRef] [PubMed]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1-42) Fibril Structure Illuminates Self-Recognition and Replication of Amyloid in Alzheimer’s Disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Barz, B.; Strodel, B. Understanding Amyloid-β Oligomerization at the Molecular Level: The Role of the Fibril Surface. Chem. Weinh. Bergstr. Ger. 2016, 22, 8768–8772. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s Disease: Genes, Proteins, and Therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Foroutanpay, B.V.; Kumar, J.; Kang, S.G.; Danaei, N.; Westaway, D.; Sim, V.L.; Kar, S. The Effects of N-Terminal Mutations on β-Amyloid Peptide Aggregation and Toxicity. Neuroscience 2018, 379, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Xi, W.-H.; Wei, G.-H. Amyloid-β Peptide Aggregation and the Influence of Carbon Nanoparticles. Chin. Phys. B 2016, 25, 018704. [Google Scholar] [CrossRef]
- Linse, S. Monomer-Dependent Secondary Nucleation in Amyloid Formation. Biophys. Rev. 2017, 9, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Laganowsky, A.; Liu, C.; Sawaya, M.R.; Whitelegge, J.P.; Park, J.; Zhao, M.; Pensalfini, A.; Soriaga, A.B.; Landau, M.; Teng, P.K.; et al. Atomic View of a Toxic Amyloid Small Oligomer. Science 2012, 335, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhao, M.; Jiang, L.; Cheng, P.-N.; Park, J.; Sawaya, M.R.; Pensalfini, A.; Gou, D.; Berk, A.J.; Glabe, C.G.; et al. Out-of-Register β-Sheets Suggest a Pathway to Toxic Amyloid Aggregates. Proc. Natl. Acad. Sci. USA 2012, 109, 20913–20918. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Tsai, C.-J.; Nussinov, R. Hollow Core of Alzheimer’s Abeta42 Amyloid Observed by CryoEM Is Relevant at Physiological PH. Proc. Natl. Acad. Sci. USA 2010, 107, 14128–14133. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.P.; Okamoto, Y.; Straub, J.E.; Brooks, B.R.; Thirumalai, D. Thermodynamic Perspective on the Dock-Lock Growth Mechanism of Amyloid Fibrils. J. Phys. Chem. B 2009, 113, 14421–14430. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Alabdullah, N.H.; Ehmedat, H.M.; Abulifa, A.R.; Taban, I.; Upadhyayula, S. NSAIDs as Potential Treatment Option for Preventing Amyloid β Toxicity in Alzheimer’s Disease: An Investigation by Docking, Molecular Dynamics, and DFT Studies. J. Biomol. Struct. Dyn. 2018, 36, 2099–2117. [Google Scholar] [CrossRef] [PubMed]
- Graff-Radford, N.R.; Crook, J.E.; Lucas, J.; Boeve, B.F.; Knopman, D.S.; Ivnik, R.J.; Smith, G.E.; Younkin, L.H.; Petersen, R.C.; Younkin, S.G. Association of Low Plasma Abeta42/Abeta40 Ratios with Increased Imminent Risk for Mild Cognitive Impairment and Alzheimer Disease. Arch. Neurol. 2007, 64, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural Conversion of Neurotoxic Amyloid-Beta(1-42) Oligomers to Fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R. Amyloid Polymorphism: Structural Basis and Neurobiological Relevance. Neuron 2015, 86, 632–645. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Gonnelli, L.; Luchinat, C.; Mao, J.; Nesi, A. A New Structural Model of Aβ40 Fibrils. J. Am. Chem. Soc. 2011, 133, 16013–16022. [Google Scholar] [CrossRef] [PubMed]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.M.; Tycko, R. Molecular Structural Basis for Polymorphism in Alzheimer’s Beta-Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Solanki, V.; Tiwari, M. In Vivo and in-Vitro Techniques Used to Investigate Alzheimer’s Disease. Front. Life Sci. 2015, 8, 332–347. [Google Scholar] [CrossRef]
- Shoghi-Jadid, K.; Small, G.W.; Agdeppa, E.D.; Kepe, V.; Ercoli, L.M.; Siddarth, P.; Read, S.; Satyamurthy, N.; Petric, A.; Huang, S.-C.; et al. Localization of Neurofibrillary Tangles and Beta-Amyloid Plaques in the Brains of Living Patients with Alzheimer Disease. Am. J. Geriatr. Psychiatry 2002, 10, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.E.; Sherman, M.A.; Grant, M.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Ashe, K.H. Brain Amyloid-β Oligomers in Ageing and Alzheimer’s Disease. Brain 2013, 136, 1383–1398. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid β-Protein Dimers Isolated Directly from Alzheimer Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Kasza, Á.; Penke, B.; Frank, Z.; Bozsó, Z.; Szegedi, V.; Hunya, Á.; Németh, K.; Kozma, G.; Fülöp, L. Studies for Improving a Rat Model of Alzheimer’s Disease: Icv Administration of Well-Characterized β-Amyloid 1-42 Oligomers Induce Dysfunction in Spatial Memory. Molecules 2017, 22, 2007. [Google Scholar] [CrossRef] [PubMed]
- Morel, B.; Varela, L.; Azuaga, A.I.; Conejero-Lara, F. Environmental Conditions Affect the Kinetics of Nucleation of Amyloid Fibrils and Determine Their Morphology. Biophys. J. 2010, 99, 3801–3810. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Shea, J.-E. Effects of Solvent on the Structure of the Alzheimer Amyloid-Beta(25-35) Peptide. Biophys. J. 2006, 91, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Tsai, M.-Y.; Wolynes, P.G. Comparing the Aggregation Free Energy Landscapes of Amyloid Beta(1-42) and Amyloid Beta(1-40). J. Am. Chem. Soc. 2017, 139, 16666–16676. [Google Scholar] [CrossRef] [PubMed]
- Barrow, C.J.; Yasuda, A.; Kenny, P.T.; Zagorski, M.G. Solution Conformations and Aggregational Properties of Synthetic Amyloid Beta-Peptides of Alzheimer’s Disease. Analysis of Circular Dichroism Spectra. J. Mol. Biol. 1992, 225, 1075–1093. [Google Scholar] [CrossRef]
- Roche, J.; Shen, Y.; Lee, J.H.; Ying, J.; Bax, A. Monomeric Aβ(1-40) and Aβ(1-42) Peptides in Solution Adopt Very Similar Ramachandran Map Distributions That Closely Resemble Random Coil. Biochemistry 2016, 55, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Colletier, J.-P.; Laganowsky, A.; Landau, M.; Zhao, M.; Soriaga, A.B.; Goldschmidt, L.; Flot, D.; Cascio, D.; Sawaya, M.R.; Eisenberg, D. Molecular Basis for Amyloid-β Polymorphism. Proc. Natl. Acad. Sci. USA 2011, 108, 16938–16943. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Sachse, C.; Richter, W.; Xu, C.; Fändrich, M.; Grigorieff, N. Comparison of Alzheimer Abeta(1-40) and Abeta(1-42) Amyloid Fibrils Reveals Similar Protofilament Structures. Proc. Natl. Acad. Sci. USA 2009, 106, 19813–19818. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W.-M.; Mattson, M.P.; Tycko, R. Self-Propagating, Molecular-Level Polymorphism in Alzheimer’s Beta-Amyloid Fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-Resolution Structure of a Disease-Relevant Aβ(1-42) Amyloid Fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.S.; Ansaloni, A.; Mezzenga, R.; Lashuel, H.A.; Dietler, G. Novel Mechanistic Insight into the Molecular Basis of Amyloid Polymorphism and Secondary Nucleation during Amyloid Formation. J. Mol. Biol. 2013, 425, 1765–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsbury, C.; Baxa, U.; Simon, M.N.; Steven, A.C.; Engel, A.; Wall, J.S.; Aebi, U.; Müller, S.A. Amyloid Structure and Assembly: Insights from Scanning Transmission Electron Microscopy. J. Struct. Biol. 2011, 173, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lyubchenko, Y. Atomic Force Microscopy Imaging and Probing of Amyloid Nanoaggregates. In Handbook of Clinical Nanomedicine: Nanoparticles, Imaging; Therapy and Clinical Applications; Pan Stanford Publishing: Singapore, 2016. [Google Scholar]
- Saraswathy, N.; Ramalingam, P. Concepts and Techniques in Genomics and Proteomics; Elsevier: Oxford, NY, USA, 2011; ISBN 978-1-908818-05-8. [Google Scholar]
- Honda, T.; Marotta, C.A. Arginine Specific Endopeptidases Modify the Aggregation Properties of a Synthetic Peptide Derived from Alzheimer Beta/A4 Amyloid. Neurochem. Res. 1992, 17, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Beyreuther, K.; Bush, A.I.; Dyrks, T.; Hilbich, C.; König, G.; Mönning, U.; Multhaup, G.; Prior, R.; Rumble, B.; Schubert, W. Mechanisms of Amyloid Deposition in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 1991, 640, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Soreghan, B.; Kosmoski, J.; Glabe, C. Surfactant Properties of Alzheimer’s A Beta Peptides and the Mechanism of Amyloid Aggregation. J. Biol. Chem. 1994, 269, 28551–28554. [Google Scholar] [PubMed]
- Huang, J.; Matthews, H.R. Application of Sodium Dodecyl Sulfate-Gel Electrophoresis to Low Molecular Weight Polypeptides. Anal. Biochem. 1990, 188, 114–117. [Google Scholar] [CrossRef]
- Weber, K.; Osborn, M. The Reliability of Molecular Weight Determinations by Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis. J. Biol. Chem. 1969, 244, 4406–4412. [Google Scholar] [PubMed]
- Kawooya, J.K.; Emmons, T.L.; Gonzalez-DeWhitt, P.A.; Camp, M.C.; D’Andrea, S.C. Electrophoretic Mobility of Alzheimer’s Amyloid-Beta Peptides in Urea-Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis. Anal. Biochem. 2003, 323, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Lewis, U.J.; Clark, M.O. Preparative methods for disk electrophoresis with special reference to the isolation of pituitary hormones. Anal. Biochem. 1963, 6, 303–315. [Google Scholar] [CrossRef]
- Racusen, D.; Calvanico, N. Preparative electrophoresis on polyacrylamide gel. Anal. Biochem. 1964, 7, 62–66. [Google Scholar] [CrossRef]
- Galland-Irmouli, A.V.; Pons, L.; Luçon, M.; Villaume, C.; Mrabet, N.T.; Guéant, J.L.; Fleurence, J. One-Step Purification of R-Phycoerythrin from the Red Macroalga Palmaria Palmata Using Preparative Polyacrylamide Gel Electrophoresis. J. Chromatogr. B Biomed. Sci. Appl. 2000, 739, 117–123. [Google Scholar] [CrossRef]
- Li, Y.; Bjorklund, S.; Jiang, Y.W.; Kim, Y.J.; Lane, W.S.; Stillman, D.J.; Kornberg, R.D. Yeast Global Transcriptional Regulators Sin4 and Rgr1 Are Components of Mediator Complex/RNA Polymerase II Holoenzyme. Proc. Natl. Acad. Sci. USA 1995, 92, 10864–10868. [Google Scholar] [CrossRef] [PubMed]
- Brinet, D.; Kaffy, J.; Oukacine, F.; Glumm, S.; Ongeri, S.; Taverna, M. An Improved Capillary Electrophoresis Method for in Vitro Monitoring of the Challenging Early Steps of Aβ1-42 Peptide Oligomerization: Application to Anti-Alzheimer’s Drug Discovery. Electrophoresis 2014, 35, 3302–3309. [Google Scholar] [CrossRef] [PubMed]
- Pryor, N.E.; Moss, M.A.; Hestekin, C.N. Unraveling the Early Events of Amyloid-β Protein (Aβ) Aggregation: Techniques for the Determination of Aβ Aggregate Size. Int. J. Mol. Sci. 2012, 13, 3038–3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prangkio, P.; Yusko, E.C.; Sept, D.; Yang, J.; Mayer, M. Multivariate Analyses of Amyloid-Beta Oligomer Populations Indicate a Connection between Pore Formation and Cytotoxicity. PLoS ONE 2012, 7, e47261. [Google Scholar] [CrossRef] [PubMed]
- Brinet, D.; Gaie-Levrel, F.; Delatour, V.; Kaffy, J.; Ongeri, S.; Taverna, M. In Vitro Monitoring of Amyloid β-Peptide Oligomerization by Electrospray Differential Mobility Analysis: An Alternative Tool to Evaluate Alzheimer’s Disease Drug Candidates. Talanta 2017, 165, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Lam, C.; Chan, M.; Cheung, R.; Law, L.; Lit, L.; Ng, K.; Suen, M.; Tai, H. Electrospray Ionisation Mass Spectrometry: Principles and Clinical Applications. Clin. Biochem. Rev. 2003, 24, 3–12. [Google Scholar] [PubMed]
- Watt, A.D.; Perez, K.A.; Rembach, A.; Sherrat, N.A.; Hung, L.W.; Johanssen, T.; McLean, C.A.; Kok, W.M.; Hutton, C.A.; Fodero-Tavoletti, M.; et al. Oligomers, Fact or Artefact? SDS-PAGE Induces Dimerization of β-Amyloid in Human Brain Samples. Acta Neuropathol. 2013, 125, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.A.; Chen, L.Y.; Plascencia-Villa, G.; Perry, G. Elongation Affinity, Activation Barrier, and Stability of Aβ42 Oligomers/Fibrils in Physiological Saline. Biochem. Biophys. Res. Commun. 2017, 487, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hashemi, M.; Lv, Z.; Lyubchenko, Y.L. Self-Assembly of the Full-Length Amyloid Aβ42 Protein in Dimers. Nanoscale 2016, 8, 18928–18937. [Google Scholar] [CrossRef] [PubMed]
- Esler, W.P.; Stimson, E.R.; Jennings, J.M.; Vinters, H.V.; Ghilardi, J.R.; Lee, J.P.; Mantyh, P.W.; Maggio, J.E. Alzheimer’s Disease Amyloid Propagation by a Template-Dependent Dock-Lock Mechanism. Biochemistry 2000, 39, 6288–6295. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Rebella, M.; Muscat, S.; Morbiducci, U.; Tuszynski, J.; Danani, A.; Deriu, M.A. Conformational Dynamics and Stability of U-Shaped and S-Shaped Amyloid β Assemblies. Int. J. Mol. Sci. 2018, 19, 571. [Google Scholar] [CrossRef] [PubMed]
- Man, V.H.; Nguyen, P.H.; Derreumaux, P. Conformational Ensembles of the Wild-Type and S8C Aβ1-42 Dimers. J. Phys. Chem. B 2017, 121, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.S.; Sawaya, M.R. Implications for Alzheimer’s Disease of an Atomic Resolution Structure of Amyloid-β(1–42) Fibrils. Proc. Natl. Acad. Sci. USA 2016, 113, 9398–9400. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.; Kang, M.; Chang, I. Polymorphism of Fibrillar Structures Depending on the Size of Assembled Aβ17-42 Peptides. Sci. Rep. 2016, 6, 38196. [Google Scholar] [CrossRef] [PubMed]
- Xi, W.; Hansmann, U.H.E. Ring-like N-Fold Models of Aβ42 Fibrils. Sci. Rep. 2017, 7, 6588. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Sun, Y.; Lei, J.; Wei, G. Dihydrochalcone Molecules Destabilize Alzheimer’s Amyloid-β Protofibrils through Binding to the Protofibril Cavity. Phys. Chem. Chem. Phys. 2018. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Characteristics | References |
|---|---|---|
| Infrared spectroscopy (IR) | IR reveals the chemical bonds, peptide interactions, and β-sheet disposition of Aβ1-42. | [5,20,21,22,23] |
| X-ray diffraction | Shows details of the fibril structure, such as sheet direction and arrangements in amyloid crystals. | [6,24,25,26,27] |
| Microscopy transmission electron microscopy (TEM) | TEM allows determination of the ultrastructure organization throughout the electron–electron interaction in the Aβ1-42 structures at molecular level and atomic resolution. | [17,28,29,30,31] |
| Atomic force microscopy (AFM) | The resolution of this technique is less than 1 nm, enabling the structural details of Aβ1-42 aggregation to be revealed. | [32,33,34,35,36] |
| Fluorescence | Monitors Aβ1-42 aggregation kinetics in real-time and detects Aβ1-42 at any state in tissue samples using fluorochromes, such as Thioflavin T (ThT). | [37,38,39,40,41] |
| Electrophoresis | This technique could be used determine molecular weight and to purify Aβ1-42. | [5,20,40,42,43,44] |
| Intramolecular Monomer | |
|---|---|
| Amino acid residue interactions | Hydrophobic regions |
| Ile41-Gly29; Ile41-Lys28; Phe19-Ile32; Phe20-Val24; Lys28-Ala42 | Ile31, Val36, Val39, Ile41, Leu17, Phe19, Phe20, Val24, Ala30, Ile32 |
| Phe19-Ala30; Val24-Gly29; Ile31-Val36; Gly29-Asn27; Gly33-Val36; Gly29-Ile41 | |
| Intermolecular Dimer | |
| Amino acid residue interactions | Hydrophobic regions |
| Met35-Leu17; Gln15-Leu31 | Val18, Ala21, Val40, Val42 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villalobos Acosta, D.M.Á.; Chimal Vega, B.; Correa Basurto, J.; Fragoso Morales, L.G.; Rosales Hernández, M.C. Recent Advances by In Silico and In Vitro Studies of Amyloid-β 1-42 Fibril Depicted a S-Shape Conformation. Int. J. Mol. Sci. 2018, 19, 2415. https://doi.org/10.3390/ijms19082415
Villalobos Acosta DMÁ, Chimal Vega B, Correa Basurto J, Fragoso Morales LG, Rosales Hernández MC. Recent Advances by In Silico and In Vitro Studies of Amyloid-β 1-42 Fibril Depicted a S-Shape Conformation. International Journal of Molecular Sciences. 2018; 19(8):2415. https://doi.org/10.3390/ijms19082415
Chicago/Turabian StyleVillalobos Acosta, Daniel Miguel Ángel, Brenda Chimal Vega, José Correa Basurto, Leticia Guadalupe Fragoso Morales, and Martha Cecilia Rosales Hernández. 2018. "Recent Advances by In Silico and In Vitro Studies of Amyloid-β 1-42 Fibril Depicted a S-Shape Conformation" International Journal of Molecular Sciences 19, no. 8: 2415. https://doi.org/10.3390/ijms19082415