Induction of Liver Steatosis in BAP31-Deficient Mice Burdened with Tunicamycin-Induced Endoplasmic Reticulum Stress


Abstract
:1. Introduction
2. Results
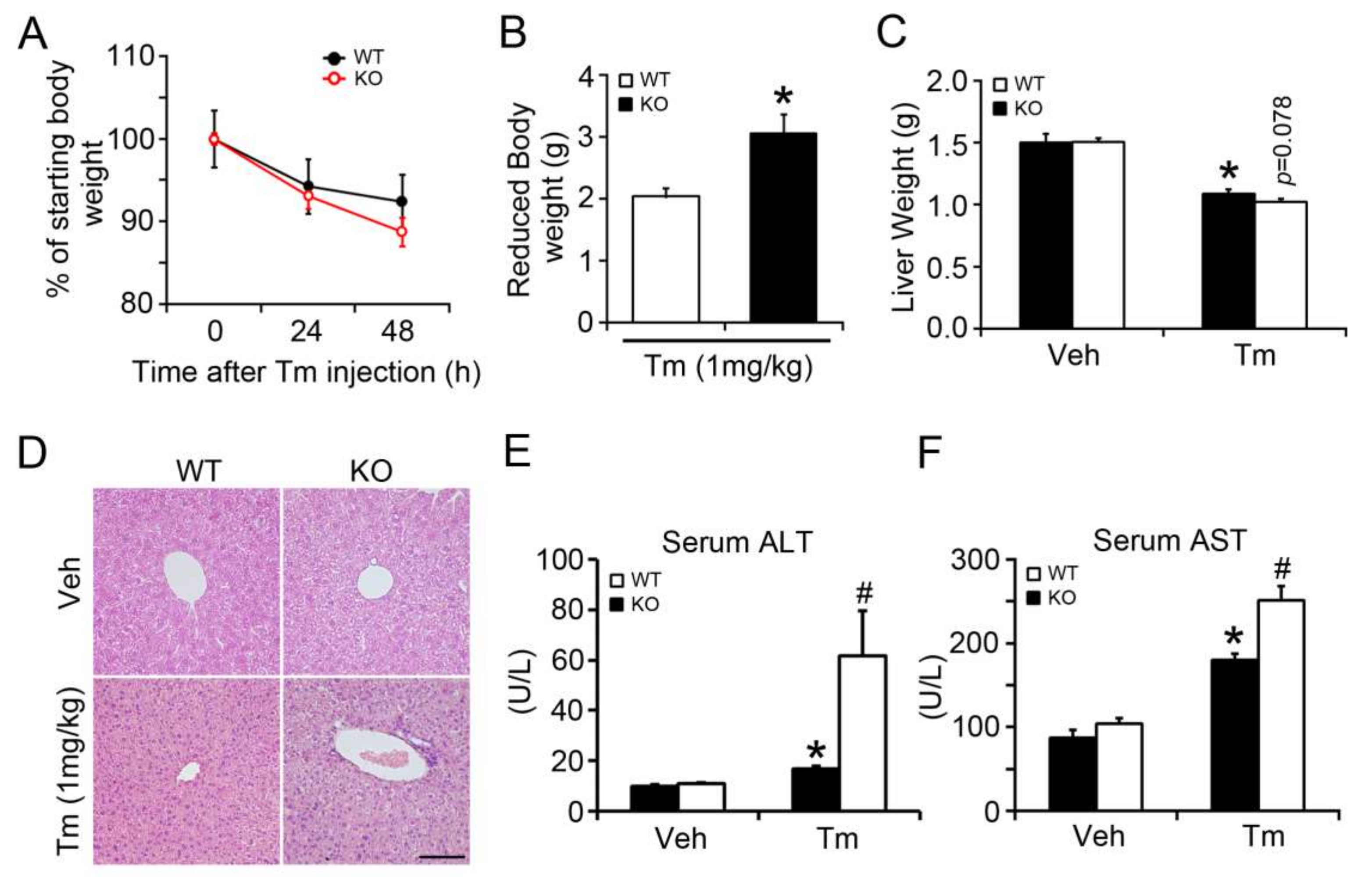
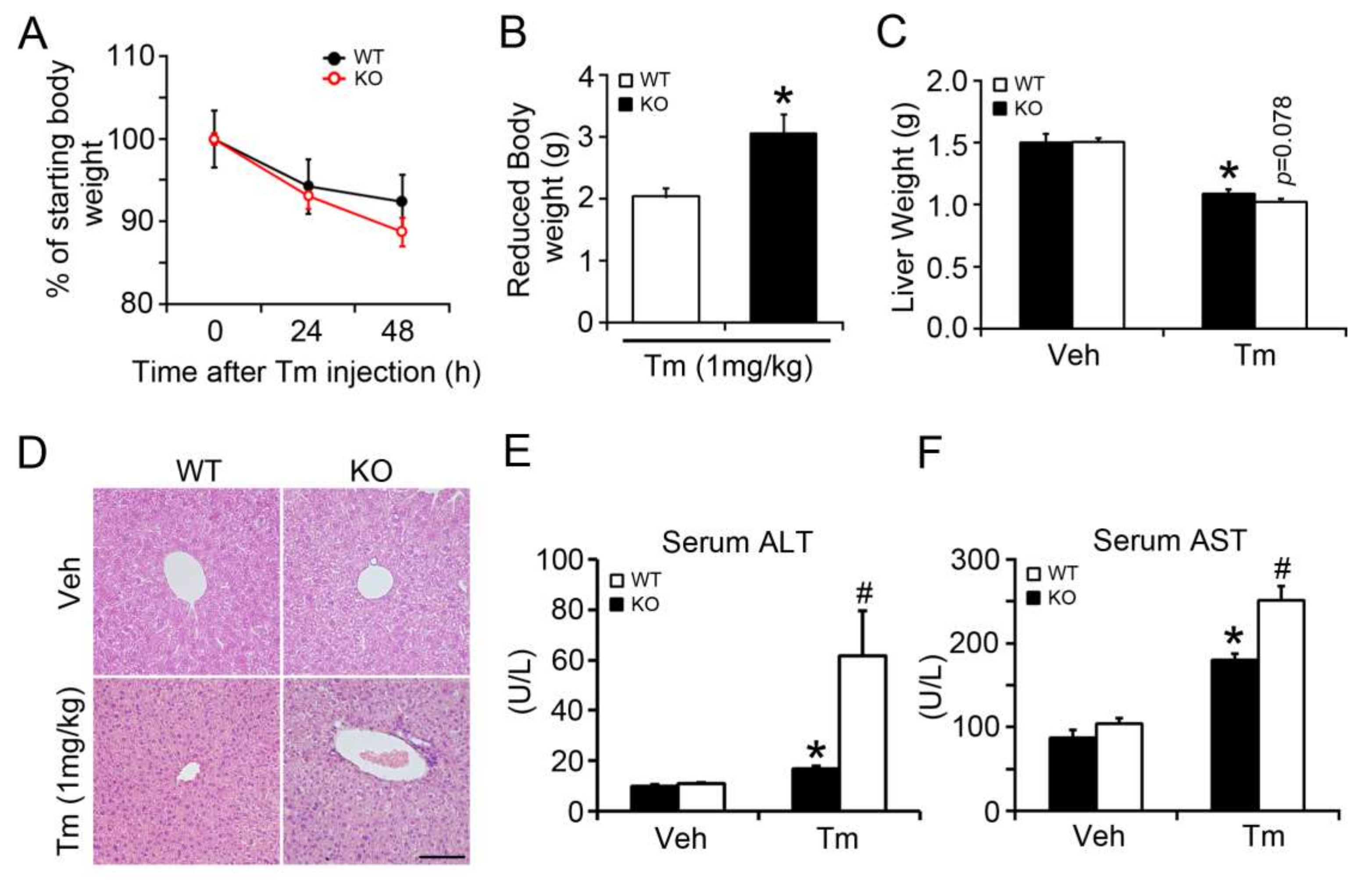
2.1. BAP31 Deficiency Promoted Liver Dysfunction after Tunicamycin Injection
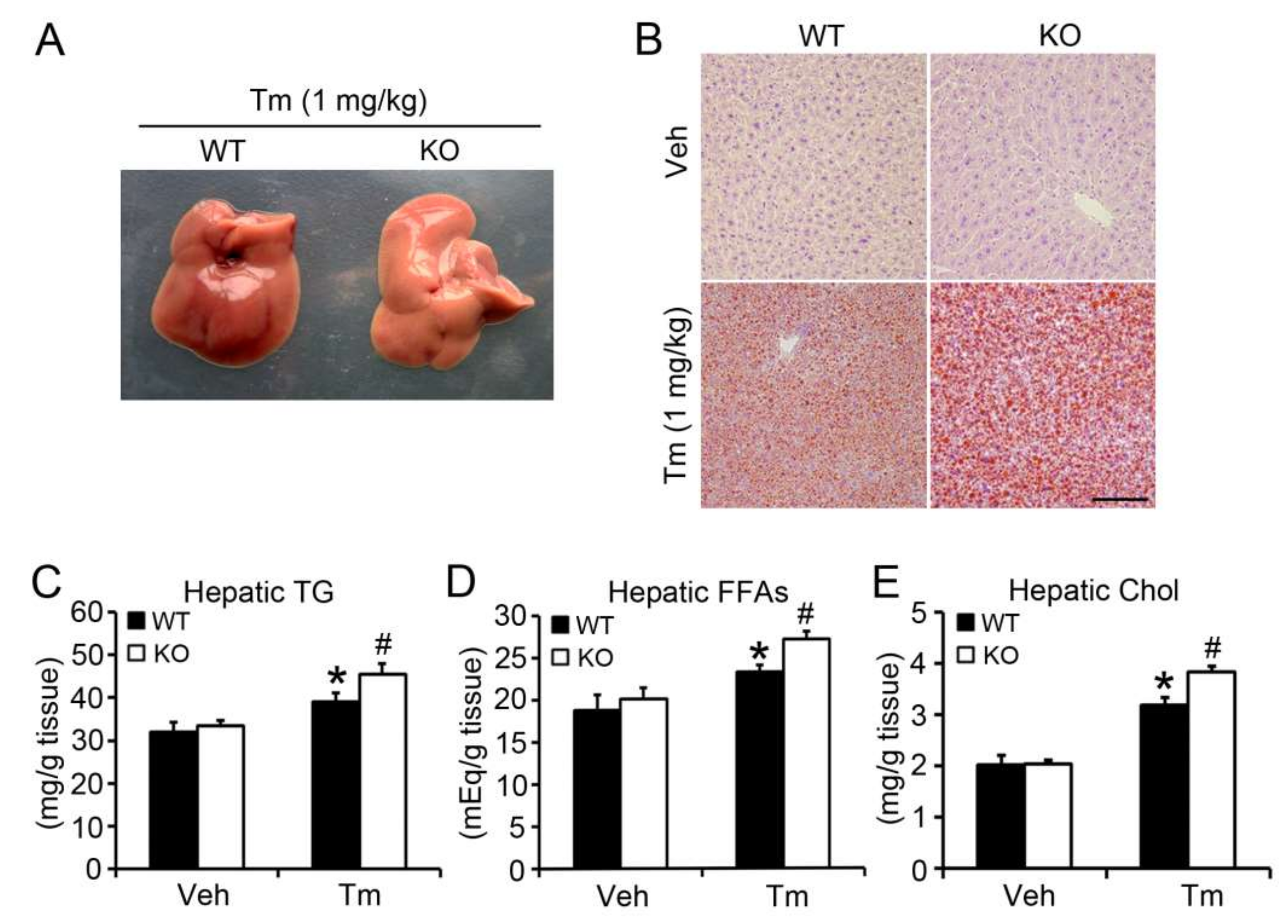
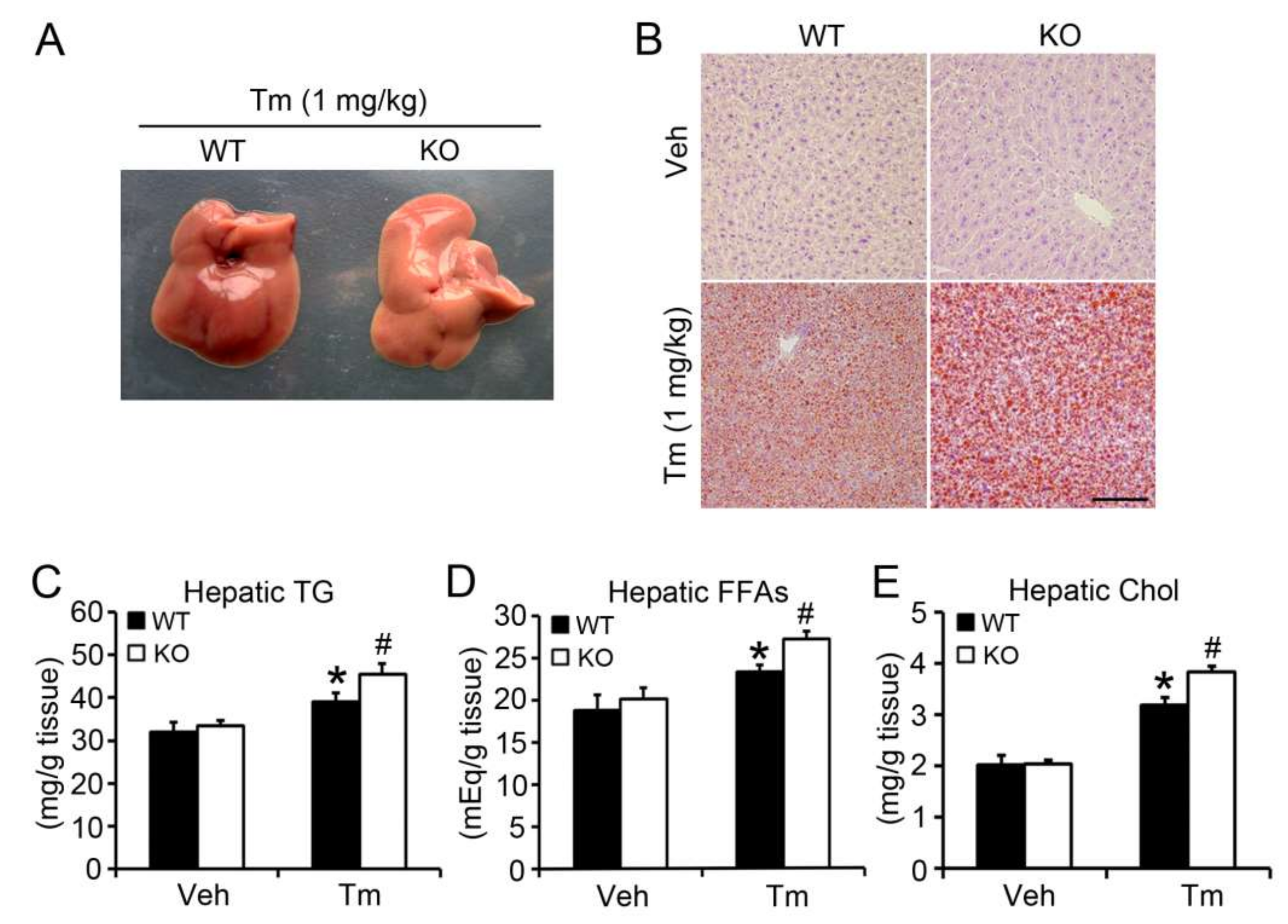
2.2. BAP31 Deficiency Promoted Tunicamycin-Induced Hepatic Lipid Accumulation
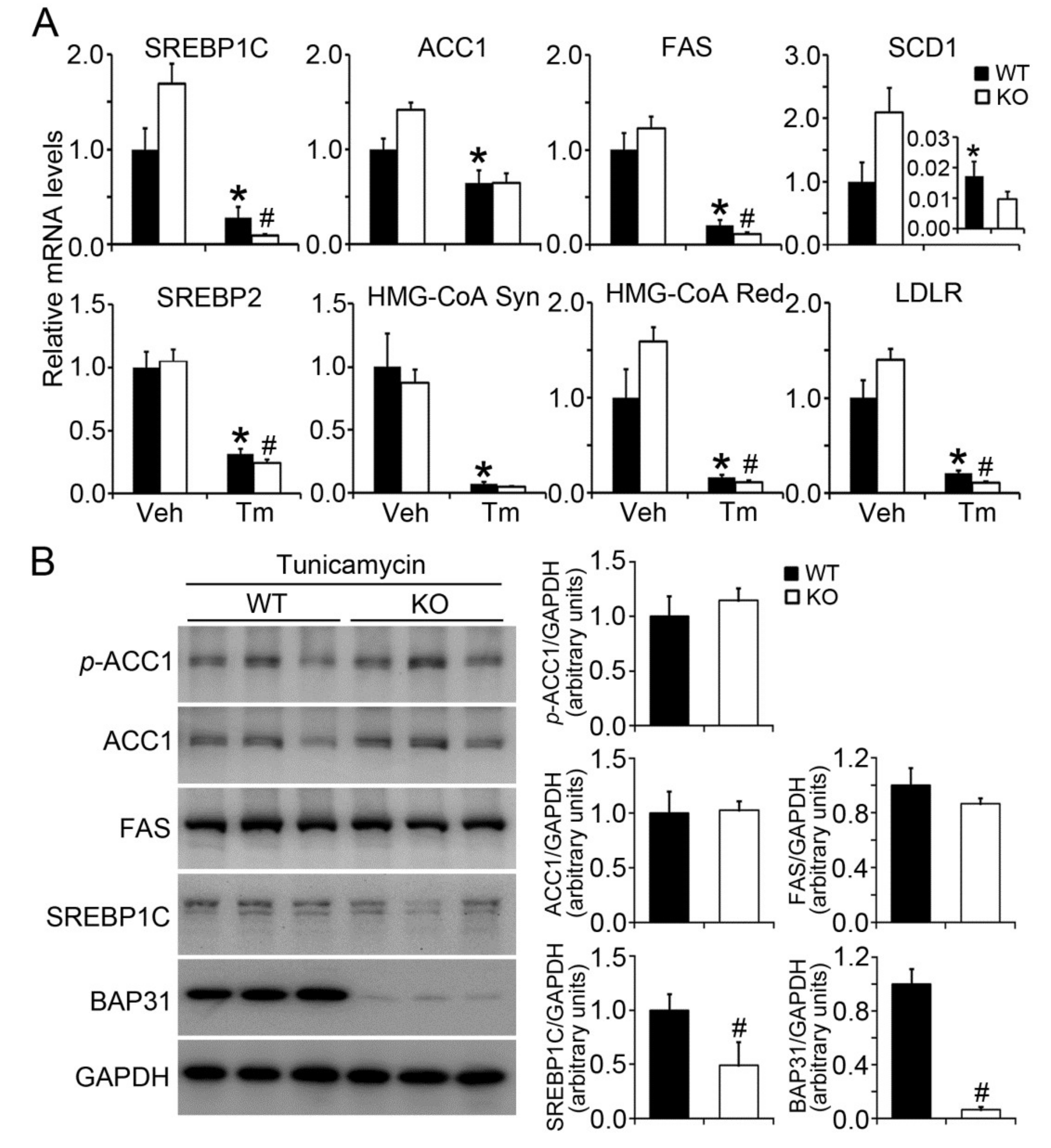
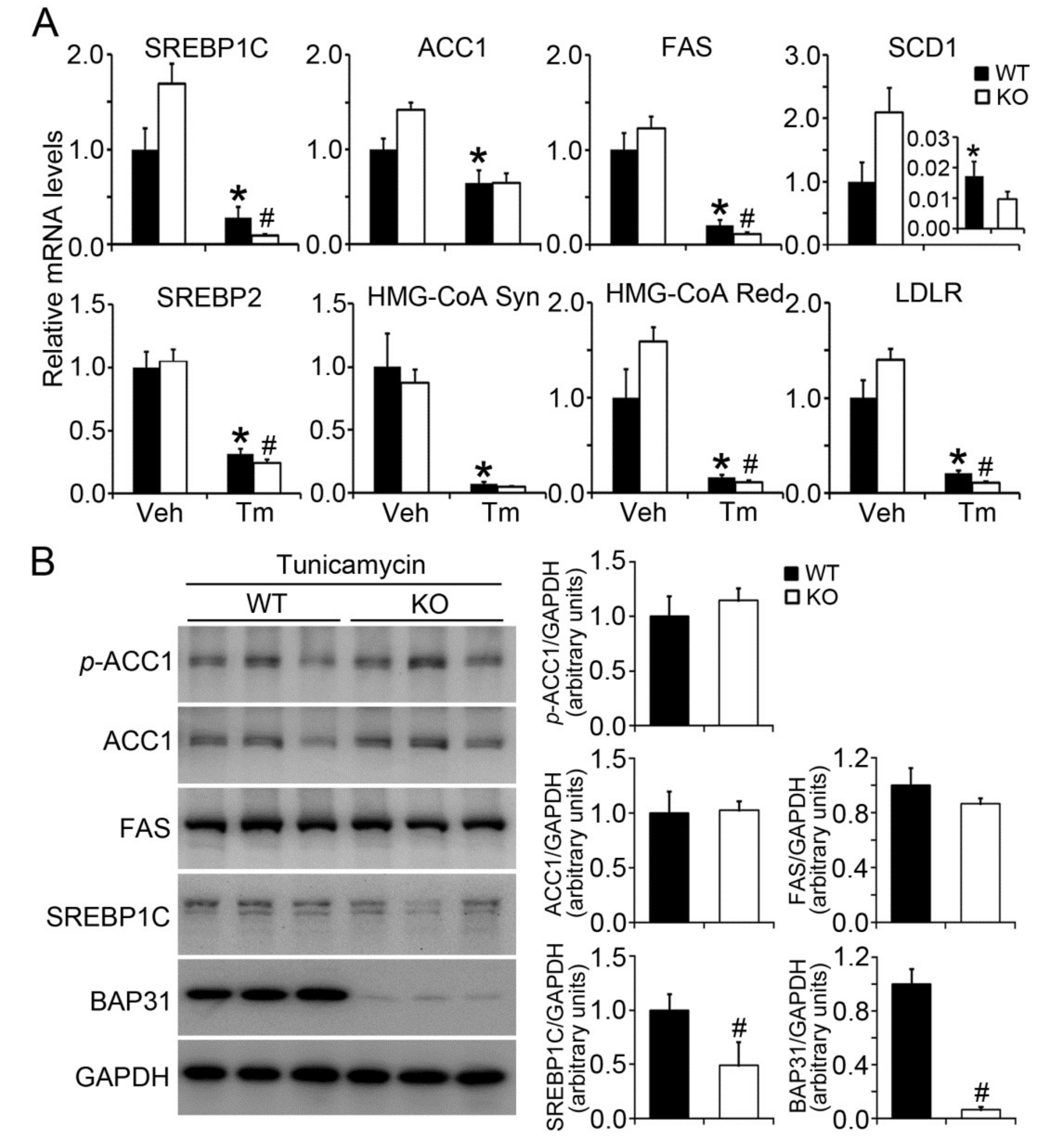
2.3. BAP31 Deficiency Reduced Lipogenic Gene Expression in Tunicamycin-Injected Mice
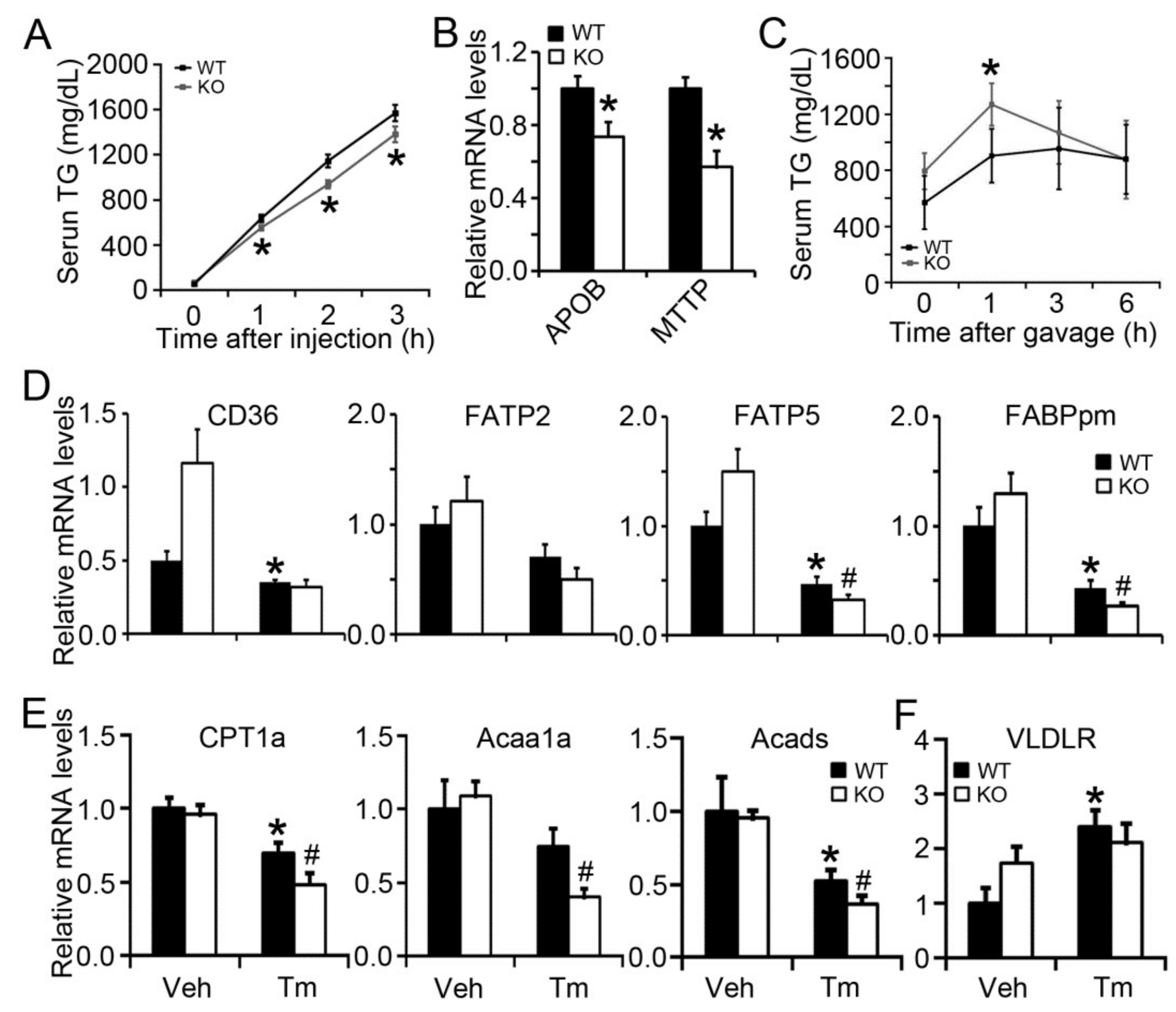
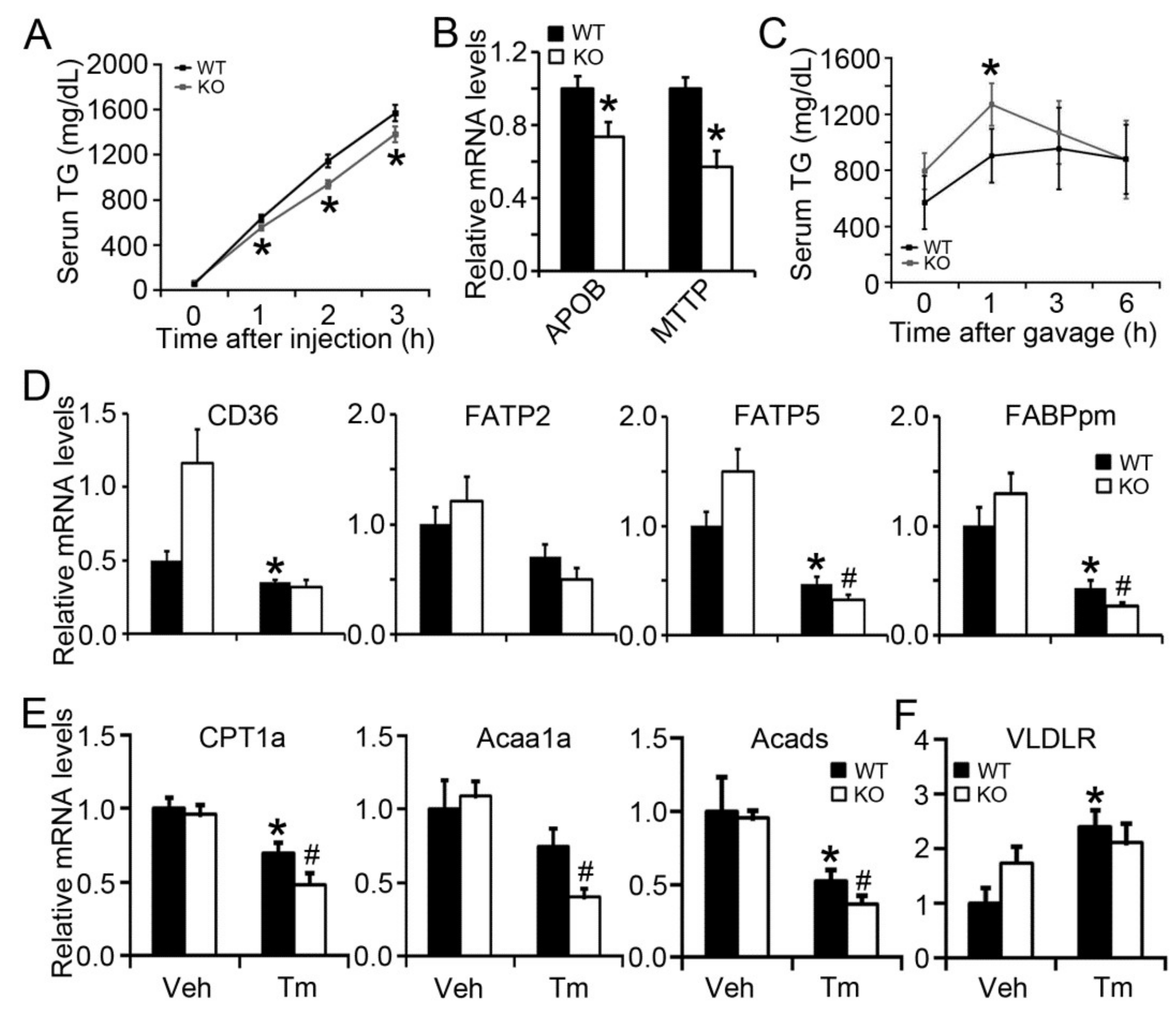
2.4. BAP31 Deficiency Impaired VLDL Secretion and Exogenous Lipid Clearance, Reduced the Gene Expression Related to Fatty Acid β-Oxidation
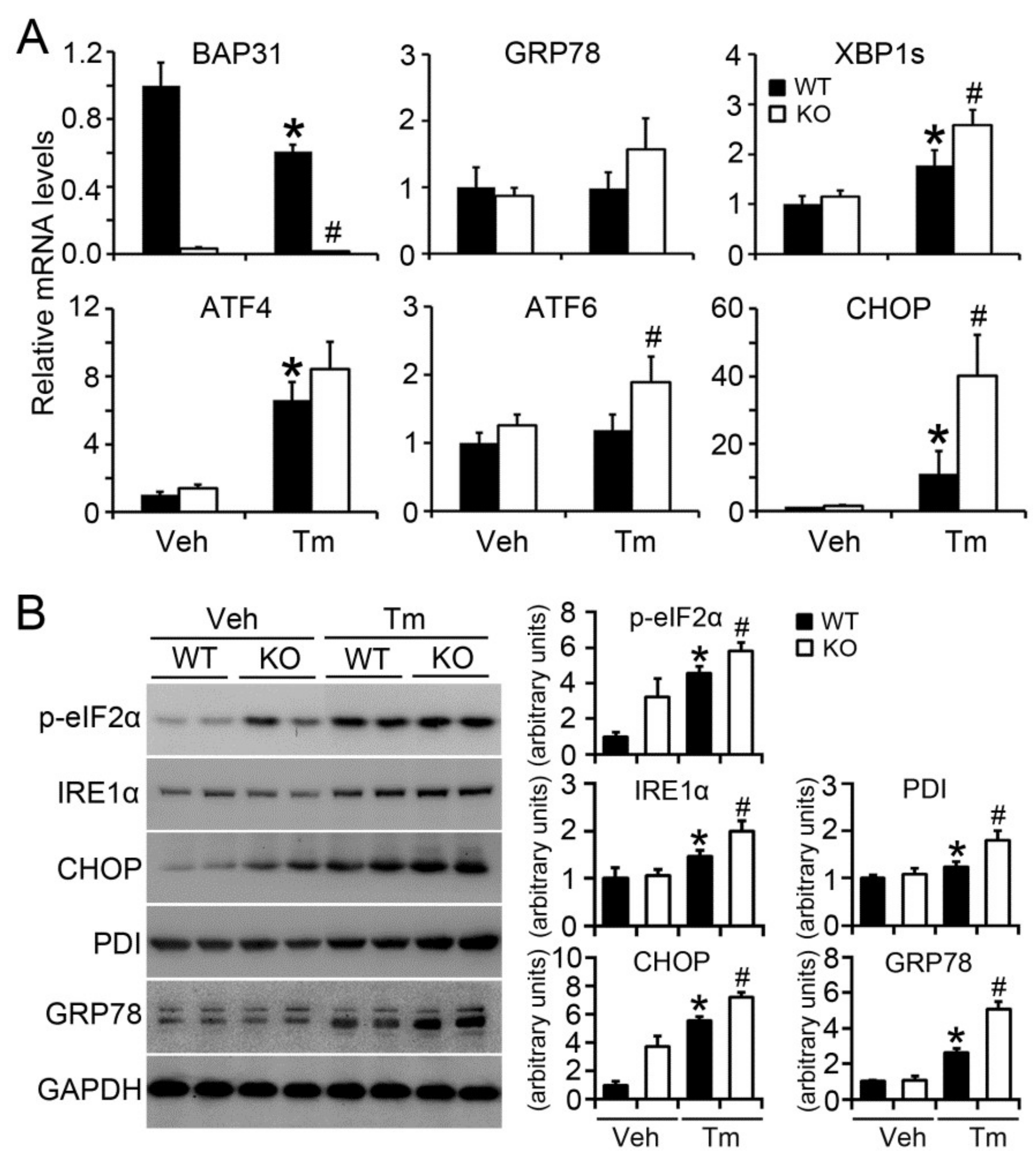
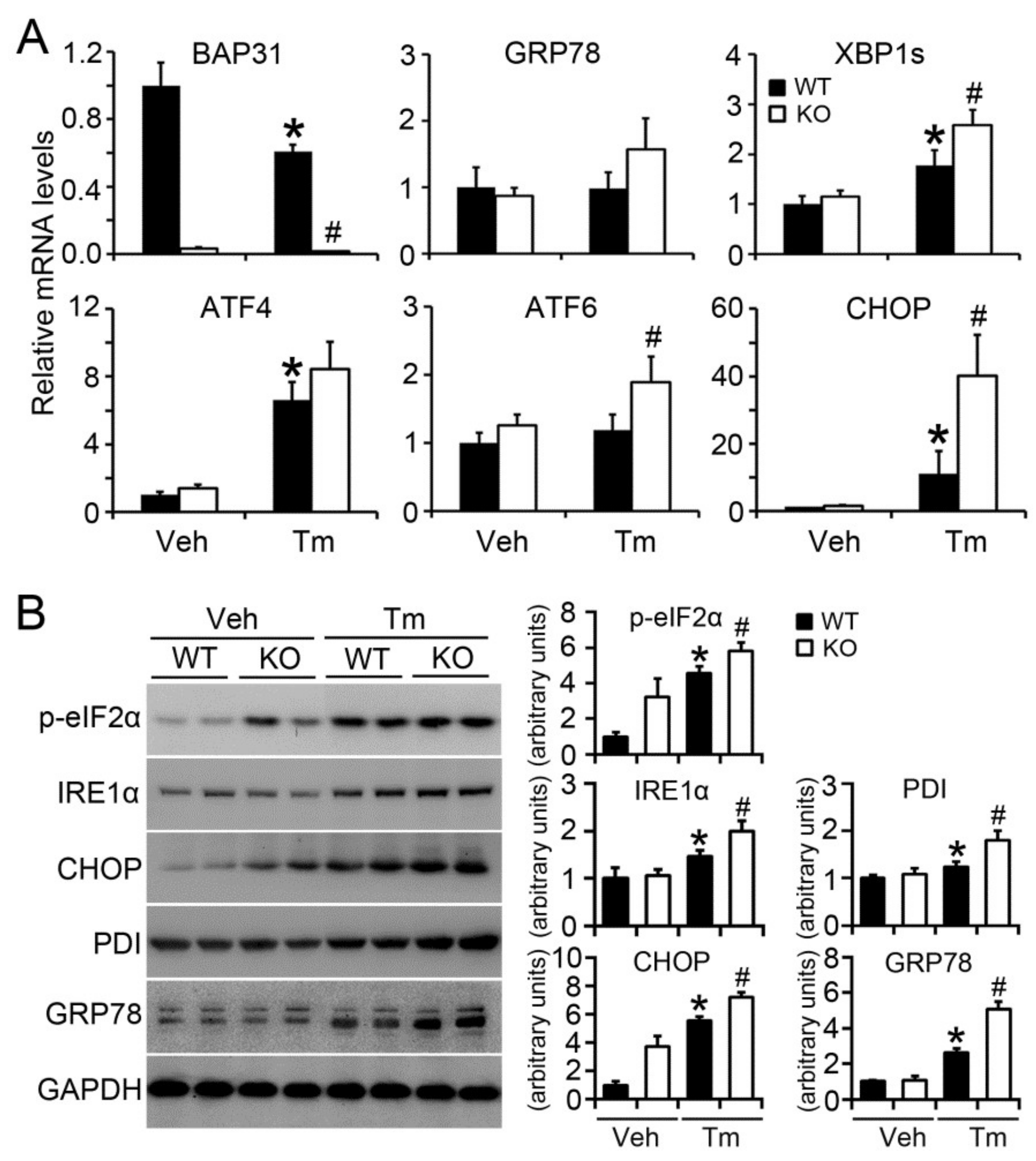
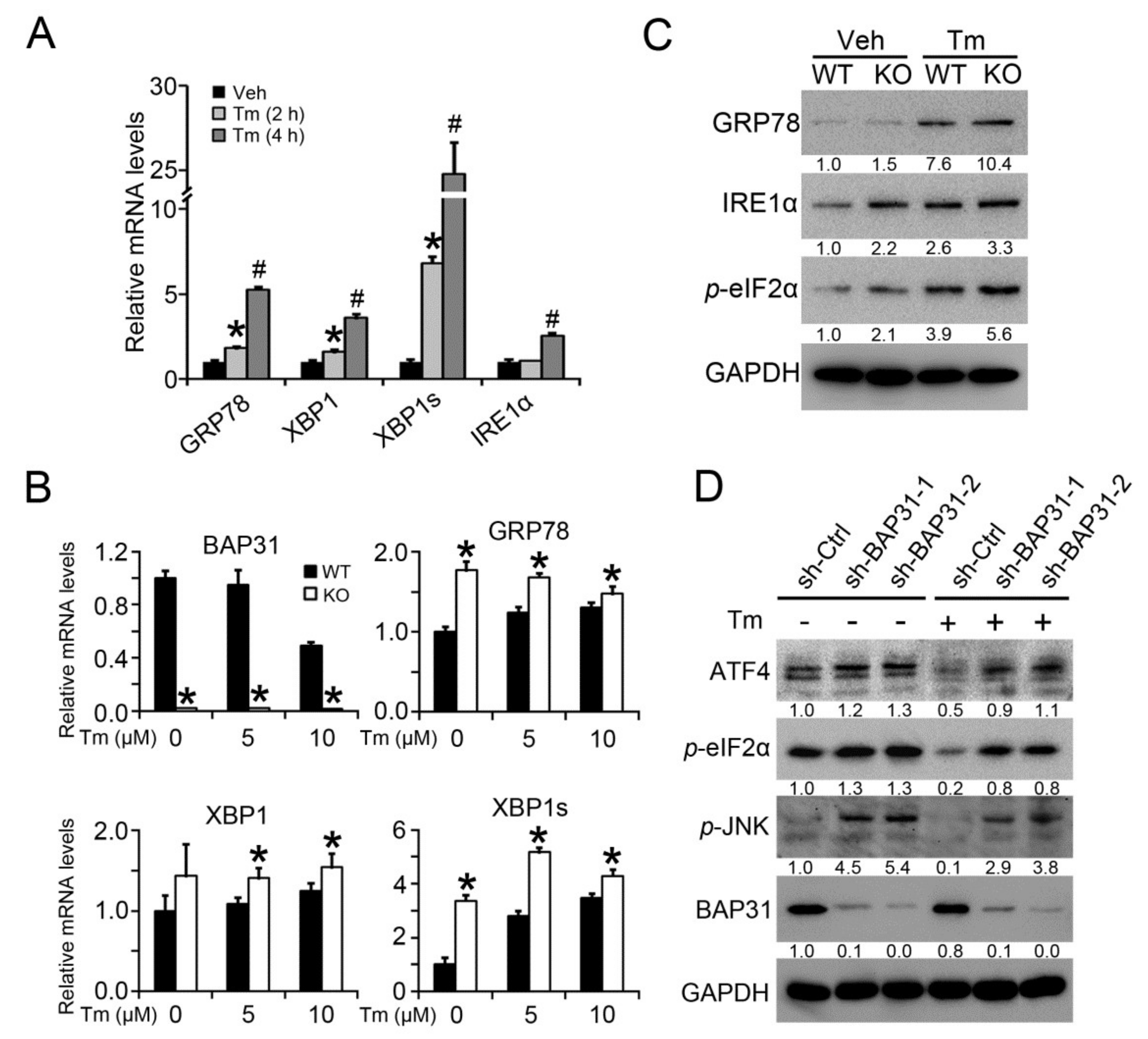
2.5. BAP31 Deficiency Increased Tunicamycin-Induced ER Stress
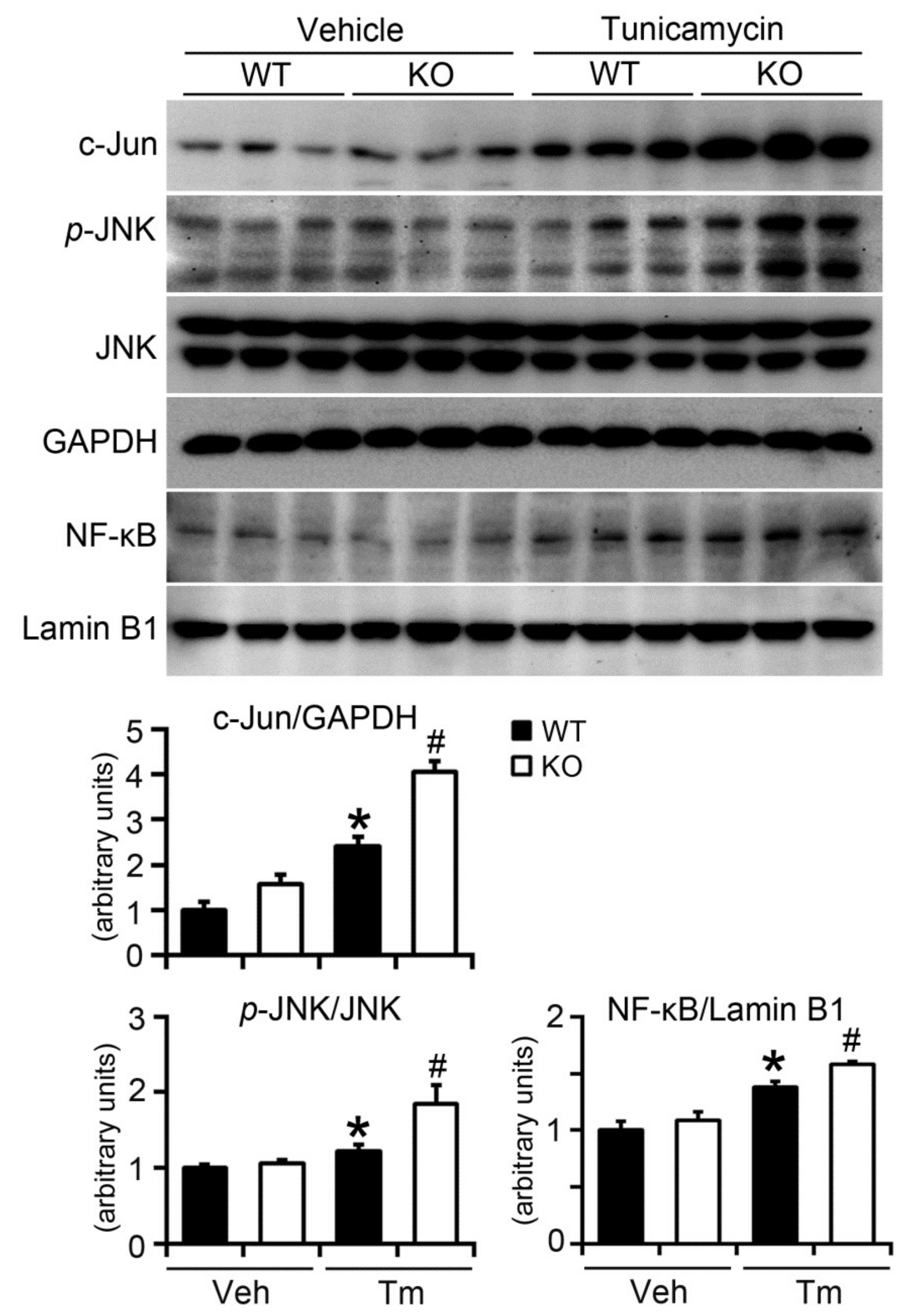
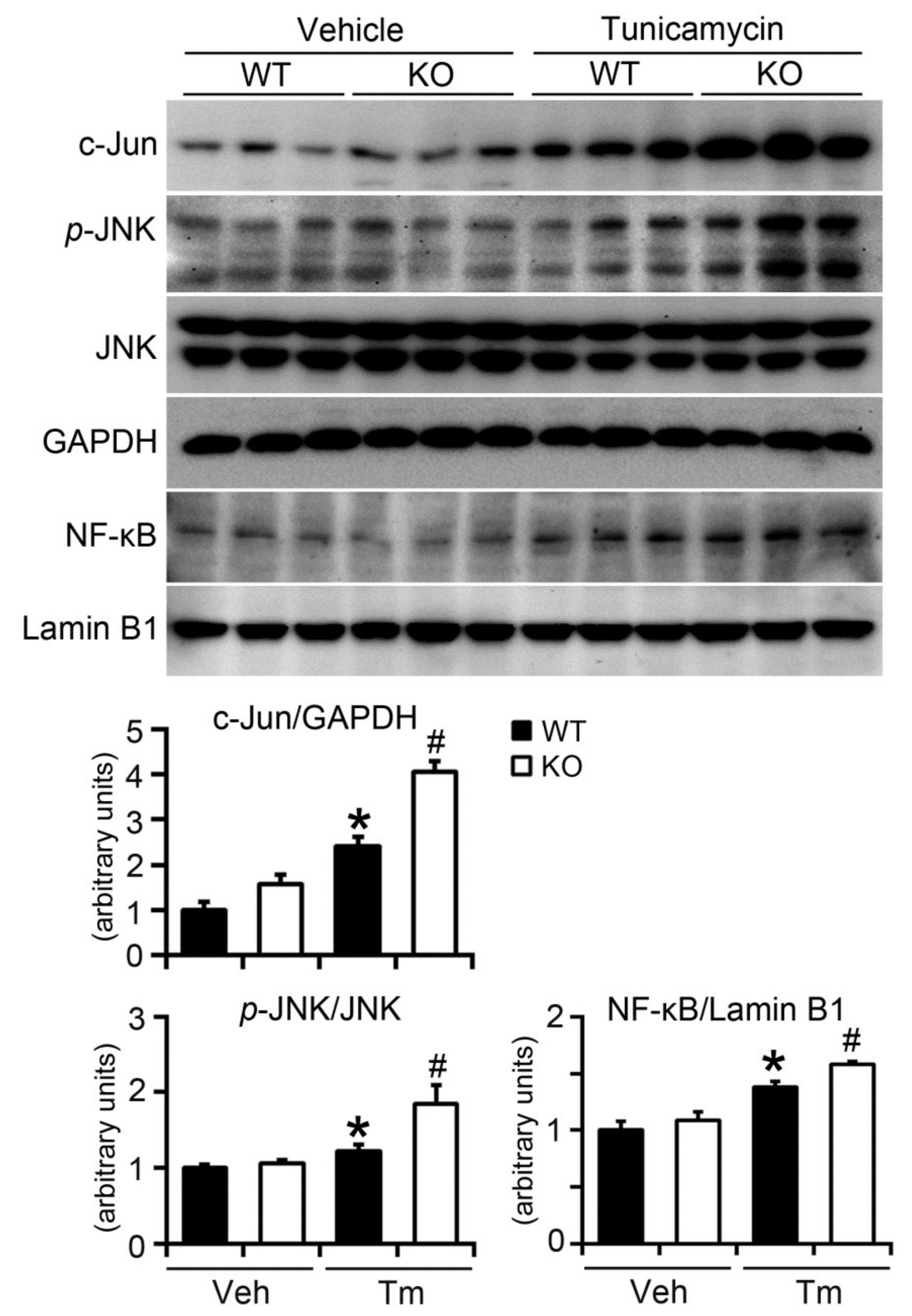
2.6. BAP31 Deficiency Increased Inflammatory Response in Mice upon Tunicamycin Injection
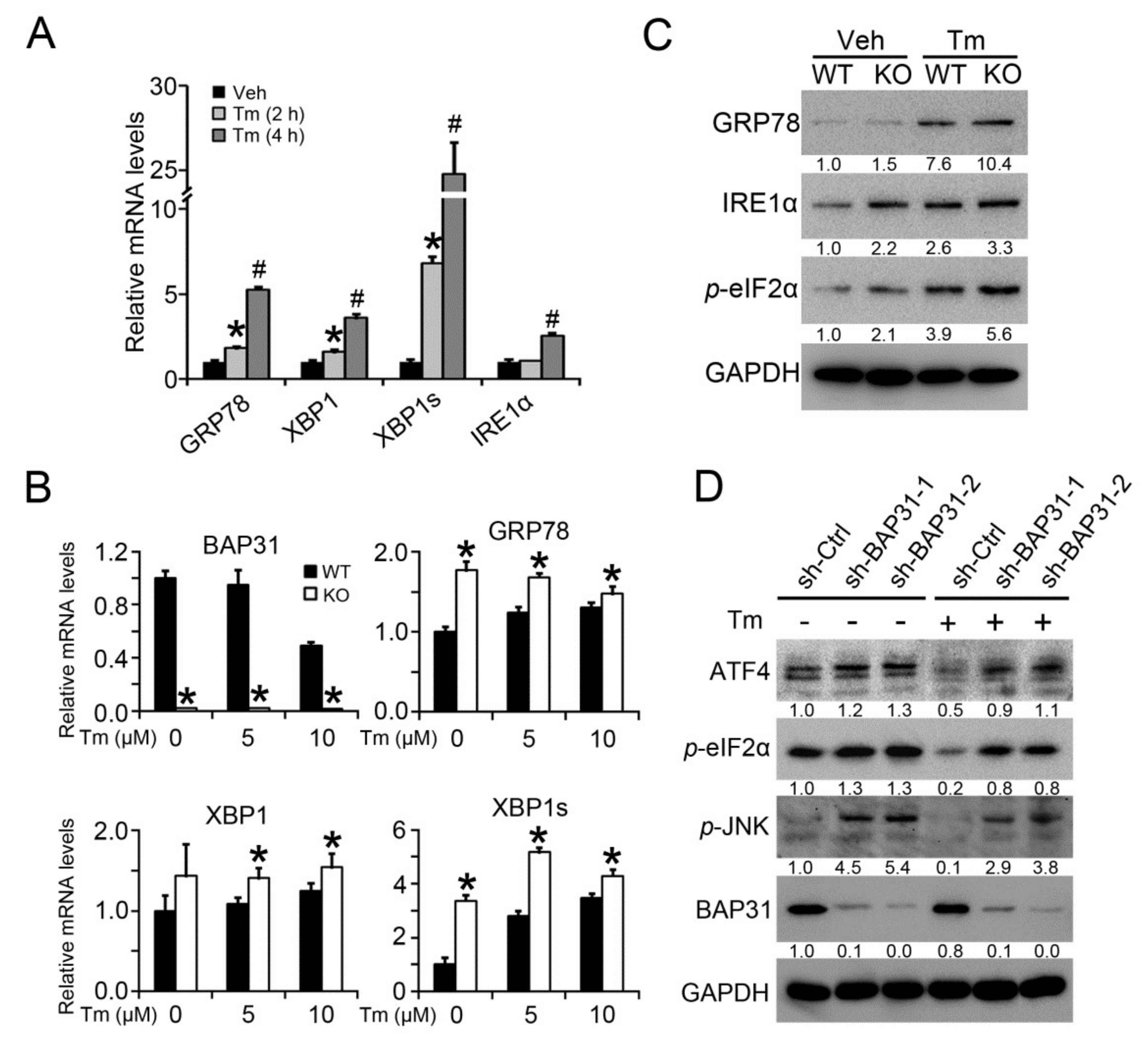
2.7. BAP31 Deficiency Induced ER Stress In Vitro
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Animals and Tunicamycin Administration
4.3. Primary Hepatocytes Isolation and Treatment
4.4. VLDL Secretion and Exogenous Lipid Clearance Measurement
4.5. HepG2 Cells Treatment
4.6. Measurement of Serum Metabolites and Liver Extracts
4.7. Histopathology
4.8. RNA Isolation and Real-Time PCR
4.9. Immunoblot Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Acaa1a | 3-ketoacyl-CoA thiolase |
| Acads | Acyl-CoA dehydrogenases |
| ACC1 | Acetyl-CoA carboxylase 1 |
| ATF | Activating transcription factor |
| ALT | Alanine transaminase |
| APOB | Apolipoprotein B |
| AST | Aspartate transaminase |
| BAP31 | B-cell receptor-associated protein 31 |
| Chol | Cholesterol |
| CD36 | Cluster of differentiation 36 |
| CHOP | C/EBP homologous protein |
| CPT1a | Carnitine palmitoyltransferase 1a |
| FABPpm | Membrane-associated fatty acid binding protein |
| FATP | Fatty acid transport protein |
| FAS | Fatty acid synthase |
| FFA | Free fatty acid |
| GRP78 | Glucose-regulated protein 78 |
| HDL-C | High-density lipoprotein cholesterol |
| HMG-CoA Syn | 3-hydroxy-3-methylglutaryl-CoA synthase |
| HMG-CoA Red | 3-hydroxy-3-methyl-glutaryl-Coenzyme A reductase |
| IRE1α | Inositol-requiring enzyme-1α |
| JNK | C-Jun N-terminal kinases |
| LDL-C | Low-density lipoprotein cholesterol |
| LDLR | Low-density lipoprotein receptor |
| MTTP | Microsomal triglyceride transfer protein |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| PDI | Protein disulfide isomerase |
| SCD1 | Stearoyl-CoA desaturase-1 |
| SREBP | Sterol regulatory element-binding protein |
| Tm | Tunicamycin |
| TG | Triglycerides |
| VLDLR | Very low-density lipoprotein receptor |
| XBP1 | X-box binding protein 1 |
References
- Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; Miele, L.; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. 2015, 47, 997–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, J.; Ning, C.; Lei, D.; Ren, J. Endoplasmic reticulum stress related molecular mechanisms in nonalcoholic fatty liver disease (NAFLD). Curr. Drug Targets 2018, 19, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Henkel, A.; Green, R.M. The unfolded protein response in fatty liver disease. Semin. Liver Dis. 2013, 33, 321–329. [Google Scholar] [PubMed]
- Yamamoto, K.; Takahara, K.; Oyadomari, S.; Okada, T.; Sato, T.; Harada, A.; Mori, K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 2010, 21, 2975–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Lang, J.Y.; Hung, M.C.; Sengupta, K.; Banerjee, S.K.; Baksi, K.; Banerjee, D.K. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J. Biol. Chem. 2011, 286, 29127–29138. [Google Scholar] [CrossRef] [PubMed]
- Basseri, S.; Austin, R.C. ER stress and lipogenesis: A slippery slope toward hepatic steatosis. Dev. Cell 2008, 15, 795–796. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Huang, X.; Jiang, D.; Hua, L.; Zhuo, Y.; Wu, D. Endoplasmic Reticulum Stress Inducer Tunicamycin Alters Hepatic Energy Homeostasis in Mice. Int. J. Mol. Sci. 2017, 18, 1710. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.W.; Nguyen, M.; Kwan, T.; Branton, P.E.; Nicholson, D.W.; Cromlish, J.A.; Shore, G.C. p28 Bap31, a Bcl-2/Bcl-XL- and procaspase-8-associated protein in the endoplasmic reticulum. J. Cell Biol. 1997, 139, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Annaert, W.G.; Becker, B.; Kistner, U.; Reth, M.; Jahn, R. Export of cellubrevin from the endoplasmic reticulum is controlled by BAP31. J. Cell Biol. 1997, 139, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- Abe, F.; Van Prooyen, N.; Ladasky, J.J.; Edidin, M. Interaction of Bap31 and MHC class I molecules and their traffic out of the endoplasmic reticulum. J. Immunol. 2009, 182, 4776–4783. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, M.; Germain, M.; Nguyen, M.; Shore, G.C. BAP31 and its caspase cleavage product regulate cell surface expression of tetraspanins and integrin-mediated cell survival. J. Biol. Chem. 2005, 280, 30018–30024. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Tian, F.; Chu, K.; Hwang, S.Y.; Yoon, K.W.; Byun, S.; Hiraki, M.; Mandinova, A.; Lee, S.W. CDIP1-BAP31 complex transduces apoptotic signals from endoplasmic reticulum to mitochondria under endoplasmic reticulum stress. Cell Rep. 2013, 5, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.L.; Li, L.Y.; Wang, Y.Q.; Li, Y.Q.; Shan, M.; Sun, S.Z.; Yu, Y.; Wang, B. Hepatocyte-specific deletion of BAP31 promotes SREBP1C activation, promotes hepatic lipid accumulation, and worsens IR in mice. J. Lipid Res. 2018, 59, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kang, H.S.; Lee, J.H.; Park, J.H.; Jung, C.H.; Bae, J.H.; Oh, B.C.; Song, D.K.; Baek, W.K.; Im, S.S. Melatonin ameliorates ER stress-mediated hepatic steatosis through miR-23a in the liver. Biochem. Biophys. Res. Commun. 2015, 458, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Welty, F.K. Hypobetalipoproteinemia and abetalipoproteinemia. Curr. Opin. Lipidol. 2014, 25, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Zhang, T.; Lee, S.; Calabuig-Navarro, V.; Yamauchi, J.; Piccirillo, A.; Fan, Y.; Uppala, R.; Goetzman, E.; Dong, H.H. FoxO6 integrates insulin signaling with MTP for regulating VLDL production in the liver. Endocrinology 2014, 155, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D.; Rodriguez-Ramiro, I.; Rushbrook, S.; Ipharraguerre, I.R.; Bevan, D.; Davies, S.; Tejera, N.; Mena, P.; de Pascual-Teresa, S.; Del Rio, D.; et al. n-3 Fatty acids combined with flavan-3-ols prevent steatosis and liver injury in a murine model of NAFLD. Biochim. Biophys. Acta 2018, 1864, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.A.; Choi, Y.K.; Jung, G.S.; Seo, H.Y.; Kim, H.S.; Jang, B.K.; Kim, J.G.; Lee, I.K.; Kim, M.K.; Park, K.G. Sitagliptin attenuates methionine/choline-deficient diet-induced steatohepatitis. Diabetes Res. Clin. Pract. 2014, 105, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Heath-Engel, H.; Zhang, D.; Nguyen, N.; Thomas, D.Y.; Hanrahan, J.W.; Shore, G.C. BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTRDeltaF508 via the derlin-1 complex. Cell 2008, 133, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Bredesen, D.E. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 2004, 16, 653–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, W.L.; Shome, K.; Wu, C.C.; Gong, X.; Frizzell, R.A.; Aridor, M. VAMP-associated Proteins (VAP) as Receptors That Couple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Proteostasis with Lipid Homeostasis. J. Biol. Chem. 2016, 291, 5206–5220. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, R.; Mahul-Mellier, A.L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Cacciagli, P.; Sutera-Sardo, J.; Borges-Correia, A.; Roux, J.C.; Dorboz, I.; Desvignes, J.P.; Badens, C.; Delepine, M.; Lathrop, M.; Cau, P.; et al. Mutations in BCAP31 cause a severe X-linked phenotype with deafness, dystonia, and central hypomyelination and disorganize the Golgi apparatus. Am. J. Hum. Genet. 2013, 93, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Andrade, J.M.; Paraiso, A.F.; de Oliveira, M.V.; Martins, A.M.; Neto, J.F.; Guimaraes, A.L.; de Paula, A.M.; Qureshi, M.; Santos, S.H. Resveratrol attenuates hepatic steatosis in high-fat fed mice by decreasing lipogenesis and inflammation. Nutrition 2014, 30, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Choe, S.S.; Shin, K.C.; Jang, H.; Lee, J.H.; Seong, J.K.; Back, S.H.; Kim, J.B. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 2013, 57, 1366–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, A.J.; Burnett, J.R. Update on primary hypobetalipoproteinemia. Curr. Atheroscler. Rep. 2014, 16, 423. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, E.; Sorensen, L.P.; Rahbek, I.; Gormsen, L.C.; Christiansen, J.S.; Nielsen, S. Postprandial VLDL-triacylglycerol secretion is not suppressed in obese type 2 diabetic men. Diabetologia 2012, 55, 2733–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.Y.; Little, H.C.; Lei, X.; Li, S.; Rodriguez, S.; Wong, G.W. Partial deficiency of CTRP12 alters hepatic lipid metabolism. Physiol. Genom. 2016, 48, 936–949. [Google Scholar] [CrossRef] [PubMed]
- DeZwaan-McCabe, D.; Sheldon, R.D.; Gorecki, M.C.; Guo, D.F.; Gansemer, E.R.; Kaufman, R.J.; Rahmouni, K.; Gillum, M.P.; Taylor, E.B.; Teesch, L.M.; et al. ER Stress Inhibits Liver Fatty Acid Oxidation while Unmitigated Stress Leads to Anorexia-Induced Lipolysis and Both Liver and Kidney Steatosis. Cell Rep. 2017, 19, 1794–1806. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R. The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J. 2016, 15, 817–828. [Google Scholar] [PubMed]
- Guo, B.; Li, Z. Endoplasmic reticulum stress in hepatic steatosis and inflammatory bowel diseases. Front. Genet. 2014, 5, 242. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [PubMed]
- Mehal, W.Z. The inflammasome in liver injury and non-alcoholic fatty liver disease. Dig. Dis. 2014, 32, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Nivala, A.M.; Reese, L.; Frye, M.; Gentile, C.L.; Pagliassotti, M.J. Fatty acid-mediated endoplasmic reticulum stress in vivo: Differential response to the infusion of Soybean and Lard Oil in rats. Metabolism 2013, 62, 753–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Kulkarni, S.R.; Li, L.; Slitt, A.L. UDP-glucuronosyltransferase expression in mouse liver is increased in obesity- and fasting-induced steatosis. Drug Metab. Dispos. 2012, 40, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kulkarni, S.R.; Donepudi, A.C.; More, V.R.; Slitt, A.L. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 2012, 61, 3208–3218. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Vehicle | Tunicamycin | ||
|---|---|---|---|---|
| WT | KO | WT | KO | |
| Glucose (mg/dL) | 160.15 ± 9.44 | 176.39 ± 6.32 | 135.37 ± 6.05 * | 123.92 ± 8.34 |
| TG (mg/dL) | 99.88 ± 11.46 | 91.84 ± 4.72 | 22.70 ± 1.63 * | 22.23 ± 3.02 |
| FFAs (mEq/L) | 0.87 ± 0.07 | 0.76 ± 0.04 | 0.24 ± 0.01 * | 0.31 ± 0.02 # |
| Chol (mg/dL) | 77.32 ± 3.36 | 88.78 ± 2.42 | 6.03 ± 0.78 * | 6.11 ± 0.87 |
| HDL-C (mg/dL) | 53.75 ± 3.10 | 57.28 ± 1.80 | 6.17 ± 0.56 * | 7.09 ± 0.75 |
| LDL-C (mg/dL) | 31.15 ± 5.90 | 40.30 ± 5.90 | n.d. | n.d. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Yang, F.; Jiang, S.; Sun, X.; Xu, J. Induction of Liver Steatosis in BAP31-Deficient Mice Burdened with Tunicamycin-Induced Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2018, 19, 2291. https://doi.org/10.3390/ijms19082291
Wu Z, Yang F, Jiang S, Sun X, Xu J. Induction of Liver Steatosis in BAP31-Deficient Mice Burdened with Tunicamycin-Induced Endoplasmic Reticulum Stress. International Journal of Molecular Sciences. 2018; 19(8):2291. https://doi.org/10.3390/ijms19082291
Chicago/Turabian StyleWu, Zhenhua, Fan Yang, Shan Jiang, Xiaoyu Sun, and Jialin Xu. 2018. "Induction of Liver Steatosis in BAP31-Deficient Mice Burdened with Tunicamycin-Induced Endoplasmic Reticulum Stress" International Journal of Molecular Sciences 19, no. 8: 2291. https://doi.org/10.3390/ijms19082291