The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease

Abstract

1. Introduction
2. HDAC Classification and Their Physiological Roles
2.1. HDAC Classification and Their Physiological Roles in Lymphoid Lineage
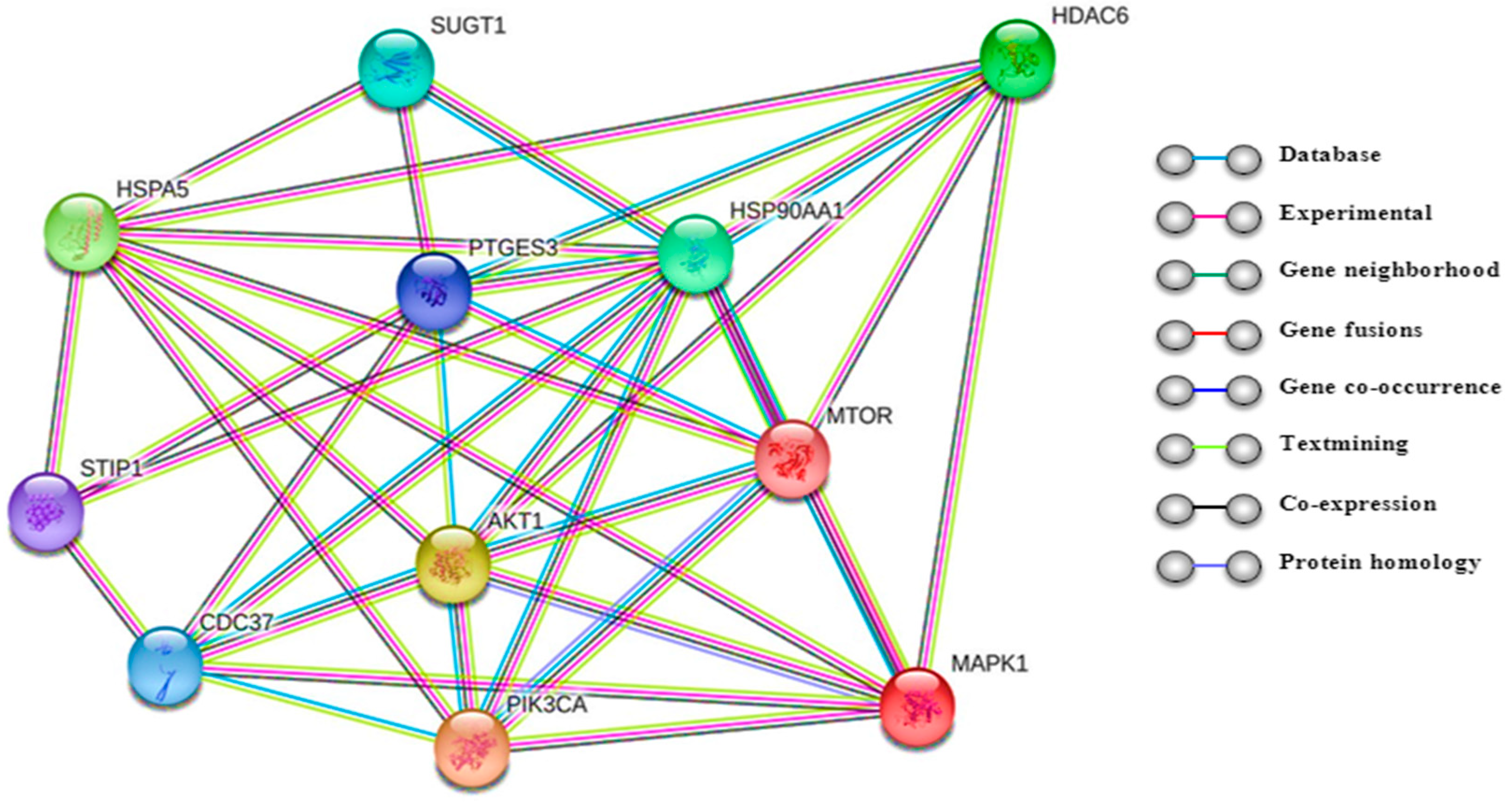
2.2. Biological Roles of Histone Deacetylase 6 (HDAC6)
3. Abnormal Expression HDAC in Lymphoproliferative Disease
4. Anticancer Effects of HDAC Inhibitors and Their Roles in Lymphoproliferative Disease
4.1. Classification of HDAC Inhibitors: Specific and Non-Specific HDACis
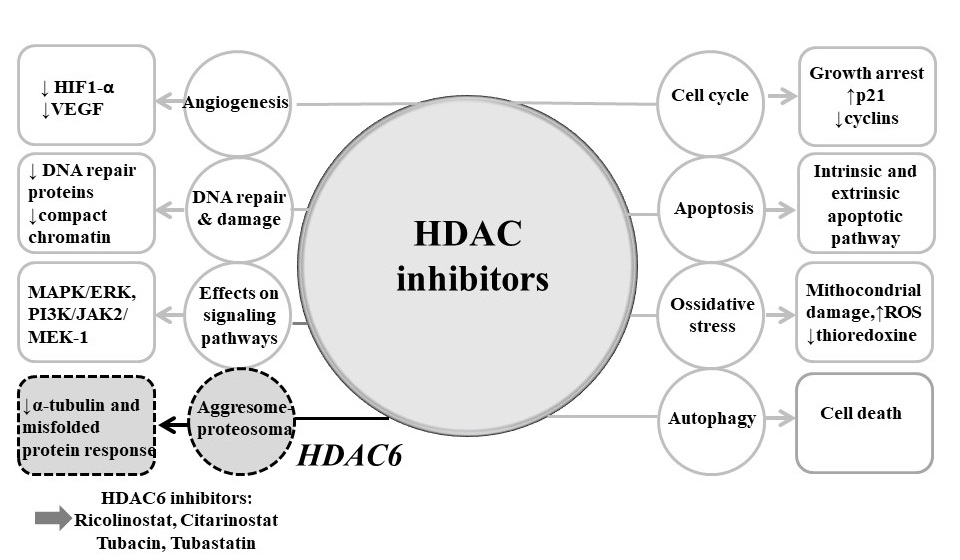
4.2. Mechanisms of Actions of HDAC Inhibitors
4.2.1. Cell Cycle Arrest
4.2.2. Apoptosis
4.2.3. Autophagy
4.2.4. Angiogenesis
4.2.5. Migration
4.2.6. Protein–Protein Interactions
4.3. Pan-HDAC Inhibitors in Lymphoproliferative Disorders: Preclinical and Clinical Data
4.4. Selective HDAC6 Inhibitors in Lymphoproliferative Diseases: Pre-Clinical and Clinical Data
4.4.1. Tubacin
4.4.2. Tubastatin
4.4.3. Ricolinostat
4.4.4. Citarinostat
5. Future Developments of HDAC Inhibitors
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Longo, F.; Giuliano, M.; Sabbatino, F.; Favia, G.; Ionna, F.; Addeo, R.; Della Vittoria Scarpati, G.; Di Lorenzo, G.; Pisconti, S. Epigenetic control of gene expression: Potential implications for cancer treatment. Crit. Rev. Oncol. Hematol. 2017, 111, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Jazirehi, A.R. Regulation of apoptosis-associated genes by histone deacetylase inhibitors: Implications in cancer therapy. Anticancer Drugs 2010, 21, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Schrump, D.S. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: Mechanisms and potential clinical implications. Clin. Cancer Res. 2009, 15, 3947–3957. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Richon, V.M.; Rifkind, R.A. Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 2000, 92, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Bassett, S.A.; Barnett, M.P.G. The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients 2014, 6, 4273–4301. [Google Scholar] [CrossRef] [PubMed]
- Villagra, A.; Cheng, F.; Wang, H.-W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Dovey, O.M.; Foster, C.T.; Conte, N.; Edwards, S.A.; Edwards, J.M.; Singh, R.; Vassiliou, G.; Bradley, A.; Cowley, S.M. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 2013, 121, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.D.W.; Cowley, S.M. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Seigneurin-Berny, D.; Verdel, A.; Curtet, S.; Lemercier, C.; Garin, J.; Rousseaux, S.; Khochbin, S. Identification of components of the murine histone deacetylase 6 complex: Link between acetylation and ubiquitination signaling pathways. Mol. Cell. Biol. 2001, 21, 8035–8044. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Kiermer, V.; Dequiedt, F.; Verdin, E. The emerging role of class II histone deacetylases. Biochem. Cell Biol. 2001, 79, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef]
- Grausenburger, R.; Bilic, I.; Boucheron, N.; Zupkovitz, G.; El-Housseiny, L.; Tschismarov, R.; Zhang, Y.; Rembold, M.; Gaisberger, M.; Hartl, A.; et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J. Immunol. 2010, 185, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.-C.; Belmonte, P.J.; Constans, M.M.; Chen, M.W.; McWilliams, D.C.; Hiebert, S.W.; Shapiro, V.S. Histone Deacetylase 3 Is Required for T Cell Maturation. J. Immunol. 2015, 195, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Pagiatakis, C.; Salma, J.; Du, M.; Andreucci, J.J.; Zhao, J.; Hou, G.; Perry, R.L.; Dan, Q.; Courtman, D.; et al. Protein kinase A-regulated assembly of a MEF2{middle dot}HDAC4 repressor complex controls c-Jun expression in vascular smooth muscle cells. J. Biol. Chem. 2009, 284, 19027–19042. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.K.; Volinia, S.; Costinean, S.; Galasso, M.; Neinast, R.; Santhanam, R.; Parthun, M.R.; Perrotti, D.; Marcucci, G.; Garzon, R.; et al. miR-155 targets histone deacetylase 4 (HDAC4) and impairs transcriptional activity of B-cell lymphoma 6 (BCL6) in the Eμ-miR-155 transgenic mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 20047–20052. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.H.; Bertos, N.R.; Vezmar, M.; Pelletier, N.; Crosato, M.; Heng, H.H.; Th’ng, J.; Han, J.; Yang, X.J. HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Mol. Cell. Biol. 1999, 19, 7816–7827. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, X.; Yin, C.; Chen, X.; Zhang, Z.; Brown, S.; Xie, H.; Zhou, L.; Mi, Q.-S. HDAC4 is expressed on multiple T cell lineages but dispensable for their development and function. Oncotarget 2017, 8, 17562–17572. [Google Scholar] [CrossRef] [PubMed]
- Kasler, H.G.; Verdin, E. Histone deacetylase 7 functions as a key regulator of genes involved in both positive and negative selection of thymocytes. Mol. Cell. Biol. 2007, 27, 5184–5200. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.N.; Goebel, J.; Feijoo-Carnero, C.; Morrice, N.; Cantrell, D.A. Phosphoproteomic analysis reveals an intrinsic pathway for the regulation of histone deacetylase 7 that controls the function of cytotoxic T lymphocytes. Nat. Immunol. 2011, 12, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Azagra, A.; Román-González, L.; Collazo, O.; Rodríguez-Ubreva, J.; de Yébenes, V.G.; Barneda-Zahonero, B.; Rodríguez, J.; Castro de Moura, M.; Grego-Bessa, J.; Fernández-Duran, I.; et al. In vivo conditional deletion of HDAC7 reveals its requirement to establish proper B lymphocyte identity and development. J. Exp. Med. 2016, 213, 2591–2601. [Google Scholar] [CrossRef] [PubMed]
- De Zoeten, E.F.; Wang, L.; Sai, H.; Dillmann, W.H.; Hancock, W.W. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 2010, 138, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Beier, U.H.; Wang, L.; Han, R.; Akimova, T.; Liu, Y.; Hancock, W.W. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci. Signal. 2012, 5, ra45. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, C.; Minucci, S.; Pelicci, P.G. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol. Res. 2010, 62, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-L.; Guan, Y.-J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, E.; Gagliostro, E.; Brancolini, C. Selective class IIa HDAC inhibitors: Myth or reality. Cell. Mol. Life Sci. 2015, 72, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fernández, A.; Cabrero, J.R.; Serrador, J.M.; Sánchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008, 18, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.-L.; Yang, W.-M. Beyond histone and deacetylase: An overview of cytoplasmic histone deacetylases and their nonhistone substrates. J. Biomed. Biotechnol. 2011, 2011, 146493. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Murphy, P.J.M.; Gaillard, S.; Zhao, X.; Wu, J.-T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.-P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Broemer, M.; Arslan, S.C.; Otto, A.; Mueller, E.-C.; Dettmer, R.; Scheidereit, C. Signal responsiveness of IkappaB kinases is determined by Cdc37-assisted transient interaction with Hsp90. J. Biol. Chem. 2007, 282, 32311–32319. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Boyault, C.; Zhang, Y.; Fritah, S.; Caron, C.; Gilquin, B.; Kwon, S.H.; Garrido, C.; Yao, T.-P.; Vourc’h, C.; Matthias, P.; Khochbin, S. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007, 21, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gonzalez, A.; Lin, T.; Ikeda, A.K.; Simms-Waldrip, T.; Fu, C.; Sakamoto, K.M. Role of the aggresome pathway in cancer: Targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 2008, 68, 2557–2560. [Google Scholar] [CrossRef] [PubMed]
- Boyault, C.; Sadoul, K.; Pabion, M.; Khochbin, S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007, 26, 5468–5476. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, H.; Ali, Y.O.; Ravichandran, M.; Dong, A.; Qiu, W.; MacKenzie, F.; Dhe-Paganon, S.; Arrowsmith, C.H.; Zhai, R.G. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J. Biol. Chem. 2012, 287, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D. Proteasome and HDAC: Who’s zooming who? Blood 2010, 116, 308–309. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Fiskus, W.; Yang, Y.; Lee, P.; Joshi, R.; Fernandez, P.; Mandawat, A.; Atadja, P.; Bradner, J.E.; Bhalla, K. HDAC6 inhibition enhances 17-AAG-mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 2008, 112, 1886–1893. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.-P.; Zhou, D.; Ouyang, D.-Y.; Xu, L.-H.; Wang, Y.; Wang, L.-X.; Pan, H.; He, X.-H. LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem. Biophys. Res. Commun. 2013, 441, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.-W.; Shin, D.-H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Lim, K.-H.; Guo, X.; Kawaguchi, Y.; Gao, Y.; Barrientos, T.; Ordentlich, P.; Wang, X.-F.; Counter, C.M.; Yao, T.-P. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008, 68, 7561–7569. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-S.; Weng, S.-C.; Tseng, P.-H.; Lin, H.-P.; Chen, C.-S. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J. Biol. Chem. 2005, 280, 38879–38887. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.A.; No, M.; Lee, J.M.; Shin, J.H.; Oh, J.S.; Choi, E.J.; Kim, I.H.; Atadja, P.; Bernhard, E.J. Epigenetic modulation of radiation response in human cancer cells with activated EGFR or HER-2 signaling: Potential role of histone deacetylase 6. Radiother. Oncol. 2009, 92, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Tien, S.-C.; Chang, Z.-F. Oncogenic Shp2 disturbs microtubule regulation to cause HDAC6-dependent ERK hyperactivation. Oncogene 2014, 33, 2938–2946. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.-W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Xu, W.-S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Rao, R.; Fernandez, P.; Herger, B.; Yang, Y.; Chen, J.; Kolhe, R.; Mandawat, A.; Wang, Y.; Joshi, R.; et al. Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor-resistant acute myeloid leukemia cells. Blood 2008, 112, 2896–2905. [Google Scholar] [CrossRef] [PubMed]
- Marquard, L.; Gjerdrum, L.M.; Christensen, I.J.; Jensen, P.B.; Sehested, M.; Ralfkiaer, E. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology 2008, 53, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Marquard, L.; Poulsen, C.B.; Gjerdrum, L.M.; de Nully Brown, P.; Christensen, I.J.; Jensen, P.B.; Sehested, M.; Johansen, P.; Ralfkiaer, E. Histone deacetylase 1, 2, 6 and acetylated histone H4 in B- and T-cell lymphomas. Histopathology 2009, 54, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Kafeel, M.I.; Avezbakiyev, B.; Chen, C.; Sun, Y.; Rathnasabapathy, C.; Kalavar, M.; He, Z.; Burton, J.; Lichter, S. Histone deacetylase in chronic lymphocytic leukemia. Oncology 2011, 81, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Gloghini, A.; Buglio, D.; Khaskhely, N.M.; Georgakis, G.; Orlowski, R.Z.; Neelapu, S.S.; Carbone, A.; Younes, A. Expression of histone deacetylases in lymphoma: Implication for the development of selective inhibitors. Br. J. Haematol. 2009, 147, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Adams, H.; Fritzsche, F.R.; Dirnhofer, S.; Kristiansen, G.; Tzankov, A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin. Ther. Targets 2010, 14, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Reid, R.C.; Iyer, A.; Sweet, M.J.; Fairlie, D.P. Towards isozyme-selective HDAC inhibitors for interrogating disease. Curr. Top. Med. Chem. 2012, 12, 1479–1499. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L.; Rosenwald, A.; Staudt, L.M. Lymphoid malignancies: The dark side of B-cell differentiation. Nat. Rev. Immunol. 2002, 2, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Lemercier, C.; Brocard, M.-P.; Puvion-Dutilleul, F.; Kao, H.-Y.; Albagli, O.; Khochbin, S. Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J. Biol. Chem. 2002, 277, 22045–22052. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Maddipoti, S.; Quesada, A.; Bohannan, Z.; Cabrero Calvo, M.; Colla, S.; Wei, Y.; Estecio, M.; Wierda, W.; Bueso-Ramos, C.; et al. Analysis of class I and II histone deacetylase gene expression in human leukemia. Leuk. Lymphoma 2015, 56, 3426–3433. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Bishton, M.J.; Harrison, S.J. Clinical studies of histone deacetylase inhibitors. Clin. Cancer Res. 2009, 15, 3958–3969. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999, 260, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Ai, T.; Cui, H.; Chen, L. Multi-targeted histone deacetylase inhibitors in cancer therapy. Curr. Med. Chem. 2012, 19, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.M.; Lefebure, M.; Doyle, M.A.; Shortt, J.; Ellul, J.; Chesi, M.; Banks, K.M.; Vidacs, E.; Faulkner, D.; Atadja, P.; et al. Preclinical screening of histone deacetylase inhibitors combined with ABT-737, rhTRAIL/MD5-1 or 5-azacytidine using syngeneic Vk*MYC multiple myeloma. Cell Death Dis. 2013, 4, e798. [Google Scholar] [CrossRef] [PubMed]
- Wanczyk, M.; Roszczenko, K.; Marcinkiewicz, K.; Bojarczuk, K.; Kowara, M.; Winiarska, M. HDACi—Going through the mechanisms. Front. Biosci. (Landmark Ed.) 2011, 16, 340–359. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Cottini, F.; Ohguchi, H.; Jakubikova, J.; Gorgun, G.; Mimura, N.; Tai, Y.-T.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Rational combination treatment with histone deacetylase inhibitors and immunomodulatory drugs in multiple myeloma. Blood Cancer J. 2015, 5, e312. [Google Scholar] [CrossRef] [PubMed]
- Heider, U.; von Metzler, I.; Kaiser, M.; Rosche, M.; Sterz, J.; Rötzer, S.; Rademacher, J.; Jakob, C.; Fleissner, C.; Kuckelkorn, U.; et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in mantle cell lymphoma. Eur. J. Haematol. 2008, 80, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gao, M.; Yang, G.; Tao, Y.; Kong, Y.; Yang, R.; Meng, X.; Ai, G.; Wei, R.; Wu, H.; et al. Synergistic Activity of Carfilzomib and Panobinostat in Multiple Myeloma Cells via Modulation of ROS Generation and ERK1/2. Biomed. Res. Int. 2015, 2015, 459052. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]
- Cyrenne, B.M.; Lewis, J.M.; Weed, J.G.; Carlson, K.R.; Mirza, F.N.; Foss, F.M.; Girardi, M. Synergy of BCL2 and histone deacetylase inhibition against leukemic cells from cutaneous T-cell lymphoma patients. Blood 2017, 130, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Almeciga-Pinto, I.; Tamang, D.; Yang, M.; Jones, S.S.; Quayle, S.N. Enhancement of pomalidomide anti-tumor response with ACY-241, a selective HDAC6 inhibitor. PLoS ONE 2017, 12, e0173507. [Google Scholar] [CrossRef] [PubMed]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.-T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, G.; Lembersky, D.; Kramer, L.; Fisher, R.I.; Friedberg, J.; Dent, P.; Grant, S. The pan-HDAC inhibitor vorinostat potentiates the activity of the proteasome inhibitor carfilzomib in human DLBCL cells in vitro and in vivo. Blood 2010, 115, 4478–4487. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Bereshchenko, O.R.; Gu, W.; Dalla-Favera, R. Acetylation inactivates the transcriptional repressor BCL6. Nat. Genet. 2002, 32, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Kurland, J.F.; Tansey, W.P. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res. 2008, 68, 3624–3629. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Curet, I.; Perkins, R.S.; Bennett, R.; Feidler, K.L.; Dunn, S.P.; Krueger, L.J. c-Myc inhibition negatively impacts lymphoma growth. J. Pediatr. Surg. 2006, 41, 207–211, discussion 207–211. [Google Scholar] [CrossRef] [PubMed]
- Kretsovali, A.; Hadjimichael, C.; Charmpilas, N. Histone deacetylase inhibitors in cell pluripotency, differentiation, and reprogramming. Stem Cells Int. 2012, 2012, 184154. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Bates, S.E. Epigenetic modifiers: Basic understanding and clinical development. Clin. Cancer Res. 2009, 15, 3918–3926. [Google Scholar] [CrossRef] [PubMed]
- Peart, M.J.; Smyth, G.K.; van Laar, R.K.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 3697–3702. [Google Scholar] [CrossRef] [PubMed]
- Peart, M.J.; Tainton, K.M.; Ruefli, A.A.; Dear, A.E.; Sedelies, K.A.; O’Reilly, L.A.; Waterhouse, N.J.; Trapani, J.A.; Johnstone, R.W. Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2003, 63, 4460–4471. [Google Scholar] [PubMed]
- Kawamata, N.; Chen, J.; Koeffler, H.P. Suberoylanilide hydroxamic acid (SAHA; vorinostat) suppresses translation of cyclin D1 in mantle cell lymphoma cells. Blood 2007, 110, 2667–2673. [Google Scholar] [CrossRef] [PubMed]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagosklonny, M.V.; Bates, S.E. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Abukhdeir, A.M.; Park, B.H. P21 and p27: Roles in carcinogenesis and drug resistance. Expert Rev. Mol. Med. 2008, 10, e19. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; He, Q.; Tsai, C.; Lei, J.; Chen, J.; Vienna Makcey, L.; Coy, D.H. HDAC inhibitors suppressed small cell lung cancer cell growth and enhanced the suppressive effects of receptor-targeting cytotoxins via upregulating somatostatin receptor II. Am. J. Transl. Res. 2018, 10, 545–553. [Google Scholar] [PubMed]
- Zhou, H.; Cai, Y.; Liu, D.; Li, M.; Sha, Y.; Zhang, W.; Wang, K.; Gong, J.; Tang, N.; Huang, A.; et al. Pharmacological or transcriptional inhibition of both HDAC1 and 2 leads to cell cycle blockage and apoptosis via p21Waf1/Cip1 and p19INK4d upregulation in hepatocellular carcinoma. Cell Prolif. 2018, 51, e12447. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Yang, Y.; Liu, S.; Lu, J.; Huang, B.; Zhang, Y. HDAC inhibitor PAC-320 induces G2/M cell cycle arrest and apoptosis in human prostate cancer. Oncotarget 2018, 9, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Mensah, A.A.; Kwee, I.; Gaudio, E.; Rinaldi, A.; Ponzoni, M.; Cascione, L.; Fossati, G.; Stathis, A.; Zucca, E.; Caprini, G.; et al. Novel HDAC inhibitors exhibit pre-clinical efficacy in lymphoma models and point to the importance of CDKN1A expression levels in mediating their anti-tumor response. Oncotarget 2015, 6, 5059–5071. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.P.; Singh, M.M.; Rivera-Del Valle, N.; Manton, C.A.; Chandra, J. Therapeutic strategies to enhance the anticancer efficacy of histone deacetylase inhibitors. J. Biomed. Biotechnol. 2011, 2011, 514261. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr. Cancer Drug Targets 2008, 8, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.M.; Newbold, A.; Johnstone, R.W. Intrinsic and extrinsic apoptotic pathway signaling as determinants of histone deacetylase inhibitor antitumor activity. Adv. Cancer Res. 2012, 116, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Ruefli, A.A.; Ausserlechner, M.J.; Bernhard, D.; Sutton, V.R.; Tainton, K.M.; Kofler, R.; Smyth, M.J.; Johnstone, R.W. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2001, 98, 10833–10838. [Google Scholar] [CrossRef] [PubMed]
- Powis, G.; Mustacich, D.; Coon, A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radic. Biol. Med. 2000, 29, 312–322. [Google Scholar] [CrossRef]
- Hrebackova, J.; Hrabeta, J.; Eckschlager, T. Valproic acid in the complex therapy of malignant tumors. Curr. Drug Targets 2010, 11, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Choi, I.-K.; Kwon, H.J. Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 2008, 369, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Bánréti, A.; Sass, M.; Graba, Y. The emerging role of acetylation in the regulation of autophagy. Autophagy 2013, 9, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.-M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-T.; Chang, H.-C.; Chiang, L.-C.; Hung, W.-C. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Res. 2003, 63, 3069–3072. [Google Scholar] [PubMed]
- Khan, O.; La Thangue, N.B. HDAC inhibitors in cancer biology: Emerging mechanisms and clinical applications. Immunol. Cell Biol. 2012, 90, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Grant, S. Histone deacetylase inhibitors in cancer therapy. Cancer Biol. Ther. 2003, 2, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Zhang, X.D.; Gillespie, S.K.; Borrow, J.M.; Hersey, P. The histone deacetylase inhibitor suberic bishydroxamate: A potential sensitizer of melanoma to TNF-related apoptosis-inducing ligand (TRAIL) induced apoptosis. Biochem. Pharmacol. 2003, 66, 1537–1545. [Google Scholar] [CrossRef]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Bae, S.-C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar] [PubMed]
- Trüe, O.; Matthias, P. Interplay between histone deacetylases and autophagy--from cancer therapy to neurodegeneration. Immunol. Cell Biol. 2012, 90, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Batlevi, Y.; Baehrecke, E.H.; Taylor, J.P. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy 2007, 3, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Zain, J.; O’Connor, O.A. Targeting histone deacetyalses in the treatment of B- and T-cell malignancies. Investig. New Drugs 2010, 28 (Suppl. 1), S58–S78. [Google Scholar] [CrossRef]
- Home—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 19 June 2018).
- Duvic, M.; Vu, J. Vorinostat: A new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin. Investig. Drugs 2007, 16, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Foss, F.; Duvic, M.; Lerner, A.; Waksman, J.; Whittaker, S. Clinical Efficacy of Romidepsin in Tumor Stage and Folliculotropic Mycosis Fungoides. Clin. Lymphoma Myeloma Leuk. 2016, 16, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.-H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.F.; Hungria, V.T.M.; Yoon, S.-S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.J.; Hirte, H.; Colgan, T.; Covens, A.; MacAlpine, K.; Grenci, P.; Wang, L.; Mason, J.; Pham, P.-A.; Tsao, M.-S.; et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur. J. Cancer 2010, 46, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Pili, R.; Salumbides, B.; Zhao, M.; Altiok, S.; Qian, D.; Zwiebel, J.; Carducci, M.A.; Rudek, M.A. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br. J. Cancer 2012, 106, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Thompson, L.A.; Wenger, S.D.; O’Bryant, C.L. Romidepsin: A histone deacetylase inhibitor for refractory cutaneous T-cell lymphoma. Ann. Pharmacother. 2012, 46, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Bishton, M.J.; Harrison, S.J.; Martin, B.P.; McLaughlin, N.; James, C.; Josefsson, E.C.; Henley, K.J.; Kile, B.T.; Prince, H.M.; Johnstone, R.W. Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood 2011, 117, 3658–3668. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Hideshima, T.; Anderson, K.C. Histone deacetylase inhibitors in multiple myeloma: From bench to bedside. Int. J. Hematol. 2016, 104, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Simms-Waldrip, T.; Rodriguez-Gonzalez, A.; Lin, T.; Ikeda, A.K.; Fu, C.; Sakamoto, K.M. The aggresome pathway as a target for therapy in hematologic malignancies. Mol. Genet. Metab. 2008, 94, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.-C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.E.; Johannet, P.; Lombardo, M.; Zullo, K.; Hoehn, D.; Bhagat, G.; Scotto, L.; Jirau-Serrano, X.; Radeski, D.; Heinen, J.; et al. Dual Targeting of Protein Degradation Pathways with the Selective HDAC6 Inhibitor ACY-1215 and Bortezomib Is Synergistic in Lymphoma. Clin. Cancer Res. 2015, 21, 4663–4675. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-X.; Wan, R.-Z.; Liu, Z.-P. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 2018, 143, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Dallavalle, S.; Pisano, C.; Zunino, F. Development and therapeutic impact of HDAC6-selective inhibitors. Biochem. Pharmacol. 2012, 84, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Jarzembowski, J.A.; Opipari, A.W.; Castle, V.P.; Kwok, R.P.S. HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia 2011, 13, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Kerr, E.; Holohan, C.; McLaughlin, K.M.; Majkut, J.; Dolan, S.; Redmond, K.; Riley, J.; McLaughlin, K.; Stasik, I.; Crudden, M.; et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ. 2012, 19, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Cabrero, J.R.; Serrador, J.M.; Barreiro, O.; Mittelbrunn, M.; Naranjo-Suárez, S.; Martín-Cófreces, N.; Vicente-Manzanares, M.; Mazitschek, R.; Bradner, J.E.; Avila, J.; et al. Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Mol. Biol. Cell 2006, 17, 3435–3445. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Ping, L.; Feng, L.; Zheng, X.; Song, Y.; Zhu, J. Histone deacetylase 6 activity is critical for the metastasis of Burkitt’s lymphoma cells. Cancer Cell Int. 2014, 14, 139. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Khong, T.; Jones, S.S.; Spencer, A. Histone deacetylase (HDAC) inhibitors as single agents induce multiple myeloma cell death principally through the inhibition of class I HDAC. Br. J. Haematol. 2013, 162, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Santo, L.; Eda, H.; Cirstea, D.; Nemani, N.; Yee, A.J.; O’Donnell, E.; Selig, M.K.; Quayle, S.N.; Arastu-Kapur, S.; et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 2015, 169, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, G.; Patel, H.; Friedberg, J.; Quayle, S.N.; Jones, S.S.; Grant, S. In vitro and in vivo interactions between the HDAC6 inhibitor ricolinostat (ACY1215) and the irreversible proteasome inhibitor carfilzomib in non-Hodgkin lymphoma cells. Mol. Cancer Ther. 2014, 13, 2886–2897. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.E.; Prabhu, S.A.; Lombardo, M.; Zullo, K.; Johannet, P.M.; Gonzalez, Y.; Scotto, L.; Serrano, X.J.; Wei, Y.; Duong, J.; et al. Mechanisms of Acquired Drug Resistance to the HDAC6 Selective Inhibitor Ricolinostat Reveals Rational Drug-Drug Combination with Ibrutinib. Clin. Cancer Res. 2017, 23, 3084–3096. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cai, Y.; Yang, Y.; Li, A.; Bi, R.; Wang, L.; Shen, X.; Wang, W.; Jia, Y.; Yu, B.; et al. Activation of MET signaling by HDAC6 offers a rationale for a novel ricolinostat and crizotinib combinatorial therapeutic strategy in diffuse large B-cell lymphoma. J. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cosenza, M.; Civallero, M.; Marcheselli, L.; Sacchi, S.; Pozzi, S. Ricolinostat, a selective HDAC6 inhibitor, shows anti-lymphoma cell activity alone and in combination with bendamustine. Apoptosis 2017, 22, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Das, D.S.; Song, Y.; Hideshima, T.; Tai, Y.-T.; Chauhan, D.; Anderson, K.C. Combination of a novel HDAC6 inhibitor ACY-241 and anti-PD-L1 antibody enhances anti-tumor immunity and cytotoxicity in multiple myeloma. Leukemia 2018, 32, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Qi, J.; Paranal, R.M.; Tang, W.; Greenberg, E.; West, N.; Colling, M.E.; Estiu, G.; Mazitschek, R.; Perry, J.A.; et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167. [Google Scholar] [CrossRef] [PubMed]
- Kawada, J.; Zou, P.; Mazitschek, R.; Bradner, J.E.; Cohen, J.I. Tubacin kills Epstein-Barr virus (EBV)-Burkitt lymphoma cells by inducing reactive oxygen species and EBV lymphoblastoid cells by inducing apoptosis. J. Biol. Chem. 2009, 284, 17102–17109. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Simões-Pires, C.A.; Zwick, V.; Cretton, S.; Cuendet, M. Simultaneous Measurement of HDAC1 and HDAC6 Activity in HeLa Cells Using UHPLC-MS. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Mazitschek, R.; Santo, L.; Mimura, N.; Gorgun, G.; Richardson, P.G.; Raje, N.; Anderson, K.C. Induction of differential apoptotic pathways in multiple myeloma cells by class-selective histone deacetylase inhibitors. Leukemia 2014, 28, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Lwin, T.; Zhao, X.; Cheng, F.; Zhang, X.; Huang, A.; Shah, B.; Zhang, Y.; Moscinski, L.C.; Choi, Y.S.; Kozikowski, A.P.; et al. A microenvironment-mediated c-Myc/miR-548m/HDAC6 amplification loop in non-Hodgkin B cell lymphomas. J. Clin. Investig. 2013, 123, 4612–4626. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.S.; Bensinger, W.; Cole, C.E.; Lonial, S.; Jagannath, S.; Arce-Lara, C.E.; Valent, J.; Rosko, A.E.; Harb, W.A.; Sandhu, I.; et al. Ricolinostat (ACY-1215), the First Selective HDAC6 Inhibitor, Combines Safely with Pomalidomide and Dexamethasone and Shows Promosing Early Results in Relapsed-and-Refractory Myeloma (ACE-MM-102 Study). Blood 2015, 126, 4228. [Google Scholar]
- Niesvizky, R.; Richardson, P.G.; Gabrail, N.Y.; Madan, S.; Yee, A.J.; Quayle, S.N.; Almeciga-Pinto, I.; Jones, S.S.; Houston, L.; Hayes, D.; et al. ACY-241, a Novel, HDAC6 Selective Inhibitor: Synergy with Immunomodulatory (IMiD®) Drugs in Multiple Myeloma (MM) Cells and Early Clinical Results (ACE-MM-200 Study). Blood 2015, 126, 3040. [Google Scholar]
- Yee, A.J.; Voorhees, P.M.; Bensinger, W.; Berdeja, J.G.; Supko, J.G.; Richardson, P.G.; Tamang, D.; Jones, S.S.; Patrick, G.; Wheeler, C.; et al. Ricolinostat (ACY-1215), a Selective HDAC6 Inhibitor, in Combination with Lenalidomide and Dexamethasone: Results of a Phase 1b Trial in Relapsed and Relapsed Refractory Multiple Myeloma. Blood 2014, 124, 4772. [Google Scholar]
- Vogl, D.T.; Raje, N.; Hari, P.; Jones, S.S.; Supko, J.G.; Leone, G.; Wheeler, C.; Orlowski, R.Z.; Richardson, P.G.; Lonial, S.; et al. Phase 1B Results of Ricolinostat (ACY-1215) Combination Therapy with Bortezomib and Dexamethasone in Patients with Relapsed or Relapsed and Refractory Multiple Myeloma (MM). Blood 2014, 124, 4764. [Google Scholar]
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2011, 10, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Guerrero, E.; Danhof, S.; Schreder, M.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M. Upregulation of CD38 Expression on Multiple Myeloma Cells By the HDAC Inhibitor Ricolinostat Augments the Efficacy of Daratumumab. Blood 2017, 130, 1803. [Google Scholar]
- Vogl, D.T.; Hari, P.N.; Jagannath, S.; Jones, S.S.; Supko, J.G.; Leone, G.; Wheeler, C.; Orlowski, R.Z.; Richardson, P.G.; Lonial, S. ACY-1215, a Selective Histone Deacetylase (HDAC) 6 Inhibitor: Interim Results Of Combination Therapy With Bortezomib In Patients With Multiple Myeloma (MM). Blood 2013, 122, 759. [Google Scholar]
- Vorhees, P.; Bensinger, W.I.; Berdeja, J.; Supko, J.G.; Richardson, P.G.; Jones, S.S.; Patrick, G.; Wheeler, C.; Raje, N. ACY-1215, a Selective Histone Deacetylase (HDAC) 6 Inhibitor, In Combination With Lenalidomide and Dexamethasone (dex), Is Well Tolerated Without Dose Limiting Toxicity (DLT) In Patients (Pts) With Multiple Myeloma (MM) At Doses Demonstrating Biologic Activity: Interim Results Of a Phase 1b Trial. Blood 2013, 122, 3190. [Google Scholar]
- Huang, P.; Almeciga-Pinto, I.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Yang, M.; Jones, S.S.; Quayle, S.N. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget 2017, 8, 2694–2707. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Hideshima, T.; Tai, Y.-T.; Song, Y.; Richardson, P.; Raje, N.; Munshi, N.C.; Anderson, K.C. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.S. ACY-1215, a First-In-Class Selective Inhibitor of HDAC6, Demonstrates Significant Synergy With Immunomodulatory Drugs (IMiDs) In Preclinical Models Of Multiple Myeloma (MM). Blood 2013, 122, 1952. [Google Scholar]
- Cosenza, M.; Civallero, M.; Sacchi, S.; Pozzi, S. Momelotinib and Citarinostat: Co-Targeting JAK2/STAT3 and HDAC6 in Lymphoid Malignancies, a New Potential Therapeutic Combination. Blood 2017, 130, 5201. [Google Scholar]
- Mehrling, T.; Chen, Y. The Alkylating-HDAC Inhibition Fusion Principle: Taking Chemotherapy to the Next Level with the First in Class Molecule EDO-S101. Anticancer Agents Med. Chem. 2016, 16, 20–28. [Google Scholar] [CrossRef] [PubMed]
- López-Iglesias, A.A.; San-Segundo, L.; González-Méndez, L.; Hernández-García, S.; Primo, D.; Garayoa, M.; Hernández, A.B.; Paíno, T.; Mateos, M.-V.; Chen, Y.; et al. The Alkylating Histone Deacetylase Inhibitor Fusion Molecule Edo-S101 Displays Full Bi-Functional Properties in Preclinical Models of Hematological Malignancies. Blood 2014, 124, 2100. [Google Scholar]
- López-Iglesias, A.-A.; Herrero, A.B.; Chesi, M.; San-Segundo, L.; González-Méndez, L.; Hernández-García, S.; Misiewicz-Krzeminska, I.; Quwaider, D.; Martín-Sánchez, M.; Primo, D.; et al. Preclinical anti-myeloma activity of EDO-S101, a new bendamustine-derived molecule with added HDACi activity, through potent DNA damage induction and impairment of DNA repair. J. Hematol. Oncol. 2017, 10, 127. [Google Scholar] [CrossRef] [PubMed]
- Besse, L.; Kraus, M.; Besse, A.; Bader, J.; Silzle, T.; Mehrling, T.; Driessen, C. The first-in-class alkylating HDAC inhibitor EDO-S101 is highly synergistic with proteasome inhibition against multiple myeloma through activation of multiple pathways. Blood Cancer J. 2017, 7, e589. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Xu, K.; Lin, J.; Jayachandran, G.; Wang, B.; Watanabe, Y.; Ge, Q.; Wu, Y.; Guo, D.; Chen, Y.; et al. Abstract 2741: Synergistic inhibition of tumor growth and overcoming chemo-resistance by simultaneously targeting key components in DNA damage/repair, epigenetic, and putative cancer stem cell signaling pathways using novel dual-functional DNA-alkylating/HDAC inhibitor and tumor suppressor gene nanoparticles in lung cancer. Cancer Res. 2012, 72, 2741. [Google Scholar] [CrossRef]
- Shuttleworth, S.J. Abstract 3996: KA2237 and KA2507: Novel, oral cancer immunotherapeutics targeting PI3K-p110β/p110δ and HDAC6 with single-agent and combination activity. Cancer Res. 2016, 76, 3996. [Google Scholar] [CrossRef]
- Dallavalle, S.; Cincinelli, R.; Nannei, R.; Merlini, L.; Morini, G.; Penco, S.; Pisano, C.; Vesci, L.; Barbarino, M.; Zuco, V.; et al. Design, synthesis, and evaluation of biphenyl-4-yl-acrylohydroxamic acid derivatives as histone deacetylase (HDAC) inhibitors. Eur. J. Med. Chem. 2009, 44, 1900–1912. [Google Scholar] [CrossRef] [PubMed]
- Zuco, V.; De Cesare, M.; Cincinelli, R.; Nannei, R.; Pisano, C.; Zaffaroni, N.; Zunino, F. Synergistic antitumor effects of novel HDAC inhibitors and paclitaxel in vitro and in vivo. PLoS ONE 2011, 6, e29085. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Class | Members | Cellular Localization | Biological Functions |
|---|---|---|---|
| I | HDAC1 | Nucleus | Proliferation control, apoptosis; p21 and p27 CDK (cyclin-dependent kinase) inhibitor repression; Represses transcription; Binds to transcription factors; Resistance to chemotherapy; Suppresses cytokine production in activated T cells and during T effector cell differentiation |
| HDAC2 | Nucleus | Negatively regulates transcription by being recruited to DNA as a corepressor; Proliferation control; Apoptosis | |
| HDAC3 | Nucleus | Proliferation; Differentiation, represses transcription; Binds to transcription factors; Deacetylates FOXP3 (forkhead box P3) that reduces Treg development and suppressive function | |
| HDAC8 | Nucleus | Proliferation; Differentiation | |
| IIA | HDAC4 | Nucleus/Cytoplasm | Differentiation, angiogenesis; Deacetylates BCL6 (B-cell lymphoma 6) which activates genes for lymphocyte activation |
| HDAC5 | Nucleus/Cytoplasm | Differentiation; Deacetylates BCL6 which activates genes for lymphocyte activation | |
| HDAC7 | Nucleus/Cytoplasm | Angiogenesis; Suppresses Nur77 expression during TCR (T-cell receptor) negative selection; Regulates gene expression during TCR positive selection; Deacetylates BCL6 which activates genes for lymphocyte activation | |
| HDAC9 | Nucleus/Cytoplasm | Deacetylates FOXP3, which reduces Treg development and immunosuppressive activity | |
| IIB | HDAC6 | Cytoplasm | Regulation of protein degradation both via aggresome and the regulation of Hsp90 chaperone activity; Migration; Angiogenesis; Controls IgM and IgG levels upon antigen stimulation; T-cell migration; Immune synapse formation; Deacetylates FOXP3 that decreases Treg development and immunosuppressive activity |
| HDAC10 | Cytoplasm | Angiogenesis | |
| III | SIRT 1 | Nucleus, Cytoplasm | DNA repair/genome stability; Chromatin organization; Stress; Cancer |
| SIRT 2 | Nucleus | Mitosis; DNA repair; Chromatin condensation | |
| SIRT 3 | Mitochondria | Cancer, chromatin silencing; DNA repair; Cellular stress | |
| SIRT 4 | Mitochondria | have not yet been fully determined | |
| SIRT 5 | Mitochondria | have not yet been fully determined | |
| SIRT6 | Nucleus | DNA repair/genome stability; Telomeric chromatin/senescence | |
| SIRT7 | Nucleus | Cellular transformation | |
| IV | HDAC11 | Nucleus | Regulates the protein stability of DNA replication factor CDT1 (chromatin licensing and DNA replication factor 1) and the expression of IL-10; Suppresses IL10 expression in APCs (antigen presenting cells) |
| Class | Members | Expression of HDACs Increased in Lymphoproliferative Disease (Cell Lines and Primary Cell) | Reference |
|---|---|---|---|
| I | HDAC1 | MM, HL, MCL, DLBCL, ALCL, CLL PTCL, | [59,60,61,62,63,64] |
| HDAC2 | MM, HL, MCL, DLBCL, ALCL, PTCL | [59,60,61,63,64] | |
| HDAC3 | MCL, CLL, DLBCL, HL; MM | [59,60,61,62,63,64,65] | |
| HDAC8 | MM | [60] | |
| IIA | HDAC4 | DLBCL, PTCL | [61,63] |
| HDAC5 | MM | [60] | |
| HDAC7 | CLL, MCL | [59,61,62,63] | |
| HDAC9 | CLL, MCL | [59,62,63] | |
| IIB | HDAC6 | MM, MCL, DLBCL, PTCL, CTCL, CLL, | [59,60,61,62,63,64] |
| HDAC10 | CLL, MCL, HL | [59,61,62,63] | |
| III | SIRT 1 | CLL | [62] |
| SIRT 2 | |||
| SIRT 3 | |||
| SIRT 4 | |||
| SIRT 5 | |||
| SIRT6 | |||
| SIRT7 | CLL | [59,62,63] | |
| IV | HDAC11 | MCL, HL | [63] |
| Class | HDACis | Target HDAC | Clinical Trial Active in Lymphoproliferative Disease (clinicaltrials.gov) |
|---|---|---|---|
| Hidroxamic acids | Trichostatin A | Pan | Preclinical |
| Vorinostat/SAHA | Pan | * Phase I/II/III MM and lymphoma | |
| Belinostat | Pan | ** Phase I/II Lymphoma | |
| Panobinostat | Pan | *** Phase I/II MM and lymphoma | |
| Givinostat | Pan | Phase I/II completed for MM and lymphoma | |
| Resminostat | Pan | Phase II CTCL | |
| Abexinostat | Pan | Phase I/II completed for MM and lymphoma | |
| Quisinostat | Pan | Phase I/II completed for MM and lymphoma | |
| Ricolinostat/Acy-1215 | II selective | Phase I/II clinical trials for MM and lymphoma | |
| Citarinostat/Acy-241 | II selective | Phase I MM | |
| Practilinostat | I, II, IV | / | |
| CHR-3996 | I | / | |
| Aliphatic acid | Valproic acid | I, IIa | Phase I/II completed for lymphoma |
| Butyric acid | I, IIa | Phase I/II completed for lymphoma | |
| Phenylbutyric acid | I, IIa | Phase I/II completed for MM and lymphoma | |
| Benzamides | Entinostat | I | Phase I/II completed—MM. Phase I/II—lymphoma |
| Tacedinaline | I | Phase II completed—MM. | |
| 4SC202 | I | Phase I completed—Advanced Hematologic | |
| Malignancies | |||
| Mocetinostat | I, IV | Phase I/II clinical trials—lymphoma | |
| Cyclic tetrapepides | Romidepsin | I | Approved for * CTCL and ** PTCL |
| Several studies of phase I/II lymphoma | |||
| Phase I/II clinical trials—MM. | |||
| Sirtuins inhibitors | Nicotinamide | Class III | Phase I/II MM. Phase I lymphoma |
| Sirtinol | SIRT 1 and 2 | Preclinical | |
| Cambinol | SIRT 1 and 2 | Preclinical | |
| Ex-527 | SIRT 1 and 2 | Preclinical |
| HDAC6 Inhibitors | Lymphoproliferative Disease | Preclinical and Clinical Study (Ref.) | Clinical Trials State |
|---|---|---|---|
| Ricolinostat (Acy-1215) | MM cell | Alone [145] | Phase 1/2 combo poma and dex in MM (NCT01997840) (active) |
| + Bortezomib [135] | |||
| + Carfilzomib [146] | |||
| + Lenalidomide [78] | |||
| +Dexamethasone [78,147] | |||
| Non-NHL | + Carfilzomib [148] | Phase 1/2 combo lena e dex in MM (NCT01583283) (active) | |
| DLBCL, MCL, TCL | + Bortezomib [136] | Phase 1 combo poma and low-dose dex in relapsed-and-refractory MM (NCT02189343) (active) | |
| DLBCL | + Ibrutinib [149] | Phase 1/2 combo bort and dex in relapsed and refractory MM (NCT01323751) (termined) | |
| + Crizotinib [150] | |||
| FL, MCL, TCL | + Bendamustine [151] | Phase 1 /2 relapsed or refractory lymphoid malignancies (NCT02091063) (recruiting) | |
| Citarinostat (Acy-241) | MM and MCL | + Pomalidomide [80] | Phase 1 combo poma and dex in MM (NCT02400242) (active) |
| + Lenalidomide [80] | |||
| MM | + anti-PD-L1 [152] | ||
| Tubacin | MM and lymphoma | + Bortezomib [42,153] | Preclinical studies. Compound not tested in clinical trials: it is not optimized for oral delivery |
| Burkitt’s lymphoma | [144,154] | ||
| Tubastatin A | Lymphoma | [155,156] | Preclinical studies compound not tested in clinical trials: It is not optimized for oral delivery |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cosenza, M.; Pozzi, S. The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. Int. J. Mol. Sci. 2018, 19, 2337. https://doi.org/10.3390/ijms19082337
Cosenza M, Pozzi S. The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. International Journal of Molecular Sciences. 2018; 19(8):2337. https://doi.org/10.3390/ijms19082337
Chicago/Turabian StyleCosenza, Maria, and Samantha Pozzi. 2018. "The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease" International Journal of Molecular Sciences 19, no. 8: 2337. https://doi.org/10.3390/ijms19082337
APA StyleCosenza, M., & Pozzi, S. (2018). The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. International Journal of Molecular Sciences, 19(8), 2337. https://doi.org/10.3390/ijms19082337