Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle


Abstract
:1. Introduction
2. Approaches and Parameters to Estimate Contractility
3. Contractile Properties of HCM and DCM Hearts
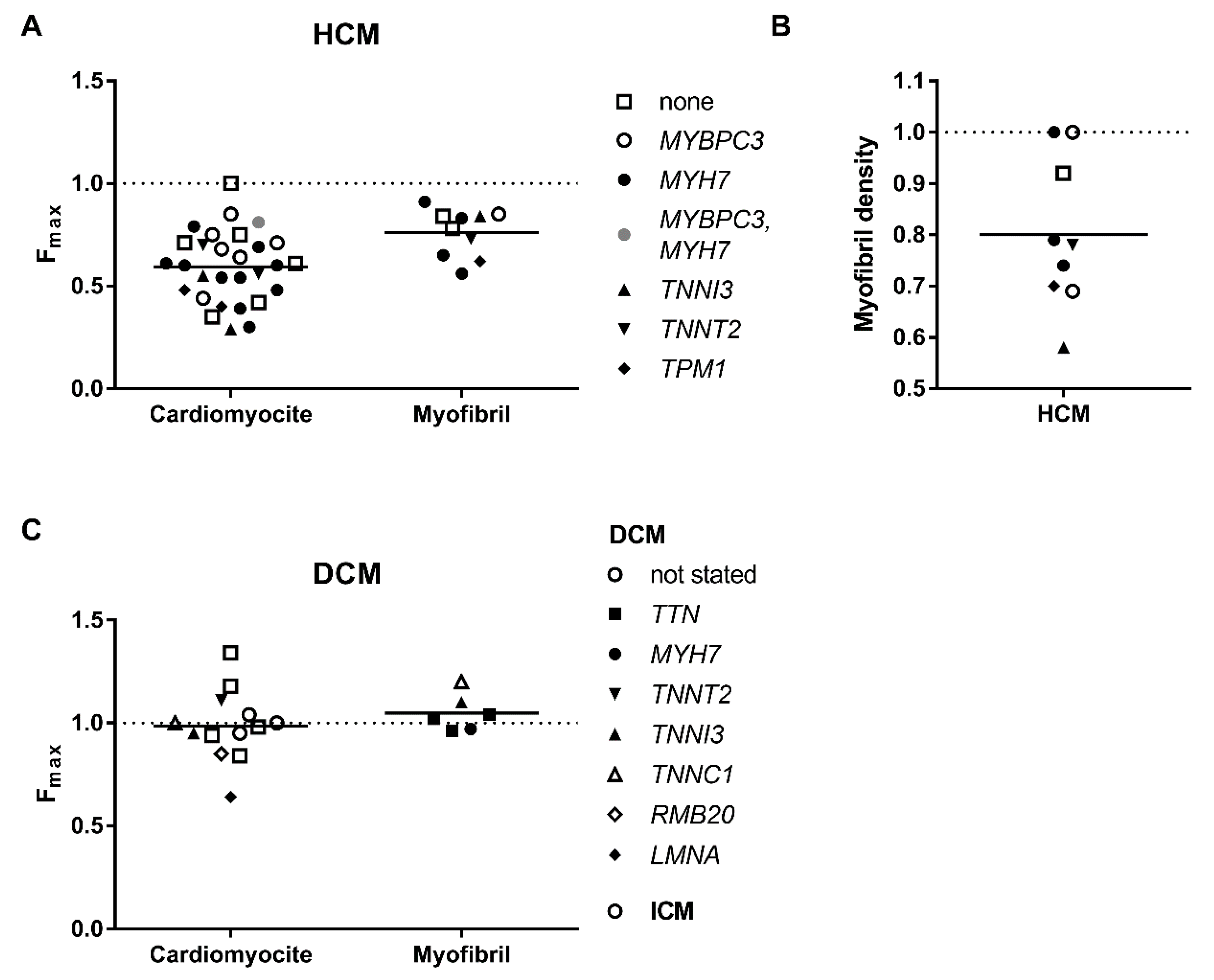
3.1. Decreased Force Capacity
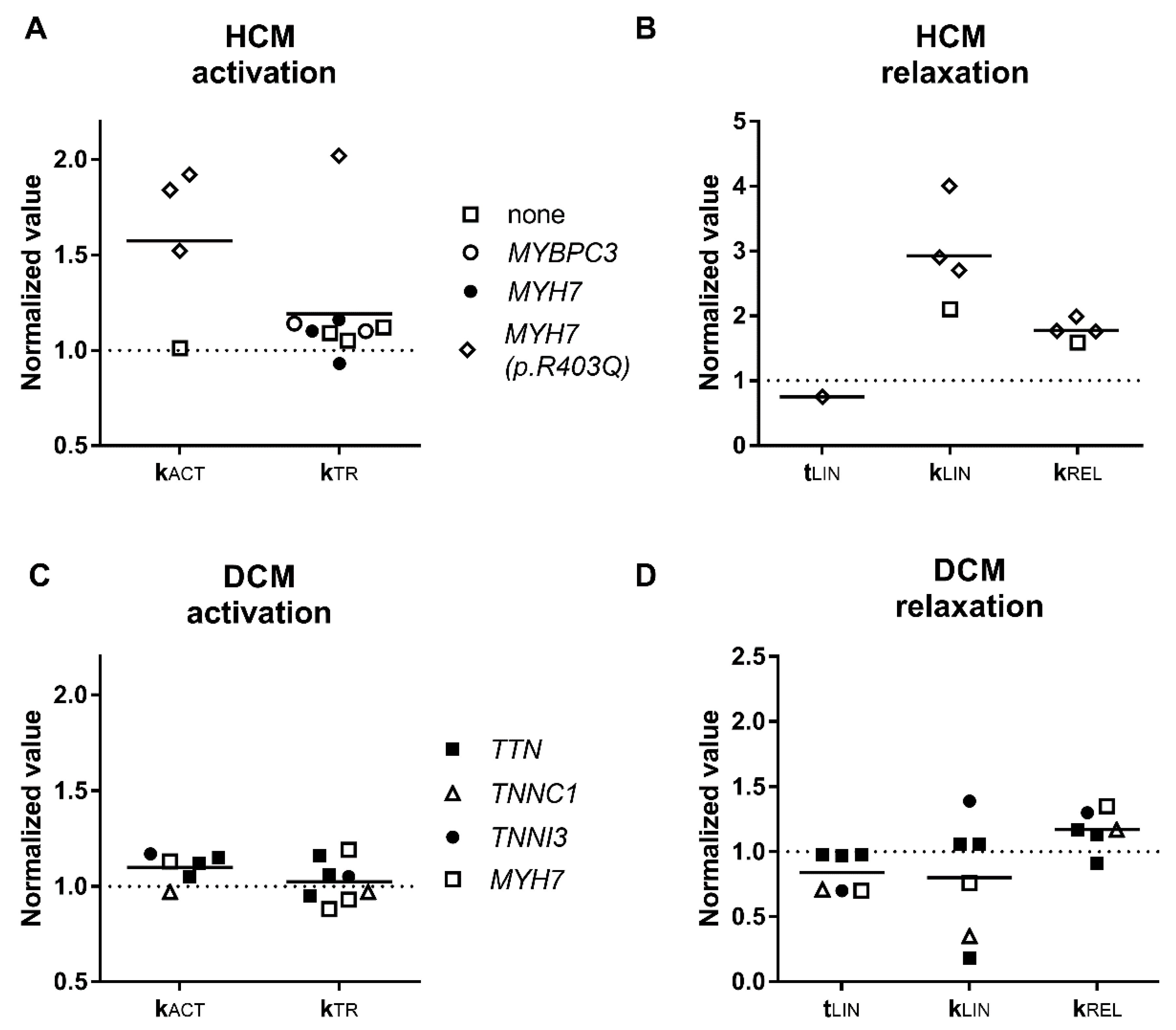
3.2. Activation and Relaxation Kinetics
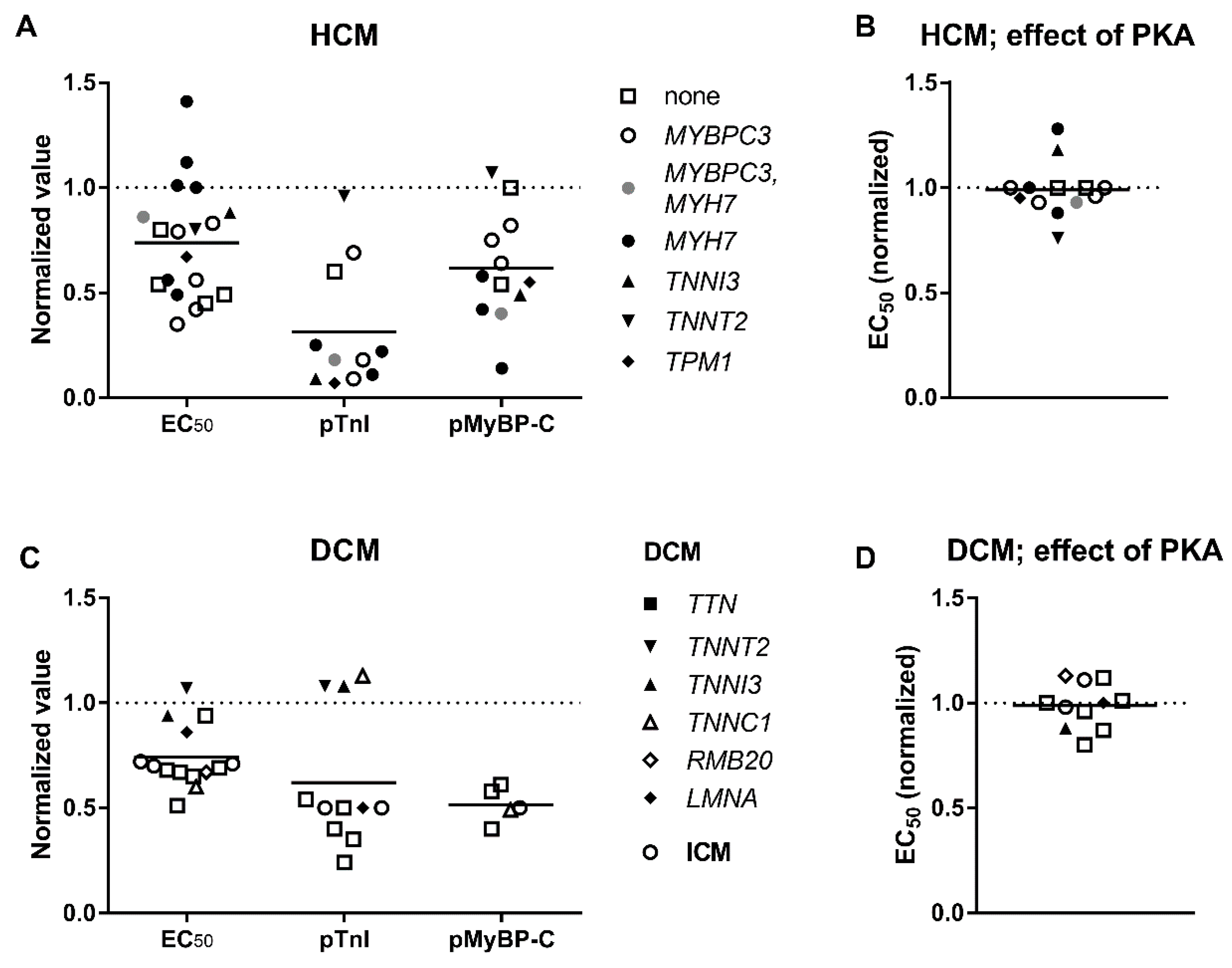
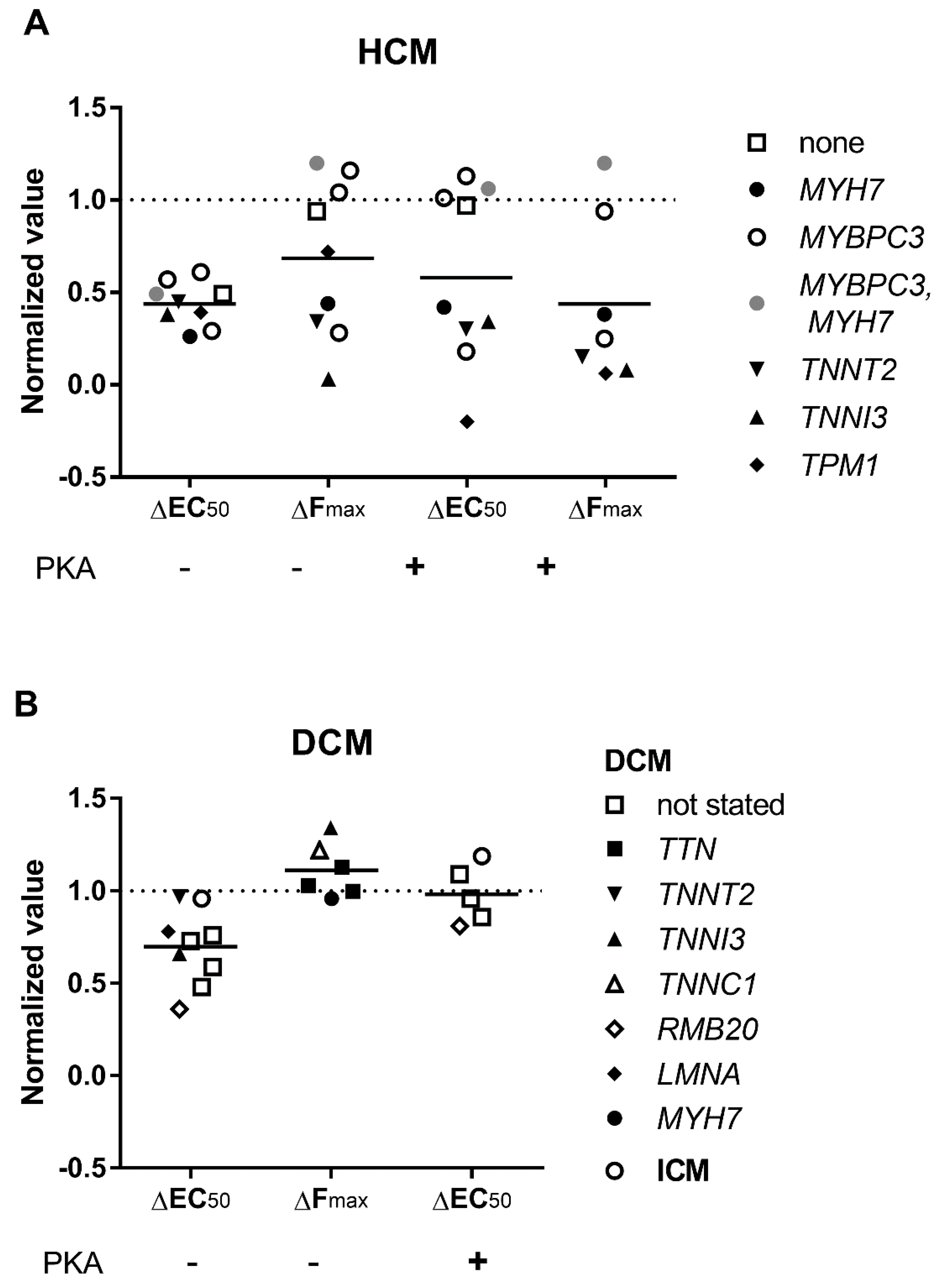
3.3. Elevated Ca2+-Sensitivity
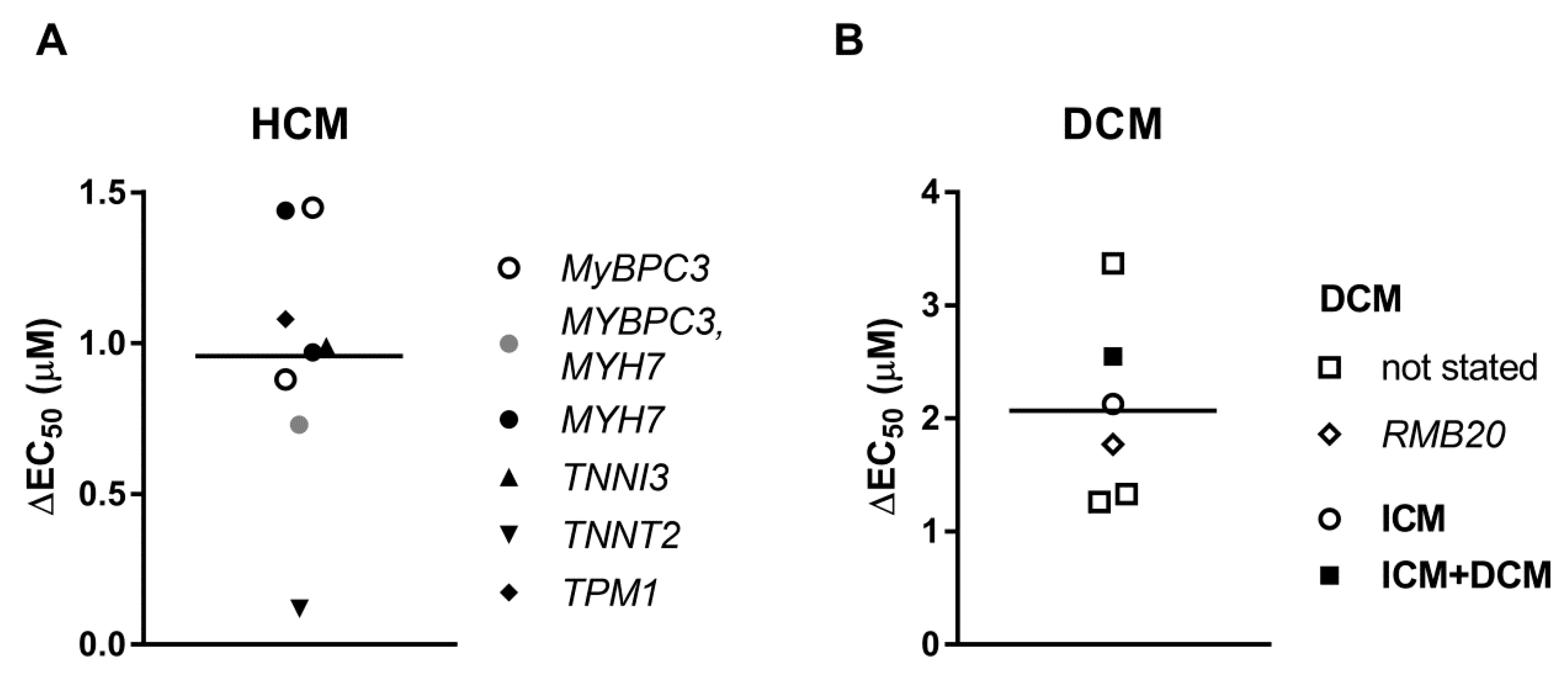
3.4. Uncoupling of TnI Phosphorylation from the Changes in Ca2+-Sensitivity
3.5. Length Dependent Activation
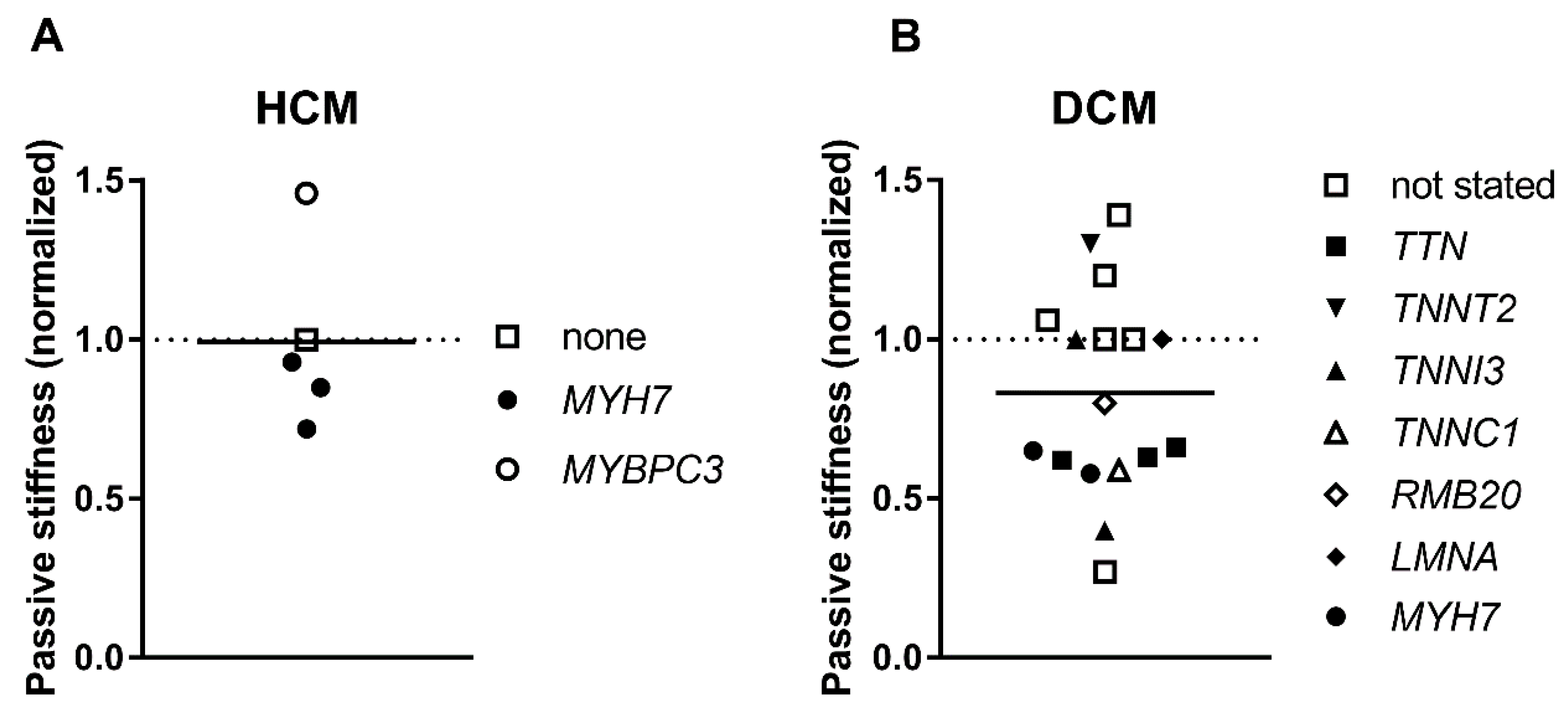
3.6. Passive Stiffness
4. Conclusions
Funding
Conflicts of Interest
References
- Gordan, R.; Gwathmey, J.K.; Xie, L.H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204–214. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, K.T. Recent advances in understanding cardiac contractility in health and disease. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.R.; Campbell, S.G.; Lehman, W. Structural determinants of muscle thin filament cooperativity. Arch. Biochem. Biophys. 2016, 594, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risi, C.; Eisner, J.; Belknap, B.; Heeley, D.H.; White, H.D.; Schroder, G.F.; Galkin, V.E. Ca(2+)-induced movement of tropomyosin on native cardiac thin filaments revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA 2017, 114, 6782–6787. [Google Scholar] [CrossRef] [PubMed]
- Layland, J.; Solaro, R.J.; Shah, A.M. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc. Res. 2005, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijnker, P.J.; Murphy, A.M.; Stienen, G.J.; van der Velden, J. Troponin I phosphorylation in human myocardium in health and disease. Neth. Heart J. 2014, 22, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marston, S.B. Why Is there a Limit to the Changes in Myofilament Ca(2+)-Sensitivity Associated with Myopathy Causing Mutations? Front. Physiol. 2016, 7, 415. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Lee, K.H.; Mun, J.Y.; Torre, I.; Luther, P.K. Structure, sarcomeric organization, and thin filament binding of cardiac myosin-binding protein-C. Pflugers Arch. 2014, 466, 425–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, S.; Szczesna-Cordary, D. Pseudophosphorylation of cardiac myosin regulatory light chain: A promising new tool for treatment of cardiomyopathy. Biophys. Rev. 2017, 9, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Kampourakis, T.; Yan, Z.; Gautel, M.; Sun, Y.B.; Irving, M. Myosin binding protein-C activates thin filaments and inhibits thick filaments in heart muscle cells. Proc. Natl. Acad. Sci. USA 2014, 111, 18763–18768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Govindan, S.; Zhang, M.; Khairallah, R.J.; Martin, J.L.; Sadayappan, S.; de Tombe, P.P. Cardiac Myosin-binding Protein C and Troponin-I Phosphorylation Independently Modulate Myofilament Length-dependent Activation. J. Biol. Chem. 2015, 290, 29241–29249. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, M.; Sharma, S.; Cox, S.; Sheppard, M.N.; Panoulas, V.F.; Behr, E.R. The magnitude of sudden cardiac death in the young: A death certificate-based review in England and Wales. Europace 2009, 11, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.E.; McKenna, W.J. New insights into the pathology of inherited cardiomyopathy. Heart 2005, 91, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.; Charron, P.; Gimeno-Blanes, J.; Helio, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Bowles, N.E. The failing heart. Nature 2002, 415, 227–233. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Abelmann, W.H.; Lorell, B.H. The challenge of cardiomyopathy. J. Am. Coll. Cardiol. 1989, 13, 1219–1239. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Muller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.A.; Cook, S.A.; Seidman, J.G.; Seidman, C.E. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2871–2886. [Google Scholar] [CrossRef] [PubMed]
- Deo, R.; Albert, C.M. Epidemiology and genetics of sudden cardiac death. Circulation 2012, 125, 620–637. [Google Scholar] [CrossRef] [PubMed]
- Konstam, M.A.; Abboud, F.M. Ejection Fraction: Misunderstood and Overrated (Changing the Paradigm in Categorizing Heart Failure). Circulation 2017, 135, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.E. The pathology of hypertrophic cardiomyopathy. Histopathology 2004, 44, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Burke, M.; Fornes, P.; Gallagher, P.J.; de Gouveia, R.H.; Sheppard, M.; Thiene, G.; van der Wal, A.; Association for European Cardiovascular, Pathology. Guidelines for autopsy investigation of sudden cardiac death. Virchows Arch. 2008, 452, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Mathew, T.; Williams, L.; Navaratnam, G.; Rana, B.; Wheeler, R.; Collins, K.; Harkness, A.; Jones, R.; Knight, D.; O’Gallagher, K.; et al. Diagnosis and assessment of dilated cardiomyopathy: A guideline protocol from the British Society of Echocardiography. Echo Res. Pract. 2017, 4, G1–G13. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, A.; Vischer, A.S.; Perez-Tome, M.C.; Castelletti, S. Diagnosis and management of hypertrophic cardiomyopathy. Echo Res. Pract. 2015, 2, R45–R53. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Fleming, E.J.; Garratt, C.J. Mimics of Hypertrophic Cardiomyopathy—Diagnostic Clues to Aid Early Identification of Phenocopies. Arrhythm. Electrophysiol. Rev. 2013, 2, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.R.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Bohm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Amor-Salamanca, A.; Tayal, U.; Govind, R.; Serrano, I.; Salazar-Mendiguchia, J.; Garcia-Pinilla, J.M.; Pascual-Figal, D.A.; Nunez, J.; Guzzo-Merello, G.; et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J. Am. Coll. Cardiol. 2018, 71, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Linschoten, M.; Teske, A.J.; Baas, A.F.; Vink, A.; Dooijes, D.; Baars, H.F.; Asselbergs, F.W. Truncating Titin (TTN) Variants in Chemotherapy-Induced Cardiomyopathy. J. Card. Fail. 2017, 23, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Bakalakos, A.; Ritsatos, K.; Anastasakis, A. Current perspectives on the diagnosis and management of dilated cardiomyopathy Beyond heart failure: A Cardiomyopathy Clinic Doctor’s point of view. Hellenic J. Cardiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Felker, G.M.; Thompson, R.E.; Hare, J.M.; Hruban, R.H.; Clemetson, D.E.; Howard, D.L.; Baughman, K.L.; Kasper, E.K. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N. Engl. J. Med. 2000, 342, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006, 296, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.; de Marvao, A.; Adami, E.; Fiedler, L.R.; Ng, B.; Khin, E.; Rackham, O.J.; van Heesch, S.; Pua, C.J.; Kui, M.; et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat. Genet. 2017, 49, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Tayal, U.; Newsome, S.; Buchan, R.; Whiffin, N.; Halliday, B.; Lota, A.; Roberts, A.; Baksi, A.J.; Voges, I.; Midwinter, W.; et al. Phenotype and Clinical Outcomes of Titin Cardiomyopathy. J. Am. Coll. Cardiol. 2017, 70, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- Begay, R.L.; Graw, S.; Sinagra, G.; Merlo, M.; Slavov, D.; Gowan, K.; Jones, K.L.; Barbati, G.; Spezzacatene, A.; Brun, F.; et al. Role of Titin Missense Variants in Dilated Cardiomyopathy. J. Am. Heart Assoc. 2015, 4, e002645. [Google Scholar] [CrossRef] [PubMed]
- Van Velzen, H.G.; Schinkel, A.F.L.; Baart, S.J.; Oldenburg, R.A.; Frohn-Mulder, I.M.E.; van Slegtenhorst, M.A.; Michels, M. Outcomes of Contemporary Family Screening in Hypertrophic Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e001896. [Google Scholar] [PubMed]
- Mathew, J.; Zahavich, L.; Lafreniere-Roula, M.; Wilson, J.; George, K.; Benson, L.; Bowdin, S.; Mital, S. Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin. Genet. 2018, 93, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Syrris, P.; Guttmann, O.P.; O’Mahony, C.; Tang, H.C.; Dalageorgou, C.; Jenkins, S.; Hubank, M.; Monserrat, L.; McKenna, W.J.; et al. Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 2015, 101, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Vasile, V.C.; Ommen, S.R.; Will, M.L.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 1903–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaski, J.P.; Syrris, P.; Esteban, M.T.; Jenkins, S.; Pantazis, A.; Deanfield, J.E.; McKenna, W.J.; Elliott, P.M. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Bouvagnet, P.; Chevalier, P.; Dauphin, C.; Jouk, P.S.; Da Costa, A.; Prieur, F.; Bresson, J.L.; Faivre, L.; Eicher, J.C.; et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur. J. Med. Genet. 2010, 53, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Mazzarotto, F.; Girolami, F.; Boschi, B.; Barlocco, F.; Tomberli, A.; Baldini, K.; Coppini, R.; Tanini, I.; Bardi, S.; Contini, E.; et al. Defining the diagnostic effectiveness of genes for inclusion in panels: The experience of two decades of genetic testing for hypertrophic cardiomyopathy at a single center. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Authors/Task Force Members; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Mademont-Soler, I.; Mates, J.; Yotti, R.; Espinosa, M.A.; Perez-Serra, A.; Fernandez-Avila, A.I.; Coll, M.; Mendez, I.; Iglesias, A.; Del Olmo, B.; et al. Additional value of screening for minor genes and copy number variants in hypertrophic cardiomyopathy. PLoS ONE 2017, 12, e0181465. [Google Scholar] [CrossRef] [PubMed]
- Dos Remedios, C.G.; Lal, S.P.; Li, A.; McNamara, J.; Keogh, A.; Macdonald, P.S.; Cooke, R.; Ehler, E.; Knoll, R.; Marston, S.B.; et al. The Sydney Heart Bank: Improving translational research while eliminating or reducing the use of animal models of human heart disease. Biophys. Rev. 2017, 9, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Ait Mou, Y.; Bollensdorff, C.; Cazorla, O.; Magdi, Y.; de Tombe, P.P. Exploring cardiac biophysical properties. Glob. Cardiol. Sci. Pract. 2015, 2015, 10. [Google Scholar] [PubMed] [Green Version]
- Poggesi, C.; Tesi, C.; Stehle, R. Sarcomeric determinants of striated muscle relaxation kinetics. Pflugers Arch. 2005, 449, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Stehle, R.; Solzin, J.; Iorga, B.; Poggesi, C. Insights into the kinetics of Ca2+-regulated contraction and relaxation from myofibril studies. Pflugers Arch. 2009, 458, 337–357. [Google Scholar] [CrossRef] [PubMed]
- Vikhorev, P.G.; Ferenczi, M.A.; Marston, S.B. Instrumentation to study myofibril mechanics from static to artificial simulations of cardiac cycle. MethodsX 2016, 3, 156–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, B. Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: Implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. USA 1988, 85, 3265–3269. [Google Scholar] [CrossRef] [PubMed]
- Witjas-Paalberends, E.R.; Piroddi, N.; Stam, K.; van Dijk, S.J.; Oliviera, V.S.; Ferrara, C.; Scellini, B.; Hazebroek, M.; ten Cate, F.J.; van Slegtenhorst, M.; et al. Mutations in MYH7 reduce the force generating capacity of sarcomeres in human familial hypertrophic cardiomyopathy. Cardiovasc. Res. 2013, 99, 432–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollen, I.A.E.; van der Meulen, M.; de Goede, K.; Kuster, D.W.D.; Dalinghaus, M.; van der Velden, J. Cardiomyocyte Hypocontractility and Reduced Myofibril Density in End-Stage Pediatric Cardiomyopathy. Front. Physiol. 2017, 8, 1103. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, G.; Zimmermann, R.; Hess, O.M.; Schneider, J.; Kubler, W.; Krayenbuehl, H.P.; Hagl, S.; Mall, G. Decreased concentration of myofibrils and myofiber hypertrophy are structural determinants of impaired left ventricular function in patients with chronic heart diseases: A multiple logistic regression analysis. J. Am. Coll. Cardiol. 1992, 20, 1135–1142. [Google Scholar] [CrossRef]
- Van Dijk, S.J.; Holewijn, R.A.; Tebeest, A.; Dos Remedios, C.; Stienen, G.J.; van der Velden, J. A piece of the human heart: Variance of protein phosphorylation in left ventricular samples from end-stage primary cardiomyopathy patients. J. Muscle Res. Cell Motil. 2009, 30, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yan, L.J. Protein Oxidative Modifications: Beneficial Roles in Disease and Health. J. Biochem. Pharmacol. Res. 2013, 1, 15–26. [Google Scholar] [PubMed]
- Wijnker, P.J.M.; Sequeira, V.; Kuster, D.W.D.; Velden, J.V. Hypertrophic Cardiomyopathy: A Vicious Cycle Triggered by Sarcomere Mutations and Secondary Disease Hits. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Predmore, J.M.; Wang, P.; Davis, F.; Bartolone, S.; Westfall, M.V.; Dyke, D.B.; Pagani, F.; Powell, S.R.; Day, S.M. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 2010, 121, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Ren, J.; Guo, W. Sarcomeric protein isoform transitions in cardiac muscle: A journey to heart failure. Biochim. Biophys. Acta 2015, 1852, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, A.C.; Jacques, A.; Bardswell, S.C.; McKenna, W.J.; Tsang, V.; dos Remedios, C.G.; Ehler, E.; Adams, K.; Jalilzadeh, S.; Avkiran, M.; et al. Normal passive viscoelasticity but abnormal myofibrillar force generation in human hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 49, 737–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marston, S.; Copeland, O.; Jacques, A.; Livesey, K.; Tsang, V.; McKenna, W.J.; Jalilzadeh, S.; Carballo, S.; Redwood, C.; Watkins, H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ. Res. 2009, 105, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, V.; Wijnker, P.J.; Nijenkamp, L.L.; Kuster, D.W.; Najafi, A.; Witjas-Paalberends, E.R.; Regan, J.A.; Boontje, N.; Ten Cate, F.J.; Germans, T.; et al. Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ. Res. 2013, 112, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, S.J.; Paalberends, E.R.; Najafi, A.; Michels, M.; Sadayappan, S.; Carrier, L.; Boontje, N.M.; Kuster, D.W.; van Slegtenhorst, M.; Dooijes, D.; et al. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ. Heart Fail. 2012, 5, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Belus, A.; Piroddi, N.; Scellini, B.; Tesi, C.; D’Amati, G.; Girolami, F.; Yacoub, M.; Cecchi, F.; Olivotto, I.; Poggesi, C. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J. Physiol. 2008, 586, 3639–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piroddi, N.; Belus, A.; Scellini, B.; Tesi, C.; Giunti, G.; Cerbai, E.; Mugelli, A.; Poggesi, C. Tension generation and relaxation in single myofibrils from human atrial and ventricular myocardium. Pflugers Arch. 2007, 454, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Witjas-Paalberends, E.R.; Ferrara, C.; Scellini, B.; Piroddi, N.; Montag, J.; Tesi, C.; Stienen, G.J.; Michels, M.; Ho, C.Y.; Kraft, T.; et al. Faster cross-bridge detachment and increased tension cost in human hypertrophic cardiomyopathy with the R403Q MYH7 mutation. J. Physiol. 2014, 592, 3257–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montag, J.; Kowalski, K.; Makul, M.; Ernstberger, P.; Radocaj, A.; Beck, J.; Becker, E.; Tripathi, S.; Keyser, B.; Muhlfeld, C.; et al. Burst-Like Transcription of Mutant and Wildtype MYH7-Alleles as Possible Origin of Cell-to-Cell Contractile Imbalance in Hypertrophic Cardiomyopathy. Front. Physiol. 2018, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Kraft, T.; Witjas-Paalberends, E.R.; Boontje, N.M.; Tripathi, S.; Brandis, A.; Montag, J.; Hodgkinson, J.L.; Francino, A.; Navarro-Lopez, F.; Brenner, B.; et al. Familial hypertrophic cardiomyopathy: Functional effects of myosin mutation R723G in cardiomyocytes. J. Mol. Cell. Cardiol. 2013, 57, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Schultz, I.; Becker, E.; Montag, J.; Borchert, B.; Francino, A.; Navarro-Lopez, F.; Perrot, A.; Ozcelik, C.; Osterziel, K.J.; et al. Unequal allelic expression of wild-type and mutated beta-myosin in familial hypertrophic cardiomyopathy. Basic Res. Cardiol. 2011, 106, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Montag, J.; Syring, M.; Rose, J.; Weber, A.L.; Ernstberger, P.; Mayer, A.K.; Becker, E.; Keyser, B.; Dos Remedios, C.; Perrot, A.; et al. Intrinsic MYH7 expression regulation contributes to tissue level allelic imbalance in hypertrophic cardiomyopathy. J. Muscle Res. Cell Motil. 2017, 38, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vikhorev, P.G.; Smoktunowicz, N.; Munster, A.B.; Copeland, O.; Kostin, S.; Montgiraud, C.; Messer, A.E.; Toliat, M.R.; Li, A.; Dos Remedios, C.G.; et al. Abnormal contractility in human heart myofibrils from patients with dilated cardiomyopathy due to mutations in TTN and contractile protein genes. Sci. Rep. 2017, 7, 14829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Spaendonck-Zwarts, K.Y.; Posafalvi, A.; van den Berg, M.P.; Hilfiker-Kleiner, D.; Bollen, I.A.; Sliwa, K.; Alders, M.; Almomani, R.; van Langen, I.M.; van der Meer, P.; et al. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur. Heart J. 2014, 35, 2165–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollen, I.A.E.; Schuldt, M.; Harakalova, M.; Vink, A.; Asselbergs, F.W.; Pinto, J.R.; Kruger, M.; Kuster, D.W.D.; van der Velden, J. Genotype-specific pathogenic effects in human dilated cardiomyopathy. J. Physiol. 2017, 595, 4677–4693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoorntje, E.T.; Bollen, I.A.; Barge-Schaapveld, D.Q.; van Tienen, F.H.; Te Meerman, G.J.; Jansweijer, J.A.; van Essen, A.J.; Volders, P.G.; Constantinescu, A.A.; van den Akker, P.C.; et al. Lamin A/C-Related Cardiac Disease: Late Onset With a Variable and Mild Phenotype in a Large Cohort of Patients With the Lamin A/C p.(Arg331Gln) Founder Mutation. Circ. Cardiovasc. Genet. 2017, 10, e001631. [Google Scholar] [CrossRef] [PubMed]
- Beqqali, A.; Bollen, I.A.; Rasmussen, T.B.; van den Hoogenhof, M.M.; van Deutekom, H.W.; Schafer, S.; Haas, J.; Meder, B.; Sorensen, K.E.; van Oort, R.J.; et al. A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc. Res. 2016, 112, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Dyer, E.C.; Jacques, A.M.; Hoskins, A.C.; Ward, D.G.; Gallon, C.E.; Messer, A.E.; Kaski, J.P.; Burch, M.; Kentish, J.C.; Marston, S.B. Functional analysis of a unique troponin c mutation, GLY159ASP, that causes familial dilated cardiomyopathy, studied in explanted heart muscle. Circ. Heart Fail. 2009, 2, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Vallins, W.J.; Brand, N.J.; Dabhade, N.; Butler-Browne, G.; Yacoub, M.H.; Barton, P.J. Molecular cloning of human cardiac troponin I using polymerase chain reaction. FEBS Lett. 1990, 270, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, R.R.; Herron, T.; Simmons, R.; Shore, D.; Kumar, P.; Sethia, B.; Chua, F.; Vassiliadis, E.; Kentish, J.C. Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circulation 2010, 121, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, J.; Papp, Z.; Zaremba, R.; Boontje, N.M.; de Jong, J.W.; Owen, V.J.; Burton, P.B.; Goldmann, P.; Jaquet, K.; Stienen, G.J. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc. Res. 2003, 57, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Bollen, I.A.E.; Ehler, E.; Fleischanderl, K.; Bouwman, F.; Kempers, L.; Ricke-Hoch, M.; Hilfiker-Kleiner, D.; Dos Remedios, C.G.; Kruger, M.; Vink, A.; et al. Myofilament Remodeling and Function Is More Impaired in Peripartum Cardiomyopathy Compared with Dilated Cardiomyopathy and Ischemic Heart Disease. Am. J. Pathol. 2017, 187, 2645–2658. [Google Scholar] [CrossRef] [PubMed]
- Kooij, V.; Saes, M.; Jaquet, K.; Zaremba, R.; Foster, D.B.; Murphy, A.M.; Dos Remedios, C.; van der Velden, J.; Stienen, G.J. Effect of troponin I Ser23/24 phosphorylation on Ca2+-sensitivity in human myocardium depends on the phosphorylation background. J. Mol. Cell. Cardiol. 2010, 48, 954–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotter, S.; Gout, L.; Von Frieling-Salewsky, M.; Muller, A.E.; Helling, S.; Marcus, K.; Dos Remedios, C.; Linke, W.A.; Kruger, M. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc. Res. 2013, 99, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp, Z.; van der Velden, J.; Borbely, A.; Edes, I.; Stienen, G.J.M. Altered myocardial force generation in end-stage human heart failure. ESC Heart Fail. 2014, 1, 160–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamidi, R.; Li, J.; Gresham, K.S.; Verma, S.; Doh, C.Y.; Li, A.; Lal, S.; Dos Remedios, C.G.; Stelzer, J.E. Dose-Dependent Effects of the Myosin Activator Omecamtiv Mecarbil on Cross-Bridge Behavior and Force Generation in Failing Human Myocardium. Circ. Heart Fail. 2017, 10, e004257. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.Y.; Lin, Y.H.; Wennersten, S.A.; Demos-Davies, K.M.; Cavasin, M.A.; Mahaffey, J.H.; Monzani, V.; Saripalli, C.; Mascagni, P.; Reece, T.B.; et al. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.; Kawai, M.; Minai, K.; Ogawa, K.; Ogawa, T.; Yoshimura, M. Associations between Left Ventricular Cavity Size and Cardiac Function and Overload Determined by Natriuretic Peptide Levels and a Covariance Structure Analysis. Sci. Rep. 2017, 7, 2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinstein, E.; Gutierrez-Fernandez, A.; Tzur, S.; Bormans, C.; Marcu, S.; Tayeb-Fligelman, E.; Vinkler, C.; Raas-Rothschild, A.; Irge, D.; Landau, M.; et al. Congenital dilated cardiomyopathy caused by biallelic mutations in Filamin C. Eur. J. Hum. Genet. 2016, 24, 1792–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messer, A.E.; Gallon, C.E.; McKenna, W.J.; Dos Remedios, C.G.; Marston, S.B. The use of phosphate-affinity SDS-PAGE to measure the cardiac troponin I phosphorylation site distribution in human heart muscle. Proteom. Clin. Appl. 2009, 3, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Bodor, G.S.; Oakeley, A.E.; Allen, P.D.; Crimmins, D.L.; Ladenson, J.H.; Anderson, P.A. Troponin I phosphorylation in the normal and failing adult human heart. Circulation 1997, 96, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Dyer, E.; Stuckey, D.J.; Copeland, O.; Leung, M.C.; Bayliss, C.; Messer, A.; Wilkinson, R.; Tremoleda, J.L.; Schneider, M.D.; et al. Molecular mechanism of the E99K mutation in cardiac actin (ACTC Gene) that causes apical hypertrophy in man and mouse. J. Biol. Chem. 2011, 286, 27582–27593. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Kooij, V.; van Dijk, S.; Merkus, D.; Paulus, W.J.; Remedios, C.D.; Duncker, D.J.; Stienen, G.J.; van der Velden, J. Sarcomeric dysfunction in heart failure. Cardiovasc. Res. 2008, 77, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Copeland, O.; Sadayappan, S.; Messer, A.E.; Steinen, G.J.; van der Velden, J.; Marston, S.B. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J. Mol. Cell. Cardiol. 2010, 49, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Jacques, A.M.; Briceno, N.; Messer, A.E.; Gallon, C.E.; Jalilzadeh, S.; Garcia, E.; Kikonda-Kanda, G.; Goddard, J.; Harding, S.E.; Watkins, H.; et al. The molecular phenotype of human cardiac myosin associated with hypertrophic obstructive cardiomyopathy. Cardiovasc. Res. 2008, 79, 481–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadaki, M.; Vikhorev, P.G.; Marston, S.B.; Messer, A.E. Uncoupling of myofilament Ca2+ sensitivity from troponin I phosphorylation by mutations can be reversed by epigallocatechin-3-gallate. Cardiovasc. Res. 2015, 108, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Messer, A.E.; Bayliss, C.R.; El-Mezgueldi, M.; Redwood, C.S.; Ward, D.G.; Leung, M.C.; Papadaki, M.; Dos Remedios, C.; Marston, S.B. Mutations in troponin T associated with Hypertrophic Cardiomyopathy increase Ca(2+)-sensitivity and suppress the modulation of Ca(2+)-sensitivity by troponin I phosphorylation. Arch. Biochem. Biophys. 2016, 601, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Messer, A.E.; Chan, J.; Daley, A.; Copeland, O.; Marston, S.B.; Connolly, D.J. Investigations into the Sarcomeric Protein and Ca(2+)-Regulation Abnormalities Underlying Hypertrophic Cardiomyopathy in Cats (Felix catus). Front. Physiol. 2017, 8, 348. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, C.R.; Jacques, A.M.; Leung, M.C.; Ward, D.G.; Redwood, C.S.; Gallon, C.E.; Copeland, O.; McKenna, W.J.; Dos Remedios, C.; Marston, S.B.; et al. Myofibrillar Ca(2+) sensitivity is uncoupled from troponin I phosphorylation in hypertrophic obstructive cardiomyopathy due to abnormal troponin T. Cardiovasc. Res. 2013, 97, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Vikhorev, P.G.; Song, W.; Wilkinson, R.; Copeland, O.; Messer, A.E.; Ferenczi, M.A.; Marston, S.B. The dilated cardiomyopathy-causing mutation ACTC E361G in cardiac muscle myofibrils specifically abolishes modulation of Ca(2+) regulation by phosphorylation of troponin I. Biophys. J. 2014, 107, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Biesiadecki, B.J.; Kobayashi, T.; Walker, J.S.; Solaro, R.J.; de Tombe, P.P. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ. Res. 2007, 100, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Messer, A.E.; Marston, S.B. Investigating the role of uncoupling of troponin I phosphorylation from changes in myofibrillar Ca(2+)-sensitivity in the pathogenesis of cardiomyopathy. Front. Physiol. 2014, 5, 315. [Google Scholar] [CrossRef] [PubMed]
- Edes, I.F.; Czuriga, D.; Csanyi, G.; Chlopicki, S.; Recchia, F.A.; Borbely, A.; Galajda, Z.; Edes, I.; van der Velden, J.; Stienen, G.J.; et al. Rate of tension redevelopment is not modulated by sarcomere length in permeabilized human, murine, and porcine cardiomyocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R20–R29. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.W.; Li, A.; Lal, S.; Bos, J.M.; Harris, S.P.; van der Velden, J.; Ackerman, M.J.; Cooke, R.; Dos Remedios, C.G. MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLoS ONE 2017, 12, e0180064. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, S.J.; Dooijes, D.; dos Remedios, C.; Michels, M.; Lamers, J.M.; Winegrad, S.; Schlossarek, S.; Carrier, L.; ten Cate, F.J.; Stienen, G.J.; et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009, 119, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Rottbauer, W.; Gautel, M.; Zehelein, J.; Labeit, S.; Franz, W.M.; Fischer, C.; Vollrath, B.; Mall, G.; Dietz, R.; Kubler, W.; et al. Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy. Characterization Of cardiac transcript and protein. J. Clin. Investig. 1997, 100, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Gilda, J.E.; Gomes, A.V. Proteasome dysfunction in cardiomyopathies. J. Physiol. 2017, 595, 4051–4071. [Google Scholar] [CrossRef] [PubMed]
- Makarenko, I.; Opitz, C.A.; Leake, M.C.; Neagoe, C.; Kulke, M.; Gwathmey, J.K.; del Monte, F.; Hajjar, R.J.; Linke, W.A. Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ. Res. 2004, 95, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Nagueh, S.F.; Shah, G.; Wu, Y.; Torre-Amione, G.; King, N.M.; Lahmers, S.; Witt, C.C.; Becker, K.; Labeit, S.; Granzier, H.L. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004, 110, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270ra6. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.J.; Winslow, R.L.; Hunter, W.C. Comparison of putative cooperative mechanisms in cardiac muscle: Length dependence and dynamic responses. Am. J. Physiol. 1999, 276, H1734–H1754. [Google Scholar] [CrossRef] [PubMed]
- Beuckelmann, D.J.; Nabauer, M.; Erdmann, E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 1992, 85, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, J.K.; Slawsky, M.T.; Hajjar, R.J.; Briggs, G.M.; Morgan, J.P. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J. Clin. Investig. 1990, 85, 1599–1613. [Google Scholar] [CrossRef] [PubMed]
- Bayer, J.D.; Narayan, S.M.; Lalani, G.G.; Trayanova, N.A. Rate-dependent action potential alternans in human heart failure implicates abnormal intracellular calcium handling. Heart Rhythm 2010, 7, 1093–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewey, S.; Xu, Q.; Gomes, A. Static and dynamic properties of the HCM myocardium. J. Mol. Cell. Cardiol. 2010, 49, 715–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poggesi, C.; Ho, C.Y. Muscle dysfunction in hypertrophic cardiomyopathy: What is needed to move to translation? J. Muscle Res. Cell. Motil. 2014, 35, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation, Gene Expression and Mutated (mut) Allele Expression | Phosphorylation Level (Cardiomyopathy/Healthy) | Titin Isoforms and Passive Stiffness (Cardiomyopathy/Healthy) | Maximal Force and Myofibril Density (Cardiomyopathy/Healthy) | Contractile Kinetics Parameters (Cardiomyopathy/Healthy) | Ca2-Sensitivity of Force (Cardiomyopathy/Healthy) | Length Dependent Activation Changes upon Stretch (Cardiomyopathy/Healthy) | Effect PKA on EC50 (in µM) | Patient Information (Sex, Age and Number of Patients) | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expression | Myocyte | Myofibril | Activation | Relaxation | +PKA | +PKA | ||||||||||||||||||||||||
| Diagnosis | Gene with Mutation | Mutation and Type: Truncated (T) or Not (NT) | Gene | Mut Allele | TnI | MyBP-C | LC2 | N2AB/N2B | Passive Stiffness | Fmax | Myofibril Density | Fmax | kACT | kTR | tLIN | kLIN | kREL | EC50 | nH | EC50 | ΔEC50 | ΔF | ΔEC50 | ΔF | ΔEC50 | Sex M/F | Age | N | Refs | |
| HCM | MYBPC3 | g.T2604A+C del at 2605 | T | 0.64 * | 0.34 * | 0.64 | 1 | 1.14 | 0.42 | M | 42 | 1 | [65,66] | |||||||||||||||||
| HCM | MYBPC3 | T | 0.18 | 0.75 | 0.85 * | 0.79 * | 0.96 | 0.57 * | 1.16 | 1.01 | 0.94 | 0.88 * | M/F | 22–69 | 17 | [67] | ||||||||||||||
| HCM | MYBPC3 | T | 0.67 | 0.69 * | 0.82 | 1.46 | 0.71 * | 0.83 * | 0.84 * | 1 | 0.61 * | 1.04 | 1.13 | M/F | 22–69 | 17 | [68] | |||||||||||||
| HCM | MYBPC3 | T/NT | 0.68 * | 0.69 * | 0.85 * | 1 | 1 | MF | 32–69 | 28 | [57] | |||||||||||||||||||
| HCM | MYBPC3 | p.R502W | NT | 0.8 * | 0.75 | 1.10 | 0.35 | M | 23 | 1 | [65] | |||||||||||||||||||
| HCM | MYBPC3 | NT | 0.09 | 0.64 | 0.44 * | 0.56 * | 0.93 | 0.29 * | 0.28 * | 0.18 * | 0.25 * | 1.45 * | M/F | 31–51 | 4 | [67] | ||||||||||||||
| HCM | MYH7 | p.R719Q | NT | 0.79 | 1.16 | 1.01 | F | 27 | 1 | [65] | ||||||||||||||||||||
| HCM | MYH7 | NT | 0.22 | 0.58 | 0.54 * | 0.56 * | 0.88 * | 0.26 * | 0.44 * | 0.42 * | 0.38 * | 0.97 * | M/F | 25–61 | 4–6 | [67] | ||||||||||||||
| fHCM | MYH7 | p.R403Q | NT | 0.35 * | 0.93 | 0.3 | 0.56 * | 1.92 * | 2.02 * | 0.75 * | 4 * | 1.76 * | M | 24 | 1 | [69,70,71] | ||||||||||||||
| fHCM | MYH7 | p.R403Q | NT | 0.5 | 0.6 | 0.83 | 1.84 | 2.9 | 1.77 | M | 35 | 1 | [69,70,71] | |||||||||||||||||
| fHCM | MYH7 | p.R403Q | NT | 0.45 | 0.6 | 0.91 | 1.52 | 2.7 | 1.99 | F | 39 | 1 | [69,70,71] | |||||||||||||||||
| fHCM | MYH7 | p.R723G | NT | 0.7 | 0.54 * | 1.41 * | M | 38, 55 | 2 | [72] | ||||||||||||||||||||
| fHCM | MYH7 | p.R723G | NT | 0.68 | 0.11 * | 0.42 * | 0.42 * | 0.72 * | 0.69 * | 0.74 * | 0.93 | 1 | 1.28 * | 1.44 * | M/F | 53, 55 | 2 | [73,74] | ||||||||||||
| fHCM | MYH7 | p.A200V | NT | 0.53 | 0.48 * | 1.12 * | F | 19 | 1 | [72,75] | ||||||||||||||||||||
| fHCM | MYH7 | NT | 0.39 * | 0.79 * | 0.65 * | 1 | M/F | 19–61 | 11 | [57] | ||||||||||||||||||||
| fHCM | TPM1 | p.I284V | NT | 0.40 * | 0.70 * | 0.62 * | M | 65 | 1 | [57] | ||||||||||||||||||||
| HCM | TPM1 | p.M281T | NT | 0.07 | 0.55 | 0.48 * | 0.67 * | 0.95 | 0.39 * | 0.72 | −0.2 * | 0.06 * | 1.08 * | M | 65 | 1 | [67] | |||||||||||||
| HCM | TNNT2 | p.K280N | NT | 0.96 | 1.07 | 0.70 | 0.80 * | 0.76 * | 0.45 * | 0.34 | 0.30 * | 0.15 * | 0.12 | M | 26 | 1 | [67] | |||||||||||||
| HCM | TNNT2 | p.K280N | NT | 0.56 | 0.78 * | 0.73 * | M | 26 | 2 | [57] | ||||||||||||||||||||
| HCM | TNNI3 | p.R145W | NT | 0.55 * | 0.58 * | 0.84 | M | 46, 66 | 2 | [57] | ||||||||||||||||||||
| HCM | TNNI3 | p.R145W | NT | 0.09 | 0.49 | 0.29 | 0.88 * | 1.18 * | 0.38 * | 0.03 * | 0.34 * | 0.08 * | 0.99 * | M | 46, 66 | 2 | [67] | |||||||||||||
| HCM | MYH7(2) MYBPC3(1) | NT | 0.25 * | 0.14 * | 0.91 | 0.70 | 0.85 | 0.61 * | 1 | 1.1 * | 0.49 * | 1.26 | M/F | 23–61 | 6 | [65,70] | ||||||||||||||
| HCM | none | 0.18 | 0.4 | 0.81 * | 0.86 * | 0.93 | 0.49 * | 1.2 | 1.06 * | 1.2 | 0.73 | M/F | 46–75 | 3–7 | [67] | |||||||||||||||
| HCM | none | 1 | 0.71 * | 0.92 * | 0.78 * | 1 | M/F | 35–75 | 3–14 | [57] | ||||||||||||||||||||
| HCM | none | 0.60 * | 0.54 * | 1 | 0.75 * | 0.8 * | 0.81 * | 1 | 0.49 * | 0.94 | 0.97 | M/F | 46–75 | 11 | [68] | |||||||||||||||
| HCM | none | 1.0 | 0.84 | 1.01 | 2.1 | 1.59 | M/F | 35–72 | 9 | [71] | ||||||||||||||||||||
| HCM | none | 0.61 | 1.09 | 0.54 | M | 59 | 1 | [65] | ||||||||||||||||||||||
| HCM | none | 0.35 | 1.05 | 0.45 | F | 61 | 1 | [65] | ||||||||||||||||||||||
| HCM | none | 0.42 | 1.12 | 0.49 | M | 58 | 1 | [65] | ||||||||||||||||||||||
| Mutation, Gene Expression and Mutated (mut) Allele Expression | Phosphorylation Level (Cardiomyopathy/Healthy) | Titin Isoforms and Passive Stiffness (Cardiomyopathy/Healthy | Maximal Force and Myofibril Density (Cardiomyopathy/Healthy) | Contractile Kinetics Parameters (Cardiomyopathy/Healthy) | Ca2-Sensitivity of Force (Cardiomyopathy/Healthy) | Length Dependent Activation Changes upon Stretch (Cardiomyopathy/Healthy) | Effect PKA on EC50 (in µM) | Patient Information (Sex, Age and Number of Patients) | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expression | Myocyte | Myofibril | Activation | Relaxation | +PKA | +PKA | |||||||||||||||||||||||
| Diagnosis | Gene with Mutation | Mutation and Type: Truncated (T) or Not (NT) | Gene | Mut Protein | TnI | MyBP-C | LC2 | N2AB/ N2B | Passive Stiffness | Fmax | Myofibril Density | Fmax | kACT | kTR | tLIN | kLIN | kREL | EC50 | nH | EC50 | ΔEC50 | ΔF | ΔEC50 | ΔEC50 | Sex M/F | Age | N | Refs | |
| IDCM | TTN | p.(R23464Tfs *41) | T | 1.0 | 0 | 1.05 | 0.63 * | 1.02 | 1.05 | 1.06 | 0.97 | 1.06 | 0.91 | 1.00 | M | 22 | 1 | [76] | |||||||||||
| fDCM | TTN | p.(R23464Tfs*41) | T | 1.0 | 0 | 1.11 | 0.66 * | 0.96 | 1.12 | 0.95 | 0.98 | 0.18 | 1.13 | 1.13 | M | 37 | 1 | [76] | |||||||||||
| fDCM | TTN | p.(Y18923 *) | T | 0.96 | 0 | 1.25 | 0.62 * | 1.04 | 1.15 | 1.16 * | 0.98 | 1.06 | 1.17 | 1.03 | F | 22 | 1 | [76] | |||||||||||
| PPCM | TTN | p.(K15664Vfs*13) | T | 1.85 | 0.4 | F | 1 | [77] | |||||||||||||||||||||
| DCM | LMNA | p.(R331Q) | NT | 0.5 | 1.98 * | 1 | 0.64 * | 0.63 | 0.86 * | 1 | 0.78 | M/F | 45.3 | 3 | [78,79] | ||||||||||||||
| fDCM | RMB20 | p.E913K | NT | 0.13 | 13.89 | 0.8 | 0.85 | 0.67 * | 0.58 | 1.13 | 0.36 | 0.81 | 1.77 | M | 19 | 1 | [80] | ||||||||||||
| fDCM | TNNC1 | p.G159D | NT | 1.13 | 0.49 | 0.13 | 0.89 | 0.59 * | 1 | 1.20 | 0.97 | 0.97 | 0.71 * | 0.35 | 1.17 | 0.60 * | 0.59 * | 1.22 | M | 3 | 1 | [76,81] | |||||||
| fDCM | TNNI3 | p.K36Q | NT | 1.0 | 0.58 * | 1.10 | 1.17 | 1.05 | 0.70 * | 1.39 | 1.30 * | 1.34 | M | 15 | 1 | [76] | |||||||||||||
| IDCM | TNNI3 | p.(R98 *) | T | 0.39 | 0 | 1.08 | 1.56 | 1 | 0.95 | 0.94 * | 0.88 * | 0.66 | M | 46 | 1 | [78,82] | |||||||||||||
| IDCM | TNNT2 | p.(K217del) | NT | 0.51 | 1.08 | 1.64 | 1.3 * | 1.11 | 1.07 | 0.97 | M | 19 | 1 | [78] | |||||||||||||||
| fDCM | MYH7 | p.E1426K | NT | 1.08 | 0.65 * | 0.97 | 1.13 | 1.19 * | 0.70 * | 0.76 | 1.35 * | 0.96 | M | 43 | 1 | [76] | |||||||||||||
| IDCM | not stated | 0.54 | 1.98 | 1 | 0.98 | 0.94 | 1 | 0.73 | M/F | 54.6 | 5 | [78] | |||||||||||||||||
| DCM | not stated | 1.24 * | 0.27 * | 51–56 | 3–4 | [83] | |||||||||||||||||||||||
| DCM | not stated | 0.5 | 0.51 | 1 | 0.96 | 3.37 | M | 45,59 | 2 | [84] | |||||||||||||||||||
| IDCM | not stated | 0.35 * | 0.58 * | 1.62 * | 1 | 0.94 | 0.69 * | 1.12 | 0.76 | 1.09 | 8 | [85] | |||||||||||||||||
| IDCM | not stated | 0.4 * | 0.4 * | 1.1 | 1.18 | 0.88 | 0.67 | 1.33 | 0.8 | 1.26 | M/F | 52–62 | 7 | [86] | |||||||||||||||
| IDCM | not stated | 1.78 | 1.2 | 1.34 | 0.68 * | 0.81 | 0.87 | 0.59 | 0.86 | 1.33 | 3 | [80] | |||||||||||||||||
| IDCM | not stated | 1.14 | F | 41–57 | 6 | [87] | |||||||||||||||||||||||
| PPCM | not stated | 0.24 * | 0.61 * | 1.58 | 1.39 * | 0.84 | 0.65 * | 1.01 | 0.48 * | 0.96 | F | 6 | [85] | ||||||||||||||||
| ICM | not stated | 0.95 | 0.93 | 0.71 * | 0.83 * | 3 | [88] | ||||||||||||||||||||||
| ICM | not stated | 0.5 | 0.72 * | 0.98 | 2.13 | M/F | 41–65 | 7 | [84] | ||||||||||||||||||||
| ICM | not stated | 0.5 | 0.5 * | 0.83 | 1 | 1.04 | 0.70 * | 1.11 | 0.96 | 1.19 | 4 | [85] | |||||||||||||||||
| ICM (7) DCM (2) | not stated | 0.5 | 0.57 * | 1.06 | 1.0 | 0.66 * | 0.99 | 0.98 | 2.55 * | M/F | 41–65 | 10 | [84] | ||||||||||||||||
| HF | not stated | 0.33 * | 0.65 * | 1 | 0.53 * | 0.53 * | 0.96 | 0.63 * | 0.87 | 0.98 | M/F | 42–57 | 4 | [89] | |||||||||||||||
| IRCM | not stated | 0.84 | 1.20 * | 0.05 * | 0.52 * | F | 66 | 1 | [90] | ||||||||||||||||||||
| IRCM | not stated | 1.16 | 1.92 * | 0.07 * | 0.33 * | M | 21 | 1 | [90] | ||||||||||||||||||||
| IDCM (9) NCC (2) | not stated | 0.27 * | 0.4–2.8 | 0.7 * | 0.64 * | 0.67 * | 1 | <18 | 11 | [58] | |||||||||||||||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vikhorev, P.G.; Vikhoreva, N.N. Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle. Int. J. Mol. Sci. 2018, 19, 2234. https://doi.org/10.3390/ijms19082234
Vikhorev PG, Vikhoreva NN. Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle. International Journal of Molecular Sciences. 2018; 19(8):2234. https://doi.org/10.3390/ijms19082234
Chicago/Turabian StyleVikhorev, Petr G., and Natalia N. Vikhoreva. 2018. "Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle" International Journal of Molecular Sciences 19, no. 8: 2234. https://doi.org/10.3390/ijms19082234
APA StyleVikhorev, P. G., & Vikhoreva, N. N. (2018). Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle. International Journal of Molecular Sciences, 19(8), 2234. https://doi.org/10.3390/ijms19082234