mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders
by
 , , , and
, , , and
Larisa Ryskalin
1 ,
,
Fiona Limanaqi
1,
Alessandro Frati
2,
Carla L. Busceti
2 and
Francesco Fornai
1,2,* 1
Human Anatomy, Department of Translational Research and New Technologies in Medicine and Surgery, University of Pisa, Via Roma 55, 56126 Pisa, Italy
2
I.R.C.C.S. Neuromed, Via Atinense 18, 86077 Isernia, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(8), 2226; https://doi.org/10.3390/ijms19082226
Submission received: 4 July 2018
/
Revised: 26 July 2018
/
Accepted: 27 July 2018
/
Published: 30 July 2018
(This article belongs to the Special Issue mTOR in Human Diseases)
Abstract
:The mammalian target of rapamycin (mTOR) is an ubiquitously expressed serine-threonine kinase, which senses and integrates several intracellular and environmental cues to orchestrate major processes such as cell growth and metabolism. Altered mTOR signalling is associated with brain malformation and neurological disorders. Emerging evidence indicates that even subtle defects in the mTOR pathway may produce severe effects, which are evident as neurological and psychiatric disorders. On the other hand, administration of mTOR inhibitors may be beneficial for a variety of neuropsychiatric alterations encompassing neurodegeneration, brain tumors, brain ischemia, epilepsy, autism, mood disorders, drugs of abuse, and schizophrenia. mTOR has been widely implicated in synaptic plasticity and autophagy activation. This review addresses the role of mTOR-dependent autophagy dysfunction in a variety of neuropsychiatric disorders, to focus mainly on psychiatric syndromes including schizophrenia and drug addiction. For instance, amphetamines-induced addiction fairly overlaps with some neuropsychiatric disorders including neurodegeneration and schizophrenia. For this reason, in the present review, a special emphasis is placed on the role of mTOR on methamphetamine-induced brain alterations.
1. Introduction
The discovery of the mammalian target of rapamycin (mTOR) dates back to early 1970s with the collection of a soil sample of Easter Island (Rapa Nui) and the serendipitous identification of a lipophilic macrolide produced by the soil bacterium Streptomyces hygroscopicus [1]. This natural compound called rapamycin was initially developed as an antifungal drug, but it soon raised considerable interest because of its unexpected and, at that time, undesired immunosuppressive side effects. The discovery of rapamycin-mediated anti-proliferative effects on immune cells was a milestone in organ transplantation [2,3,4,5,6]. However, the finding of anti-proliferative activities, way beyond the immunosuppressive properties, disclosed novel potential therapeutic uses that fueled research on its mechanisms of action [7,8,9,10]. Nowadays, it is well established that rapamycin exerts its effects by forming a complex with FK506-binding protein 12 (FKBP12), which in turn inhibits the target of rapamycin (TOR). TOR is a large (289 kDa), evolutionarily highly conserved, serine/threonine kinase, which represents the catalytic domain of a multiprotein complex named TORC (target of rapamycin complex) [5,11,12,13]. The need of FKBP-12 to mediate the effects of rapamycin on TORC is demonstrated by a lack of effects induced by rapamycin when the binding between FKBP12 and rapamycin is occluded by a missense point mutations within the FRB domain of TOR [14,15]. In mammals, TOR kinase, also known as mTOR (i.e., mammalian TOR), is ubiquitously expressed in all cell types and it assembles with several scaffolds and regulatory subunits to form two distinct multiprotein complexes, hereinafter referred as mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [16,17,18,19]. These mTOR complexes share four components that are identical; they possess (i) a catalytic subunit, along with (ii) a small protein known as mLSt8, which represent the core of both complexes. These in turn are composed of two more components, namely (iii) the Tti1/Tel2 associated regulatory proteins, which create a scaffold for recruitment of substrates; and (iv) the negative regulator Deptor, which inhibits the substrate binding [20,21,22]. In addition, there are specific subunits depending on which mTOR complex is considered. In detail, mTORC1 contains the scaffold protein Raptor and the inhibitory subunit PRAS40 as key components, while mTORC2 specifically associates with the regulatory subunit Protor 1/2 and scaffold proteins Rictor and mSIN1, which help the complex assembly [20,23,24,25,26,27,28]. The mTOR complex represents a downstream substrate of the PI3K/PTEN/Akt pathway, which controls cell growth, proliferation, metabolism, and motility in response to bioenergetics and nutritional requests [29,30]. Extracellular and environmental stimuli are conveyed through mTOR via the PI3K/PTEN/Akt pathway. The binding of insulin and growth factors to tyrosine kinase receptors (RTKs) activates the lipid kinase PI3K, which phosphorylates phosphatidylinositol-4,5-phosphate (PIP2) to generate phosphatidylinositol-3,4,5-phosphate (PIP3). This second messenger recruits Akt, which promotes mTOR activity. This occurs via the phosphorylation of the tuberous sclerosis complex (TSC), which impairs its inhibitory activity on mTOR. TSC is a heterodimer that is composed of hamartin (TSC1) and tuberin (TSC2). In this way, the activation of PI3K/Akt signaling leads to the activation of mTOR through TSC inhibition. Once activated, mTOR promotes various activities including protein synthesis, ribosome, and lipid biogenesis [21,31] (Figure 1). Among these activities, mTOR inhibits autophagy [32]. In mammalian cells, three main autophagy pathways are described, all providing the lysosomal degradation of intracellular components: (i) micro-autophagy; (ii) macro-autophagy; and (iii) chaperone-mediated autophagy (CMA). In detail, micro-autophagy enwraps small portions of cytosol and proteins into lysosomes [33], while macro-autophagy sequesters “in bulk” cytosolic cargoes, including organelles, within autophagosomes to merge with lysosomes [34]. Finally, CMA is a rather selective process where proteins are bound to cytosolic chaperones (e.g., LAMP-2) to be recognized and translocated across the lysosomal membrane for degradation [35]. In the present review, we focus on macro-autophagy (hereinafter referred to as autophagy), which is mostly related to mTOR activity. In detail, mTOR inhibits autophagy by suppressing the ULK1 complex, which consists of several autophagy-related proteins (ULK1, Atg13, FIP200). In fact, by phosphorylating the ULK1 complex, mTOR inhibits early steps in the biogenesis of autophagosomes [36,37,38,39] (Figure 1). Conversely, rapamycin-induced mTOR inhibition strongly activates autophagy. In eukaryotic cells, autophagy represents the main cell clearing system. The autophagy pathway is initiated through a nascent double-layered membrane vacuole, which, at early stages, is not yet complete and is named phagophore. The maturation of the phagophore leads to seal the vacuole, which is then named autophagosome. At this stage, the vacuole stains for beclin-1 (the ortholog of yeast Atg6) and LC3 (Atg8), which are thus considered gold standard autophagy markers [40]. The autophagosome carries a variety of substrates to the lysosomal compartment, which possesses a rich enzymatic activity. In detail, when the autophagosome merges with the lysosome, the catalytic organelle autophagolysosome is generated, where degradation and recycling of sequestered cytosolic cargoes occurs. Autophagy can be tuned very finely to obtain either slight or robust effects based upon specific cell needs. For instance, autophagy is strongly induced by nutrient depletion, which occurs during cell starvation. In these conditions, enhanced protein degradation within lysosomes results in the generation of single amino acids. This is the prototype for extreme cell conditions when cell survival is jeopardized; however, a slight autophagy activation is needed in baseline conditions to keep steady the level of misfolded proteins that naturally occur within a living cell. An appropriate tuning of autophagy avoids the burden of aged structures within the cell. Along with degrading misfolded proteins, autophagy degrades altered subcellular organelles (mitochondria, endoplasmic reticulum, ribosomes, and even synaptic vesicles). Thus, when a mild failure in the autophagy pathway occurs, the cell still survives, though such a decreased protein turnover during prolonged time intervals is detrimental. In fact, this may alter a variety of cell activities and may even produce toxicity. This is why it takes time to appreciate the effects of a slight relenting of protein and organelle turnover due to a mild autophagy deficiency. In fact, in these conditions, the cell may easily cope with moderate energy demands. In contrast, when the production of altered structures exceeds a reduced autophagy activity, these accumulate and become visible over the years through intracellular deposits that contain altered protein aggregates. This is probably why an autophagy defect eventually leads to slowly developing neuronal inclusions. This is facilitated by the inner nature of specific proteins such as alpha synuclein, which is prone to aggregate because 30% of its native form undergoes spontaneous oligomerization independently of the metabolic conditions [41]. Thus, autophagy defects are expected to generate protein aggregates, which in turn promote toxicity and cell death [42]. This is kind of an oversimplification, but it helps to schematize the concept of autophagy as a cell clearing system beyond its powerful energetic effects. Unlike most cell types, neurons are extremely vulnerable to autophagy impairment. This is not surprising when considering that adult neuronal cells are post-mitotic. Therefore, neurons cannot profit from mitosis to dilute potential toxic waste within daughter cells. In fact, mice deficient for autophagy-related proteins, such as Atg5 or Atg7, show inclusion bodies and marked neuronal loss [43,44]. In line with this, mTOR-dependent impairment of autophagy is implicated in various neuropsychiatric disorders such as dementia, movement disorders, motor neuron disease, seizures, brain ischemia, autism, affective disorders, addiction, and schizophrenia [45,46,47,48,49,50,51,52,53,54,55,56,57]. While the involvement of autophagy in neurological disorders is intensely investigated, the evidence about an involvement of autophagy in psychiatric disorders is less clear. Therefore, the aim of the present manuscript is to mention the role of mTOR-dependent autophagy in neurodegeneration while emphasizing its role in methamphetamine (METH) addiction and psychiatric disorders, namely schizophrenia. Remarkably, a growing evidence shows that mTOR dysfunction may underlie a variety of psychiatric syndromes, including mood disorders, drug addiction, and schizophrenia. In fact, many psychotropic drugs including mood stabilizers and neuroleptics are powerful autophagy inducers [58,59,60,61,62,63,64,65,66,67]. In such an attempt, we hint to the role of mTOR-dependent autophagy as a hub in etiologically distinct brain disorders from neurodegeneration to METH abuse and schizophrenia.
2. A Short Overview on Autophagy Impairment in Neurodegenerative Disorders
Dysfunctional autophagy appears as a recurring feature in neurodegenerative disorders (NDDs), such as Parkinson’s disease (PD) and Alzheimer’s disease (AD), where a defect along the autophagy pathway occurs at different stages [68,69,70,71,72,73,74,75,76]. The accumulation of aggregate-prone proteins triggers the formation of cytoplasmic and/or extracellular neuronal inclusions within specific brain areas. This occurs in PD, where aggregate-prone alpha synuclein accumulates in the so-called Lewy bodies, which are mostly found within spared dopaminergic (DA) neurons of the substantia nigra pars compacta (SNpc) [77,78], as well as within extra-nigral neuronal populations [79]. Remarkably, genetic ablation of Atg7 specifically within dopamine (DA) neurons fully reproduces PD pathology, including the formation of Lewy bodies, which stain for alpha synuclein, pointing at a key role for autophagy in DA-related disorders [80]. Likewise, AD cortical pathology features abnormal intracellular hyperphosporylated tau protein, which forms fibrils known as neurofibrillary tangles (NFT) along with extracellular amyloid-β (Aβ) plaques [81]. Pathological TAR DNA-binding protein 43 (TDP-43) is a major component of inclusions that are found in most cases of amyotrophic lateral sclerosis (ALS) and in frontotemporal lobar degeneration (FTLD) [82,83]. Remarkably, further investigation on the autophagy pathway revealed that all these misfolded proteins are autophagy substrates depending on mTOR activity [84,85,86,87,88]. In an attempt to attribute a specific protein accumulation to the onset of a specific disorder within the aim of describing a sort of “precision medicine”, investigators faced the hurdle that these protein aggregates may indeed be shared by different disorders and different proteins may aggregate in the same disease. In fact, TDP-43 positive inclusions are found within the very same neurons containing tau-positive NFTs and alpha synuclein-positive Lewy bodies of post-mortem brains from patients with AD and dementia with Lewy bodies (DLB), respectively [89]. In addition, most DLB patients show most features of AD (i.e., hyperphosphorylated tau deposits and Aβ) to various extents [90]. Moreover, alpha synuclein immunoreactivity often co-localizes within huntingtin polyglutamine-positive aggregates in brain sections from patients with late-stage Huntington’s disease (HD) patients [91], or even within SOD1-positive inclusions, as revealed by immunohistochemical analysis performed in post-mortem brain and spinal cord from cases of familial ALS [92]. All this evidence challenges the concept of a protein-specific vulnerability to characterize each disease leading to clinically distinct neurological phenotypes. Such a contamination even overcomes the clear cut between neurodegenerative disorders and acute cerebral ischemia/chronic hypoperfusion [93]. A failure in autophagy-dependent handling of misfolded proteins impedes the clearance of these substrates that are likely to accumulate within the cell. Therefore, a common pathogenesis underlying all these NDDs disorders has been linked to autophagy inhibition due to mTOR hyperactivation [52,54,94,95,96,97]. For instance, an increased mTOR activity correlates with accumulation of Aβ and hyperphosphorylated tau in AD brains [98,99]. On the other hand, some evidence indicates that suppressing mTOR activity ameliorates AD cognitive defects by decreasing Aβ and tau pathology [100]. Again, rapamycin and rapalogs protect against toxicity produced by a number of misfolded proteins encompassing alpha synuclein, TDP43, and hyperphosphorylated tau [101,102,103,104]. Therefore, mTOR inhibitors, as such autophagy inducers, may be useful to boost relented cell clearing mechanisms by decreasing abnormal aggregate-prone proteins, which is supposed to ameliorate neurodegeneration.
3. Beyond Classic Neurodegeneration
The beneficial effects of mTOR inhibitors have been demonstrated in patients affected by neurodevelopmental disorders [105,106,107,108]. In fact, compounds belonging to the macrolides family, such as rapamycin and rapalogs, and ameliorate cognitive, affective, and overall psychiatric symptoms, which is in line with an Akt–mTOR-dependent antidepressant and mood stabilizing effect [105,109,110]. This is further supported by mounting evidence obtained in rodent models, which demonstrate that rapamycin normalizes impaired social interactions and reverses behavioral defects [61,109,111,112,113,114]. This appears indeed as a continuum rather than a concomitance of different effects, as patients affected by neurodegenerative disorders frequently develop psychiatric symptoms like mood alterations, depression, and schizophrenia, which may appear early during disease development and then may persist throughout the disease course [115,116,117,118,119,120]. As briefly reported, despite a high number of studies correlating mTOR and autophagy with neurological disorders [45,46,47,48,49,50,51,52,53,54,55,56,57], only a few studies addressed such an issue in psychiatric disorders such as schizophrenia [121,122]. This is likely to depend on the lower amount of biological and pathological investigations that are carried out in these patients and the scarce knowledge about the molecular neurobiology of disease-concerning psychiatric disorders. In schizophrenia, a progressive synaptic disorder is likely to promote neurodegeneration [123]. In support of this view, autoptic studies on schizophrenic brains have revealed the presence of neuronal inclusions (see Section 5), which may depend on dysfunctional mTOR-related cell clearing systems. Similarly, neuronal inclusions occur in METH abusers [124], confirming what was previously demonstrated in animal models [125,126]. As detailed in the following paragraph, METH exerts disruptive effects on DA neurotransmission, which translate into abnormal stimulation of post-synaptic DA receptors, mainly D1-type DA receptors (D1R), thus leading to non-canonical signaling cascades sustaining behavioral alterations that overlap with schizophrenia-like symptoms (i.e., visual and auditory hallucinations and delusions) [127,128,129,130]. Increased activity of D1R is considered as a major determinant of neuropsychiatric alterations occurring in both METH models/abusers and in schizophrenia [130,131,132]. This is key, because abnormal stimulation of D1R and subsequent signaling cascades were recently shown to produce an over-activation of mTOR and inhibition of the autophagy machinery [133]. In addition, several susceptibility genes for schizophrenia (e.g., DISC1, NRG1/ErbB4, and CRMP2), which are involved in either pre-synaptic DA release or post-synaptic D1R-related cascades, are similarly dysregulated by METH. Interestingly, they all converge on mTOR signaling (see Section 6, Table 1). In fact, mTOR-induced autophagy inhibition exacerbates the ultrastructural effects of METH [126,134,135,136], while rapamycin administration reverts both behavioral and morphological alterations induced by METH [137]. Such an issue will be further dealt with in the next paragraph. Here, we wish to point out that the detrimental effects of METH on both DA neurotransmission and mTOR-dependent cell clearing systems produce behavioral alterations that are reminiscent of schizophrenia. Thus, METH addicted brains may represent a bridge that connects neurodegenerative and psychiatric disorders. Understanding the molecular and cellular mechanisms that operate during METH toxicity is expected to increase our insight into the neurobiology of schizophrenia. Thus, in the next paragraph we discuss evidence on how altered mTOR and an impaired autophagy pathway may indeed represent a common hub between drug addiction and schizophrenia.
4. Bridging Neurodegeneration and Psychiatric Disorders: The Paradigm of Methamphetamine-Addicted Brains
METH is a widely abused drug that rapidly enters and persists within the central nervous system (CNS), thereby exerting powerful addictive effects [138,139,140]. In humans, the sensitizing effects of prolonged chronic METH intake are considered a major determinant to the occurrence and relapse of psychoses, which mirror those occurring in schizophrenic patients. In fact, METH-addicted patients commonly develop psychoses with positive symptoms similar to those of schizophrenia, which led to the use of METH as an experimental model of schizophrenia (Figure 2). In fact, psychotic patients are oversensitive to amphetamines [141,142]. Accordingly, clinical evidence points towards an elevation of pre-synaptic DA synthesis and release as a key event for schizophrenia [141,143,144]. Likewise, the psychostimulant effects experienced by METH-addicted patients rely on increased DA synthesis and massive DA release from nerve terminals within limbic areas as occurring in the schizophrenic brain [145,146,147,148,149]. Such an abnormal DA release produces peaks of extracellular DA, which cannot be taken up within nerve terminals, because METH inhibits and reverts the direction of the dopamine transporter (DAT). In line with this, recent studies suggest that DAT expression is significantly reduced in the midbrain of postmortem schizophrenic samples [150], which is reminiscent of the METH-addicted brain [151,152]. Upon METH administration, the massive amount of extracellular DA is followed by DA depletion, which translates into a pulsatile stimulation of post-synaptic DA receptors. This triggers non-canonical transduction pathways driving phenotypic changes at the level of post-synaptic neurons. The abnormal activity of DA receptors is an additional commonality between METH and schizophrenia, which is likely to represent the molecular mechanism underlying behavioral alterations [130,131,132]. Remarkably, mTOR over-activation was recently linked to METH-induced behavioral sensitization, while rapamycin prevents such an effect [137]. Likewise, rapamycin was found to be beneficial for ameliorating psychotic symptoms [109,112,114,153,154]. The relevance of autophagy for sustaining these mTOR-induced effects is confirmed by drugs inducing autophagy independently of mTOR activation. In fact, lithium is able to delay METH-induced sensitization, while being a powerful treatment in schizophrenia [155]. Behavioral alterations driven by abnormal DA receptor activity are widely dependent on the amount of DA released from pre-synaptic terminals. Noteworthy, genetic ablation of autophagy was shown to produce an extremely powerful DA release upon electrical stimuli, suggesting that autophagy is key to restrain DA release both upon basal neural activity and mostly after rapamycin-induced autophagy [156]. These findings strongly suggest that an autophagy dysfunction acts both at pre- and post-synaptic level to alter DA neurotransmission during both METH administration and schizophrenia (Figure 2). This confirms our previous studies showing that both genetic and pharmacological autophagy inhibition worsen the effects of METH administration [126]. Given the paucity of studies showing such a role in schizophrenia, dissecting the molecular mechanisms underlying autophagy dysfunction in METH may provide insights in the pathophysiology of schizophrenia. In line with this, METH produces ultrastructural alterations reflecting dysfunctional autophagy flux, which are DA-dependent. In fact, METH produces a massive increase of endogenous intra-cytosolic DA levels by inhibiting and reverting the direction of the vesicular monoamine transporter type 2 (VMAT-2), thus disrupting the physiological storage of DA. A reduction in VMAT-2 gene expression and protein levels in DA neurons occurs in both METH models and schizophrenic patients, marking quite impressively the overlap between these disorders [150,157,158]. It is worth mentioning that freely diffusible intra-cytosolic DA can readily undergo auto-oxidation and produce a cascade of oxidative-related damage, which is bound to the neurotoxic effects of high doses of METH [125,159,160,161]. In fact, DA auto-oxidation generates several toxic and highly reactive species such as DA-quinones, hydrogen peroxide, and superoxide radicals. Beyond METH, redox-related changes that result from an imbalance between reactive oxygen species (ROS) production and ROS clearance are implicated in schizophrenia [122]. As a result of an altered intracellular redox environment, proteins lose their native conformational fold and assume an aberrant, misfolded conformation with an abnormal tendency to aggregate into larger, often insoluble, inclusions [148,159,162]. This excessive amount of aggregate-prone proteins within DA axon terminals leads to an autophagy engulfment, which fuels a vicious cycle of oxidative stress, protein misfolding, and aggregation [125,126,148,162] (Figure 2). In fact, when administered both in vitro and in vivo, METH generates multi-lamellar whorls corresponding to stagnant autophagy vacuoles, which further develop into eosinophilic cytoplasmic inclusions within both nigral DA neurons and striatal cells [125,162,163]. These inclusions are reminiscent of PD-like Lewy bodies because they stain for typical PD markers such alpha synuclein [125,126,162,164,165]. The occurrence of analogous nigral inclusions was confirmed in human chronic METH abusers [124]. In addition, the occurrence of alpha synuclein gene (SNCA) polymorphisms is associated with human METH psychosis [166]. This is in line with genetic studies associating psychotic symptoms with SNCA multiplications [167]. As both DA neurotransmission and handling of misfolded proteins are directly bound to autophagy, it is likely that autophagy alterations represent a causative mechanism underling ongoing synaptic pathology, which could predispose to neurodegeneration. In the light of these findings, in the next paragraph, we provide evidence that ultrastructural changes related to autophagy alterations occur in schizophrenia as well.
5. Cytoskeletal Abnormalities and Neuronal Inclusions in Schizophrenia
Neuropathological evidence showing cytoarchitectural abnormalities and neuronal inclusions in schizophrenic patients date back to the late 1990s from post-mortem studies. In detail, ultrastructural alterations encompassing reduction and swelling of DA terminals, mitochondrial alterations, and multi-lamellar structures were reported within DA terminals of the SNpc [168]. Interestingly, these ultrastructural abnormalities are highly reminiscent of those induced by METH. In addition, cytoskeletal derangement appears as a prominent feature of the ultrastructural pathology of schizophrenia [169]. Cytoskeleton organization and dynamics depend on the fine control of microtubule assembly, which relies on the interaction of microtubules with a specific class of proteins known as microtubule-associated protein (MAP). These proteins represent a sort of cytoskeletal regulatory elements that bind microtubules to ensure their stability and integrity. Among various identified MAPs, microtubule-associated protein 2 (MAP2), which belongs to the MAP2/Tau family, is enriched in the brain and especially in dendrites, where it contributes to microtubule stabilization and overall dendritic architecture. Alterations in MAP2 immunoreactivity within the subiculum, entorhinal cortex, hippocampus, and prefrontal cortex have been suggested as the primary array of cytoskeletal abnormalities, which in turn result in impaired neurotransmission observed in schizophrenia [170,171,172,173]. Notably, a marked reduction in MAP2 immunoreactivity, along with a decrease in dendritic arbor, is reported in the primary auditory cortex (BA41) of schizophrenic subjects compared with healthy controls [174]. These structural abnormalities observed in the post-mortem auditory cortex of schizophrenic individuals may underlie altered auditory information processing, which in turn may manifest as auditory hallucinations. Moreover, pathological deposition of hyperphosphorylated MAP-tau (MAPT), which is the hallmark of several neurodegenerative disorders such as AD and frontotemporal dementia (FTD), has been described in elderly subjects with schizophrenia [175,176,177]. However, this issue is still under debate, because some autopsy studies do not report any significant difference in the prevalence of AD pathology between elderly schizophrenic patients and age-matched healthy controls [178,179,180,181]. Momeni and colleagues (2010) recently reported two relatives with an early age at onset (27 and 29 years) of schizophrenic symptoms showing a marked neuronal tau deposition, as confirmed at pathological examination [182]. Remarkably, this familial behavioral variant of frontotemporal lobar degeneration (FTLD) was associated with a novel exon 12 mutation in the conserved microtubule binding region of microtubule-associated protein tau (MAPT) gene, thus suggesting that disturbances in proteins involved in regulation of microtubule stability and overall cytoskeletal dynamics may accelerate tau deposition, leading to early disease onset. Notably, post-mortem analysis performed in the prefrontal cortex of schizophrenic patients revealed oligodendrocyte ultrastructural abnormalities [183]. Similarly, cytoskeletal derangements within nigro-striatal DA neurons and axons were recently evidenced in another cohort of schizophrenic brains [184]. Remarkably, a very recent neuropathological examination provided evidence for TDP-43-positive cytosolic inclusions and dystrophic neurites in the brain of a patient diagnosed with FTLD presenting brief psychotic episodes and catatonia, which is a syndrome related to schizophrenia [120]. Preliminary in vitro studies demonstrated that two proteins, namely DISC1 and dysbindin-1, which are encoded by two susceptibility genes for schizophrenia, can form insoluble protein aggregates that are reminiscent of those occurring in neurodegenerative disorders [185,186,187,188]. Intriguingly, the interactome analysis of both DISC1, dysbindin-1, and CRMP2, which is another susceptibility gene for schizophrenia, revealed common protein interactions with microtubules, actin cytoskeleton, and proteins involved in intracellular transport [189,190]. In particular, CRMP2 is a cytosolic protein enriched in the CNS, which has been implicated in microtubule stabilization, and thus in the regulation of cytoskeletal dynamics and vesicle trafficking. Remarkably, multiple proteomic studies of postmortem brains show altered CRMP2 protein levels in schizophrenic patients compared with healthy subjects [191,192,193,194]. These findings suggest that all these schizophrenia risk genes may encompass cytoskeletal stability and organization. It is worth mentioning that the reciprocal modulation between cytoskeleton dynamics and autophagy is emerging as a crucial point for neuronal homeostasis. Inhibition of the physiological microtubule transport is known to associate with an impaired shuttling of protein aggregates towards autophagy vacuoles, as well as impaired intracellular vesicle trafficking. Such an effect contributes to dysfunction in both the autophagy and secretory pathway leading to altered transmission of axonal information to and from the somato-dendritic domain [195]. The biological implication behind an impairment of microtubule dynamics is confirmed in post-mortem schizophrenic brain samples, as well as in mouse models of schizophrenia, where mTOR-dependent autophagy dysfunction is accompanied by an altered gene expression and protein levels of the microtubule-associated protein 6 (MAP6) [196,197]. These findings suggest that a close interplay between cytoskeletal dynamics and mTOR signaling is key in early axonal transport defects and altered synaptic transmission, a common pathological hallmark in schizophrenia [144,198,199,200,201].
6. mTOR Modulation of Dopamine Transmission in Methamphetamine and Schizophrenia
Despite some epidemiological evidence concerning risk factors for schizophrenia, which represents a chronic debilitating condition, the identification of the molecular mechanisms underlying its pathogenesis is still challenging. Interestingly, as has emerged from review of the literature published in the last few years, a novel scenario begins to delineate in which a dysfunctional mTOR pathway may be a key mechanism in the chain of events for the development of schizophrenia (Figure 3). In line with this, a number of genetic studies linked mTOR-related genetic alterations to schizophrenia. This is the case of the disrupted in schizophrenia 1 (DISC1) gene, a schizophrenia-related gene, originally discovered in a large Scottish family with a high incidence of psychiatric symptoms [202]. This gene encodes for the DISC1 ubiquitous protein, which is implicated in neurogenesis, neuronal migration, axon/dendrite, and synapse formation [203,204,205,206]. Remarkably, DISC1 plays a key role in DA neurotransmission [207]. In line with this, experimental models of DISC1 deficiency treated with METH show a significant potentiation of DA release, along with increased expression of D1R in the ventral striatum when compared with controls [208]. Mutations of DISC1 in the striatum associate with increased METH-induced behavioral sensitization suggesting that DISC1 represents a hub underlying alterations in those DA-dependent molecular mechanisms that modulate reward and sensitization in both drug abuse and mental disorders [209]. In addition, these findings strongly suggest that DISC1 alterations may increase the risk of schizophrenia by dysregulating DA release. In support of this view, DISC1 alterations are associated with pathological stress and converge in producing early alterations in DA neurotransmission during adolescence. This is a critical life-time for the development of schizophrenia [210,211]. Noteworthy, both DISC1 deficiency and over-stimulated D1Rs up-regulate the Akt–mTOR pathway [133,153,212], which highlights the impressive overlap between pathways that modulate DA neurotransmission and cell clearing systems. In particular, DISC1 acts by blocking KIAA1212, an Akt-binding partner, which directly interacts with Akt and strengthens the activation of this kinase, which represents a major mediator of the mTOR pathway. Therefore, the binding between DISC1 and KIAA1212 prevents KIAA1212-dependent Akt activation (Figure 3). This decreases Akt activity, which in turn dampens mTOR signaling [212]. Therefore, disruption of DISC1 activity, due to genetic rearrangements (i.e., balanced (1;11) (q42;q14) chromosomal translocation) or missense mutations, produces schizophrenic-like behavior, which is bound to enhanced Akt activity, over-activation of mTOR signaling, and depressed autophagy [213,214,215]. Likewise, administration of either D1R agonists or METH enhances Akt activity and over-activates mTOR signaling [133,137,216]. Remarkably, inhibition of mTOR with rapamycin reverses the effects occurring in both DISC1-shRNA and METH-treated mice, while ameliorating behavioral alterations [137,153,212]. Again, an impaired Akt signaling, achieved by neuronal deletion of rictor, a key regulatory subunit of mTORC2, contributes to schizophrenia-like phenotypes in rictor-null (KO) mice [217]. Dysregulation in Akt signaling and altered Akt protein levels were found in the frontal cortex and hippocampus of post-mortem brain samples from individuals affected by schizophrenia [218]. Since the first report by Emamian et al. (2004) [218], numerous subsequent studies further confirmed the genetic association of AKT1 gene variants with schizophrenia, supporting the key role of impaired Akt–mTOR signaling in the pathogenesis of this psychiatric disorder [219,220,221,222,223,224]. In line with this, increased AKT1 gene expression, due to increased hypomethylation of AKT1 gene promoter, was detected in human METH abusers [225].
Furthermore, genetic linkage and association studies led to the identification of two additional susceptibility factors related to schizophrenia, such as neuregulin-1 (NRG1) and its receptor ErbB4 [226,227]. NRG1 is a family of trophic factors that is synthesized as trans-membrane proteins displaying an extracellular epidermal growth factor (EGF)-like domain that is essential for ErbB4 receptor binding. Upon proteolytic processing, the soluble N-terminal moieties that contain the EGF-like domain is released and it acts by stimulating the ErbB4 receptor. Alterations in the NRG1/ErbB4 signaling are reported in schizophrenic brains [228]. In particular, NRG1, which is mostly involved in regulating neurodevelopment and neurotransmission, acts by binding ErbB4, a type I transmembrane receptor tyrosine kinase belonging to the family of ErbB proteins, which contain a binding site for PI3K kinase, an AKT upstream effector, in the C-terminal cytoplasmic tail (CYT) [229]. The binding between ErbB4 and PI3K activates this latter kinase, which in turn can phosphorylate and activate its downstream target Akt. Thus, changes in NRG1/ErbB4 signaling leads to a cascade of events that culminate in a dysregulation of the Akt–mTOR pathway (Figure 3). Moreover, it has been demonstrated that NRG1 also regulates DISC1 expression [230], thus further worsening the aberrancy of the Akt–mTOR pathway and the pathogenesis of schizophrenia and related behaviors. Noteworthy, NRG1/ErbB4 signaling plays a key role in DA-related behaviors by increasing DA release within the hippocampus, striatum, and prefrontal cortex [231]. In mice harboring mutated ErbB4, D1R’s levels and binding activity are significantly increased [232], suggesting that ErbB4 may be another putative protein linking increased DA activity to increased mTOR activity and depressed autophagy.
Another identified susceptibility gene for schizophrenia is the dihydropyrimidinase-like 2 (DPYSL2) gene. This gene is located on chromosome 8p21 and it encodes the cytosolic microtubule-associated protein CRMP2 (collapsin response mediator protein-2), which is highly expressed in the CNS and plays a role in axonal growth [233]. Several DPYSL2 single-nucleotide polymorphisms (SNPs) have been associated with the development to schizophrenia [234,235]. Although in mammals there are three DPYSL2 transcripts (i.e., DPYSL2A, DPYSL2B, and DPYSL2C), which differ in their first exon sequence, most of the studies have focused on the DPYSL2B transcript, also known as “short” transcript [236]. Multiple functional sequence variants were identified both in and around DPYSL2B, including three single-nucleotide polymorphisms (SNPs) in the proximal promoter and two SNPs in intron 1, which were significantly associated with schizophrenia [236]. In vitro functional luciferase assays in both neuronal (mouse primary cortical neurons) and non-neuronal (HEK293) cell types demonstrated that in the presence of increasing concentrations of rapamycin, a polymorphic di-nucleotide repeat (DNR) in the 5′-UTR of the DPYSL2B gene dose-dependently decreases allele expression at translation level, suggesting a functional link between this schizophrenia high-risk allele (13 DNRs) and mTOR signaling [236]. The relationship between the DPYSL2 gene and susceptibility to schizophrenia was recently confirmed in vivo in rats exposed to prenatal stress (PNS), which indeed is frequently reported as an environmental risk factor for developing schizophrenia in adults [237]. Remarkably, immunohistochemical and Western blot analyses performed in the prefrontal cortex and hippocampus revealed a decreased DPYSL2 expression in the PNS group compared with non-stressed control offspring [238]. CRMP2 protein levels are significantly altered by METH administration [239]. In turn, CRMP2-KO mice show altered levels of proteins and genes involved in GABA-, glutamate-, and neurotrophin-signaling pathways, which are related to both schizophrenia and METH-induced sensitization [240]. Once again, such an overlap between altered molecular mechanisms occurring in both schizophrenia and METH may be key to decipher those early events linking CRMP2 and DA activity. In line with this, the dendritic spine-regulating activity of CRMP2 is under the control of the cyclin-dependent kinase 5 (CDK5) [241]. CDK5 is a key second messenger participating in METH-induced behavioral sensitization [130,242,243]. Both administration of amphetamines and stimulation of D1R induce a significant increase of CDK5 gene expression and protein levels, which, at molecular level, associates with increased dendritic spine density and hyper-phosphorylation of the cytoskeletal tau protein [244,245,246]. In detail, the activation of CDK5 by D1R occurs via proteolysis of p35, the binding partner of CDK5. Remarkably, the marked reduction of p35 levels in schizophrenic brains, which mirrors enhanced CDK5 activity [247], suggests a role for CDK5-CRMP2-dependent alterations of cytoskeleton architecture and psychiatric behavior.
7. A Step Forward about a Role of Autophagy in the Pathophysiology of Schizophrenia
While recent advances in molecular psychiatry have identified several mTOR-related schizophrenia risk genes, the role of autophagy in schizophrenia has been recently investigated. Remarkably, the identification of rare genetic variants of ULK1 in a cohort of schizophrenic patients by means of exome sequence analysis strengthens the idea of a key role of both disrupted mTOR signaling and autophagy in the pathophysiology and susceptibility to schizophrenia [248] (Figure 3). The first evidence of a dysregulation of autophagy in schizophrenia was provided in 2011 by the Horesh group, who performed gene expression profile analysis in different brain areas of post-mortem schizophrenic patients compared with healthy controls, with no evidence of concomitant dementia [249]. The study revealed profound differences between the two groups, especially when looking at Broadman area 22 (BA 22), which is associated with positive symptoms, mainly auditory-verbal hallucinations or “hearing voices” [250,251]. In particular, at BA 22, the vast majority of abnormally expressed genes referred to key autophagy genes (i.e., BECN1, ULK2, ATG3), which were significantly down-regulated compared with controls [249]. A few months later, another transcriptomic study reported a BA 22-specific down-regulation in several autophagy-related genes, thus strengthening the link between impaired autophagy and schizophrenia positive symptoms [252]. Furthermore, the transcriptional analysis performed on the very same post-mortem samples demonstrated no substantial changes in the mRNA levels of the above-mentioned autophagy-related genes within the anterior prefrontal cortex (BA 10), which is mainly involved in schizophrenic negative symptoms and cognitive dysfunction, thus reinforcing the involvement of an impaired autophagy in mediating positive symptoms. Later on, further analysis reported a disruption of the autophagy pathway also in the hippocampus of post-mortem schizophrenic patients [197]. In detail, the analysis of mRNA expression of a key protein for autophagy initiation, namely beclin1, revealed a significant region-specific reduction in hippocampal samples from 12 schizophrenic patients compared with 12 age-matched healthy controls. The deficiency in hippocampal beclin1 transcript levels matches those observed in haploinsufficient mice for the activity-dependent neuroprotective protein (ADNP) (ADNP+/− mice), a transgenic model of schizophrenia [197]. ADNP is an essential protein for brain development and it has been shown to physically interact with a key protein in autophagosome biogenesis and maturation, namely LC3 [253]. Remarkably, a co-immunoprecipitation assay performed in a hippocampal protein fraction from ADNP+/− mice showed a dramatic reduction in the ADNP–LC3 protein interaction, which correlates with decreased ADNP expression [197]. A reduction of ADNP and its homologous protein, ADNP2, is observed in schizophrenic patients [254], and it is recapitulated in Map6-deficient (Map6+/−) mice, another transgenic model of schizophrenia [196]. Immuno-histochemical analysis showed a three-fold decrease in the number of beclin1-positive cells in Map6+/− mice. These results were confirmed at transcriptional level by demonstrating a reduced expression of BECN1 mRNA. On the other hand, chronic treatment with an eight-amino-acid peptide snippet from ADNP (NAP), also known as davunetide, restored both Beclin1 and ADNP mRNA levels along with ADNP-LC3 interaction, thus providing neuroprotection while ameliorating schizophrenic-like behavioral and cognitive deficits in Map6+/− mice [196]. A recent phase II, multicenter, double-blind, randomized clinical trial has shown an improvement in cognitive performance of schizophrenic patients treated with NAP (AL-108; 5 and 30 mg/day, intranasally) versus placebo-treated patients [255]. These pieces of evidence corroborate findings showing that several autophagy inducers, such as lithium, rapamycin, and Food and Drug Administration (FDA) approved antipsychotic drugs are effective to treat psychosis including schizophrenia [59,60,61,62,109,256,257,258,259,260]. Notably, high-throughput image-based screens performed by Zhang et al. (2007) [60] on a human glioblastoma H4 cell line expressing human LC3 coupled with green fluorescent protein (GFP) led us to disclose that three typical antipsychotic drugs (fluspirilene, trifluoperazine, and pimozide) are effective autophagy inducers. In particular, pimozide provides an mTOR-independent autophagy induction, because it directly activates AMPK1, which in turn promotes autophagy through the phosphorylation of ULK1 [260]. In contrast, chlorpromazine, which is a typical antipsychotic agent, induces autophagy by inhibiting the Akt/mTOR pathway [59]. Recently, in vitro studies on the effects of second-generation, atypical antipsychotics demonstrated that sertindole and clozapine are potent autophagy inducers in both neuronal and non-neuronal cell lines [257,261]. Similar to pimozide, clozapine activates the autophagy process via the AMPK–ULK1–Beclin1 pathway, as evidenced by increased levels of autophagy markers (i.e., LC3-II and Atg5–Atg12 conjugate); increased phosphorylation of AMPK and its downstream substrates, namely ULK1 and beclin1; and an increased number of autophagosomes in the frontal cortex in clozapine-treated rats [259]. Most reports evidenced autophagy induction by neuroleptics indirectly, only by measuring the degradation of autophagy-dependent substrates. For instance, the increase in autophagy flux induced by pimozide occurs along with a depression of phosphorylated tau in a transgenic mouse model of AD [260]. Again, the effects of two typical antipsychotics, trifluoperazine and haloperidol, on autophagy have been demonstrated indirectly [262,263]. For instance, haloperidol occludes huntingtin aggregation [262]. Chronic clozapine treatment (20 mg/kg/day) reduces Aβ deposition [264]. Although typical and atypical antipsychotics may alleviate diseases featuring aberrant protein misfolding and accumulation, in vivo systematic investigations regarding the efficacy and the molecular mechanisms of these drugs on autophagy have been questioned [265]. This issue is biased by the routine intake of neuroleptics by most schizophrenic patients for long time intervals, sometimes lasting decades.
8. Conclusions and Future Perspectives
The exponential development in the past few years of genome-wide linkage studies and high-throughput genotyping technologies has led to the identification of many other susceptibility genes for schizophrenia, and this list is expected to grow further. However, the molecular mechanisms underlying schizophrenia are far from being deciphered. Up-to-date neuropathological studies performed on post-mortem schizophrenic brains appear to be scattered and they have not yielded to the identification of a distinct neuropathological hallmark. This is due to the limited sample availability and confounding interpretation of pathological data when comparing antipsychotic-treated and non-treated patients. This contrasts with classic neurodegenerative disorders where the accumulation of misfolded or aggregated proteins within neurons may imply a dysregulation of autophagy. Again, most experimental models available so far fail at large to reproduce most features of schizophrenia. Thus, methamphetamine remains an appropriate model compared with genetic manipulation to decipher the molecular progression underlying the pathophysiology of schizophrenia. In fact, METH bridges autophagy alterations with altered DA transmission and degeneration that is reminiscent of schizophrenia.
The present manuscript reviewed genetic and biochemical evidence that suggests that autophagy impairment may be involved in early DA neurotransmission, leading to synaptic dysfunction, which underlies some psychiatric disorders. An ongoing and persistent autophagy dysfunction that occludes handling of misfolded proteins while fueling synaptic alterations predisposes to the onset of degeneration. This scenario, depicting schizophrenia as an autophagy-dependent progressive synaptic pathology, may be a ground for planning the use of mTOR inhibitors and autophagy inducers as early treatment intervention.
Funding
This research was funded by MINISTERO DELLA SALUTE (RICERCA CORRENTE 2018).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AD | Alzheimer’s disease |
| ADNP | activity-dependent neuroprotective protein |
| Akt | protein kinase B |
| ALS | Amyotrophic lateral sclerosis |
| Aβ | amyloid-β |
| BA 22 | Broadman area 22 |
| BA41 | Broadman area 41 |
| CDK5 | cyclin-dependent kinase 5 |
| CMA | chaperone-mediated autophagy |
| CNS | central nervous system |
| CRMP2 | collapsin response mediator protein 2 |
| D1R | dopamine receptor type 1 |
| DA | dopamine/dopaminergic |
| DAT | dopamine transporter |
| DISC1 | disrupted in schizophrenia 1 |
| DLB | Dementia with Lewy bodies |
| DNR | di-nucleotide repeat |
| DPYSL2 | dihydropyrimidinase-like 2 |
| ErbB4 | avian erythroblastosis oncogene B4-like protein |
| FRKBP12 | FK506-binding protein 12 |
| FTD | Frontotemporal dementia |
| FTLD | Frontotemporal lobar degeneration |
| GFP | green fluorescent protein |
| GPCRs | G-protein coupled receptors |
| HD | Huntington’s disease |
| MAP | microtubule-associated protein |
| MAPT | microtubule-associated protein tau |
| METH | methamphetamine |
| mTOR | mammalian Target Of Rapamycin |
| mTORC1 | mammalian Target Of Rapamycin complex 1 |
| mTORC2 | mammalian Target Of Rapamycin complex 2 |
| NDDs | neurodegenerative disorders |
| NFT | neurofibrillary tangles |
| NRG1 | neuregulin-1 |
| PD | Parkinson’s disease |
| PI3k | phosphatidylinositol-3-Kinase |
| PIP2 | phosphatidylinositol-4,5-phosphate |
| PIP3 | phosphatidylinositol-3,4,5-phosphate |
| PNS | prenatal stress |
| ROS | reactive oxygen species |
| RTKs | receptor tyrosine kinase receptors |
| SNARE | Soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein receptor |
| SNCA | alpha synuclein gene |
| SNpc | substantia nigra pars compacta |
| SNPs | single-nucleotide polymorphisms |
| SOD1 | superoxide dismutase 1 |
| TDP-43 | TAR DNA-binding protein 43 |
| TEM | transmission electron microscopy |
| TORC | target of rapamycin complex |
| TSC | tuberous sclerosis complex |
| TSC1 | hamartin |
| TSC2 | tuberin |
| VMAT-2 | vesicular monoamine transporter type 2 |
References
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Calne, R.Y.; Collier, D.S.; Lim, S.; Pollard, S.G.; Samaan, A.; White, D.J.; Thiru, S. Rapamycin for immunosuppression in organ allografting. Lancet 1989, 2, 227. [Google Scholar] [CrossRef]
- Starzl, T.E.; Schreiber, S.L.; Albers, M.W.; Porter, K.A.; Foglieni, C.S.; Francavilla, A. Hepatotrophic properties in dogs of human FKBP, the binding protein for FK506 and rapamycin. Transplantation 1991, 52, 751–753. [Google Scholar] [PubMed]
- Watson, C.J.; Friend, P.J.; Jamieson, N.V.; Frick, T.W.; Alexander, G.; Gimson, A.E.; Calne, R. Sirolimus: A potent new immunosuppressant for liver transplantation. Transplantation 1999, 67, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Sehgal, S.N. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant. Proc. 2003, 35, 7S–14S. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Stallock, J.P.; Ng, J.C.; Reinhard, C.; Neufeld, T.P. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000, 14, 2712–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, K. Rapamycin’s resurrection: A new way to target the cancer cell cycle. J. Natl. Cancer Inst. 2001, 93, 1517–1519. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fingar, D.C.; Blenis, J. Target of rapamycin (TOR): An integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004, 23, 3151–3171. [Google Scholar] [CrossRef] [PubMed]
- Soulard, A.; Cohen, A.; Hall, M.N. TOR signaling in invertebrates. Curr. Opin. Cell Biol. 2009, 21, 825–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, L.A. Origin and diversification of eukaryotes. Annu. Rev. Microbiol. 2012, 66, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Koltin, Y.; Faucette, L.; Bergsma, D.J.; Levy, M.A.; Cafferkey, R.; Koser, P.L.; Johnson, R.K.; Livi, G.P. Rapamycin sensitivity in Saccharomyces cerevisiae is mediated by a peptidyl-prolyl cis-trans isomerase related to human FK506-binding protein. Mol. Cell. Biol. 1991, 11, 1718–1723. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.C.; Heitman, J. TOR mutations confer rapamycin resistance by preventing interaction with FKBP12-rapamycin. J. Biol. Chem. 1995, 270, 27531–27537. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. An expanding role for mTOR in cancer. Trends Mol. Med. 2005, 11, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. MTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawłowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.M.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of protor as a novel Rictor-binding component ofmTOR complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C., Jr. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Hara, T.; Oshiro, N.; Kikkawa, U.; Yonezawa, K.; Takehana, K.; Iemura, S.; Natsume, T.; Mizushima, N. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J. Biol. Chem. 2010, 285, 20109–20116. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haissaguerre, M.; Saucisse, N.; Cota, D. Influence of mTOR in energy and metabolic homeostasis. Mol. Cell. Endocrinol. 2014, 397, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Signalling and autophagy regulation in health, aging and disease. Mol. Asp. Med. 2006, 27, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Ahlberg, J.; Marzella, L.; Glaumann, H. Uptake and degradation of proteins by isolated rat liver lysosomes. Suggestion of a microautophagic pathway of proteolysis. Lab. Investig. 1982, 47, 523–532. [Google Scholar] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Chaperone-mediated autophagy. Autophagy 2007, 3, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK–Atg13–FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Bernard, A.; Klionsky, D.J. Defining the membrane precursor supporting the nucleation of the phagophore. Autophagy 2014, 10, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of native brain α-synuclein. Nature 2013, 498, E4–E6; discussion E6–E7. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; López-Otín, C. Autophagy: Molecular mechanisms, physiological functions and relevance in human pathology. Cell. Mol. Life Sci. 2004, 61, 1439–1454. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Longone, P.; Ferrucci, M.; Lenzi, P.; Isidoro, C.; Ruggieri, S.; Paparelli, A. Autophagy and amyotrophic lateral sclerosis: The multiple roles of lithium. Autophagy 2008, 4, 527–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P.; et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Cloughesy, T.F.; Mischel, P.S. mTOR signaling in glioblastoma: Lessons learned from bench to bedside. Neuro Oncol. 2010, 12, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Weiss, W.A. Inhibition of PI3K-Akt-mTOR signaling in glioblastoma by mTORC1/2 inhibitors. Methods Mol. Biol. 2012, 821, 349–359. [Google Scholar] [PubMed]
- Arcella, A.; Biagioni, F.; Antonietta Oliva, M.; Bucci, D.; Frati, A.; Esposito, V.; Cantore, G.; Giangaspero, F.; Fornai, F. Rapamycin inhibits the growth of glioblastoma. Brain Res. 2013, 1495, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.S.; Biagioni, F.; Lenzi, P.; Frati, A.; Fornai, F. The role of autophagy in epileptogenesis and in epilepsy-induced neuronal alterations. J. Neural Transm. 2015, 122, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Biagioni, F.; Lenzi, P.; Gambardella, S.; Ferese, R.; Calierno, M.T.; Falleni, A.; Grimaldi, A.; Frati, A.; Esposito, V.; et al. Rapamycin promotes differentiation increasing βIII-tubulin, NeuN, and NeuroD while suppressing nestin expression in glioblastoma cells. Oncotarget 2017, 8, 29574–29599. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Gambardella, S.; Frati, A.; Fornai, F. mTOR-Dependent Cell Proliferation in the Brain. Biomed. Res. Int. 2017, 2017, 7082696. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Limanaqi, F.; Biagioni, F.; Frati, A.; Esposito, V.; Calierno, M.T.; Lenzi, P.; Fornai, F. The emerging role of m-TOR up-regulation in brain Astrocytoma. Histol. Histopathol. 2017, 32, 413–431. [Google Scholar] [PubMed]
- Williams, R.S.; Cheng, L.; Mudge, A.W.; Harwood, A.J. A common mechanism of action for three mood-stabilizing drugs. Nature 2002, 417, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yu, J.; Pan, H.; Hu, P.; Hao, Y.; Cai, W.; Zhu, H.; Yu, A.D.; Xie, X.; Ma, D.; et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc. Natl. Acad. Sci. USA 2007, 104, 19023–19028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kara, N.Z.; Toker, L.; Agam, G.; Anderson, G.W.; Belmaker, R.H.; Einat, H. Trehalose induced antidepressant-like effects and autophagy enhancement in mice. Psychopharmacology 2013, 229, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Lee, K.S.; Choi, Y.K.; Lim, H.J.; Lee, H.G.; Lim, Y.; Lee, Y.H. The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells. Carcinogenesis 2013, 34, 2080–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vucicevic, L.; Misirkic-Marjanovic, M.; Paunovic, V.; Kravic-Stevovic, T.; Martinovic, T.; Ciric, D.; Maric, N.; Petricevic, S.; Harhaji-Trajkovic, L.; Bumbasirevic, V.; et al. Autophagy inhibition uncovers the neurotoxic action of the antipsychotic drug olanzapine. Autophagy 2014, 10, 2362–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; McGreal, S.; Zhao, J.; Huang, R.; Zhou, Y.; Zhong, H.; Xia, M.; Ding, W.X. A cell-based quantitative high-throughput image screening identified novel autophagy modulators. Pharmacol. Res. 2016, 110, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, T.D.; O’Donnell, K.C.; Dow, E.R.; Du, J.; Chen, G.; Manji, H.K. Involvement of AMPA receptors in the antidepressant-like effects of lithium in the mouse tail suspension test and forced swim test. Neuropharmacology 2008, 54, 577–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einat, H.; Yuan, P.; Szabo, S.T.; Dogra, S.; Manji, H.K. Protein kinase C inhibition by tamoxifen antagonizes manic-like behavior in rats: Implications for the development of novel therapeutics for bipolar disorder. Neuropsychobiology 2007, 55, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A., Jr.; Singh, J.B.; Carlson, P.J.; Quiroz, J.; Jolkovsky, L.; Luckenbaugh, D.A.; Manji, H.K. Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: A pilot study. Bipolar Disord. 2007, 9, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, D.D.; Pletnikova, O.; Vonsattel, J.P.; Ross, C.A.; Margolis, R.L. A comparison of huntington disease and huntington disease-like 2 neuropathology. J. Neuropathol. Exp. Neurol. 2008, 67, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Eisenberg, T.; Kroemer, G. Autophagy for the avoidance of neurodegeneration. Genes Dev. 2009, 23, 2253–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquali, L.; Longone, P.; Isidoro, C.; Ruggieri, S.; Paparelli, A.; Fornai, F. Autophagy, lithium, and amyotrophic lateral sclerosis. Muscle Nerve 2009, 40, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, L.; Ruggieri, S.; Murri, L.; Paparelli, A.; Fornai, F. Does autophagy worsen or improve the survival of dopaminergic neurons? Parkinsonism Relat. Disord. 2009, 15, S24–S27. [Google Scholar] [CrossRef]
- Ferrucci, M.; Fulceri, F.; Toti, L.; Soldani, P.; Siciliano, G.; Paparelli, A.; Fornai, F. Protein clearing pathways in ALS. Arch. Ital. Biol. 2011, 149, 121–149. [Google Scholar] [PubMed]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Natale, G.; Lenzi, P.; Lazzeri, G.; Falleni, A.; Biagioni, F.; Ryskalin, L.; Fornai, F. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front. Cell. Neurosci. 2015, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 2015, 138, 1642–1657. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Hattori, N. Dopaminergic Neuron-Specific Autophagy-Deficient Mice. Methods Mol. Biol. 2018. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Grossman, M.; Wood, E.M.; Moore, P.; Neumann, M.; Kwong, L.; Forman, M.S.; Clark, C.M.; McCluskey, L.F.; Miller, B.L.; Lee, V.M.; et al. TDP-43 pathologic lesions and clinical phenotype in frontotemporal lobar degeneration with ubiquitin-positive inclusions. Arch. Neurol. 2007, 64, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Chang, J.C.; Fan, E.Y.; Flajolet, M.; Greengard, P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 17071–17076. [Google Scholar] [CrossRef] [PubMed]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef] [PubMed]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007, 1184, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Gelpi, E.; Charif, S.; Belbin, O.; Blesa, R.; Martí, M.J.; Clarimón, J.; Lleó, A. Confluence of α-synuclein, tau, and β-amyloid pathologies in dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 2013, 72, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Charles, V.; Mezey, E.; Reddy, P.H.; Dehejia, A.; Young, T.A.; Polymeropoulos, M.H.; Brownstein, M.J.; Tagle, D.A. α-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models. Neurosci. Lett. 2000, 289, 29–32. [Google Scholar] [CrossRef]
- Takei, Y.; Oguchi, K.; Koshihara, H.; Hineno, A.; Nakamura, A.; Ohara, S. α-Synuclein coaggregation in familial amyotrophic lateral sclerosis with SOD1 gene mutation. Hum. Pathol. 2013, 44, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gong, C.X. From chronic cerebral hypoperfusion to Alzheimer-like brain pathology and neurodegeneration. Cell. Mol. Neurobiol. 2015, 35, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef] [PubMed]
- Bové, J.; Martínez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Roscic, A.; Baldo, B.; Crochemore, C.; Marcellin, D.; Paganetti, P. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J. Neurochem. 2011, 119, 398–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryor, W.M.; Biagioli, M.; Shahani, N.; Swarnkar, S.; Huang, W.C.; Page, D.T.; MacDonald, M.E.; Subramaniam, S. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington’s disease. Sci. Signal. 2014, 7, ra103. [Google Scholar] [CrossRef] [PubMed]
- An, W.L.; Cowburn, R.F.; Li, L.; Braak, H.; Alafuzoff, I.; Iqbal, K.; Iqbal, I.G.; Winblad, B.; Pei, J.J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 2003, 163, 591–607. [Google Scholar] [CrossRef]
- Li, X.; Alafuzoff, I.; Soininen, H.; Winblad, B.; Pei, J.J. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005, 272, 4211–4220. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Maldonado, M.A.; Majumder, S.; Medina, D.X.; Holbein, W.; Magri, A.; Oddo, S. Naturally secreted amyloid-β increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J. Biol. Chem. 2011, 286, 8924–8932. [Google Scholar] [CrossRef] [PubMed]
- Ozcelik, S.; Fraser, G.; Castets, P.; Schaeffer, V.; Skachokova, Z.; Breu, K.; Clavaguera, F.; Sinnreich, M.; Kappos, L.; Goedert, M.; et al. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS ONE 2013, 8, e62459. [Google Scholar] [CrossRef] [PubMed]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Tan, M.S.; Wang, H.F.; Cao, L.; Zhang, Q.Q.; Shi, J.Q.; Gao, L.; Qin, H.; et al. Temsirolimus promotes autophagic clearance of amyloid-β and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol. Res. 2014, 81, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Frederick, C.; Ando, K.; Leroy, K.; Heraud, C.; Suain, V.; Buee, L.; Brion, J.P. Rapamycin ester analog CCI-779/Temsirolimus alleviates tau pathology and improves motor deficit in mutant tau transgenic mice. J. Alzheimers Dis. 2015, 44, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Lang, U.E.; Heger, J.; Willbring, M.; Domula, M.; Matschke, K.; Tugtekin, S.M. Immunosuppression using the mammalian target of rapamycin (mTOR) inhibitor everolimus: Pilot study shows significant cognitive and affective improvement. Transplant. Proc. 2009, 41, 4285–4288. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Silva, A.J. Rapamycin for treating Tuberous sclerosis and Autism spectrum disorders. Trends Mol. Med. 2011, 17, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.K.; Lee, J.H.; Yang, J.E.; Lim, C.S.; Lee, J.A.; Lee, Y.S.; Lee, K.; Kaang, B.K. Everolimus improves neuropsychiatric symptoms in a patient with tuberous sclerosis carrying a novel TSC2 mutation. Mol. Brain 2016, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Kilincaslan, A.; Kok, B.E.; Tekturk, P.; Yalcinkaya, C.; Ozkara, C.; Yapici, Z. Beneficial Effects of Everolimus on Autism and Attention-Deficit/Hyperactivity Disorder Symptoms in a Group of Patients with Tuberous Sclerosis Complex. J. Child Adolesc. Psychopharmacol. 2017, 27, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Cleary, C.; Linde, J.A.; Hiscock, K.M.; Hadas, I.; Belmaker, R.H.; Agam, G.; Flaisher-Grinberg, S.; Einat, H. Antidepressive-like effects of rapamycin in animal models: Implications for mTOR inhibition as a new target for treatment of affective disorders. Brain Res. Bull. 2008, 76, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Bou Khalil, R. Is there any place for macrolides in mood disorders? Med. Hypotheses 2012, 78, 86–87. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Han, S.; Shilyansky, C.; Zhou, Y.; Li, W.; Kwiatkowski, D.J.; Ramesh, V.; Silva, A.J. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med. 2008, 14, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Kasai, S.; Kobayashi, T.; Takamatsu, Y.; Hino, O.; Ikeda, K.; Mizuguchi, M. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat. Commun. 2012, 3, 1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Kara, N.Z.; Flaisher-Grinberg, S.; Anderson, G.W.; Agam, G.; Einat, H. Mood-stabilizing effects of rapamycin and its analog temsirolimus: Relevance to autophagy. Behav. Pharmacol. 2018, 29, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H.; National Institute for Clinical Excellence. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef]
- Velakoulis, D.; Walterfang, M.; Mocellin, R.; Pantelis, C.; McLean, C. Frontotemporal dementia presenting as schizophrenia-like psychosis in young people: Clinicopathological series and review of cases. Br. J. Psychiatry 2009, 194, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Lyketsos, C.G.; Carrillo, M.C.; Ryan, J.M.; Khachaturian, A.S.; Trzepacz, P.; Amatniek, J.; Cedarbaum, J.; Brashear, R.; Miller, D.S. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement. 2011, 7, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornai, F.; Frati, A.; Gesi, M.; Fulceri, F.; Paparelli, S.; Falleni, A.; Ruggieri, S. Neurobiology and neuroanatomy of psychiatric symptoms in parkinsonism. Arch. Ital. Biol. 2013, 151, 179–191. [Google Scholar] [PubMed]
- Aarsland, D.; Kramberger, M.G. Neuropsychiatric Symptoms in Parkinson’s Disease. J. Parkinsons Dis. 2015, 5, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Kawakami, I.; Onaya, M.; Higashi, S.; Arai, N.; Akiyama, H.; Hasegawa, M.; Arai, T. Frontotemporal dementia with trans-activation response DNA-binding protein 43 presenting with catatonic syndrome. Neuropathology 2018, 38, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Gururajan, A.; van den Buuse, M. Is the mTOR-signalling cascade disrupted in Schizophrenia? J. Neurochem. 2014, 129, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Maas, D.A.; Vallès, A.; Martens, G.J.M. Oxidative stress, prefrontal cortex hypomyelination and cognitive symptoms in schizophrenia. Transl. Psychiatry 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Khan, B.K.; Woolley, J.D.; Chao, S.; See, T.; Karydas, A.M.; Miller, B.L.; Rankin, K.P. Schizophrenia or neurodegenerative disease prodrome? Outcome of a first psychotic episode in a 35-year-old woman. Psychosomatics 2012, 53, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.; Ishikawa, T.; Michiue, T.; Li, D.R.; Zhao, D.; Oritani, S.; Zhu, B.L.; Maeda, H. Ubiquitin-immunoreactive structures in the midbrain of methamphetamine abusers. Leg. Med. 2005, 3, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Lenzi, P.; Gesi, M.; Soldani, P.; Ferrucci, M.; Lazzeri, G.; Capobianco, L.; Battaglia, G.; De Blasi, A.; Nicoletti, F.; et al. Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells. J. Neurochem. 2004, 88, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Castino, R.; Lazzeri, G.; Lenzi, P.; Bellio, N.; Follo, C.; Ferrucci, M.; Fornai, F.; Isisoro, C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J. Neurochem. 2008, 106, 1426–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yui, K.; Ikemoto, S.; Goto, K.; Nishijima, K.; Yoshino, T.; Ishiguro, T. Spontaneous recurrence of methamphetamine-induced paranoid-hallucinatory states in female subjects: Susceptibility to psychotic states and implications for relapse of schizophrenia. Pharmacopsychiatry 2002, 35, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Fasihpour, B.; Molavi, S.; Shariat, S.V. Clinical features of inpatients with methamphetamine induced psychosis. J. Ment. Health 2013, 22, 341–349. [Google Scholar] [CrossRef] [PubMed]
- McKetin, R. Methamphetamine psychosis: Insights from the past. Addiction 2018, 113, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Gambardella, S.; Biagioni, F.; Busceti, C.; Fornai, F. Epigenetic effects induced by methamphetamine and methamphetamine-dependent oxidative stress. Oxid. Med. Cell. Longev. 2018, 2018, 4982453. [Google Scholar] [CrossRef]
- Abi-Dargham, A.; Moore, H. Prefrontal DA transmission at D1 receptors and the pathology of schizophrenia. Neuroscientist 2003, 9, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Perreault, M.L.; Hasbi, A.; Alijaniaram, M.; Fan, T.; Varghese, G.; Fletcher, P.J.; Seeman, P.; O’Dowd, B.F.; George, S.R. The dopamine D1-D2 receptor heteromer localizes in dynorphin/enkephalin neurons: Increased high affinity state following amphetamine and in schizophrenia. J. Biol. Chem. 2010, 285, 36625–36634. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ji, X.; Liu, J.; Li, Z.; Zhang, X. Dopamine Receptor Subtypes Differentially Regulate Autophagy. Int. J. Mol. Sci. 2018, 19, 1540. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, L.; Lazzeri, G.; Isidoro, C.; Ruggieri, S.; Paparelli, A.; Fornai, F. Role of autophagy during methamphetamine neurotoxicity. Ann. N. Y. Acad. Sci. 2008, 1139, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Chandramani-Shivalingappa, P.; Jin, H.; Ghosh, A.; Anantharam, V.; Ali, S.; Kanthasamy, A.G.; Kanthasamy, A. Methamphetamine-induced neurotoxicity linked to ubiquitin-proteasome system dysfunction and autophagy-related changes that can be modulated by protein kinase C delta in dopaminergic neuronal cells. Neuroscience 2012, 210, 308–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aki, T.; Funakoshi, T.; Unuma, K.; Uemura, K. Impairment of autophagy: From hereditary disorder to drug intoxication. Toxicology 2013, 311, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Wu, W.R.; Lee, L.M.; Huang, P.R.; Chen, J.C. mTOR signaling in the nucleus accumbens mediates behavioral sensitization to methamphetamine. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 86, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Meredith, C.W.; Jaffe, C.; Ang-Lee, K.; Saxon, A.J. Implications of chronic methamphetamine use: A literature review. Harv. Rev. Psychiatry 2005, 13, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Homer, B.D.; Solomon, T.M.; Moeller, R.W.; Mascia, A.; DeRaleau, L.; Halkitis, P.N. Methamphetamine abuse and impairment of social functioning: A review of the underlying neurophysiological causes and behavioral implications. Psychol. Bull. 2008, 134, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Fowler, J.S.; Wang, G.J.; Shumay, E.; Telang, F.; Thanos, P.K.; Alexoff, D. Distribution and Pharmacokinetics of Methamphetamine in the Human Body: Clinical Implications. PLoS ONE 2010, 5, e15269. [Google Scholar] [CrossRef] [PubMed]
- Laruelle, M.; Abi-Dargham, A.; Gil, R.; Kegeles, L.; Innis, R. Increased dopamine transmission in schizophrenia: Relationship to illness phases. Biol. Psychiatry 1999, 46, 56–72. [Google Scholar] [CrossRef]
- Weidenauer, A.; Bauer, M.; Sauerzopf, U.; Bartova, L.; Praschak-Rieder, N.; Sitte, H.H.; Kasper, S.; Willeit, M. Making Sense of: Sensitization in Schizophrenia. Int. J. Neuropsychopharmacol. 2016, 20, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, B. Neurochemistry of schizophrenia: The contribution of neuroimaging postmortem pathology and neurochemistry in schizophrenia. Curr. Top. Med. Chem. 2012, 12, 2375–2392. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Kambeitz, J.; Kim, E.; Stahl, D.; Slifstein, M.; Abi-Dargham, A.; Kapur, S. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch. Gen. Psychiatry 2012, 69, 776–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abekawa, T.; Ohmori, T.; Koyama, T. Effects of repeated administration of a high dose of methamphetamine on dopamine and glutamate release in rat striatum and nucleus accumbens. Brain Res. 1994, 643, 276–281. [Google Scholar] [CrossRef]
- Stephans, S.E.; Yamamoto, B.Y. Effect of repeated methamphetamine administrations on dopamine and glutamate efflux in rat prefrontal cortex. Brain Res. 1995, 700, 99–106. [Google Scholar] [CrossRef]
- Nishijima, K.; Kashiwa, A.; Hashimoto, A.; Iwama, H.; Umino, A.; Nishikawa, T. Differential effects of phencyclidine and methamphetamine on dopamine metabolism in rat frontal cortex and striatum as revealed by in vivo dialysis. Synapse 1996, 22, 304–312. [Google Scholar] [CrossRef]
- Larsen, K.E.; Fon, E.A.; Hastings, T.G.; Edwards, R.H.; Sulzer, D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J. Neurosci. 2002, 22, 8951–8960. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Sumiyoshi, T.; Itoh, H.; Kurachi, M. Inhibition of dopamine synthesis with alpha-methyl-p-tyrosine abolishes the enhancement of methamphetamine-induced extracellular dopamine levels in the amygdala of rats with excitotoxic lesions of the entorhinal cortex. Neurosci. Lett. 2004, 356, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Purves-Tyson, T.D.; Owens, S.J.; Rothmond, D.A.; Halliday, G.M.; Double, K.L.; Stevens, J.; McCrossin, T.; Shannon Weickert, C. Putative presynaptic dopamine dysregulation in schizophrenia is supported by molecular evidence from post-mortem human midbrain. Transl. Psychiatry 2017, 7, e1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkow, N.D.; Chang, L.; Wang, G.J.; Fowler, J.S.; Leonido-Yee, M.; Franceschi, D.; Sedler, M.J.; Gatley, S.J.; Hitzemann, R.; Ding, Y.S.; et al. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am. J. Psychiatry 2001, 158, 377–382. [Google Scholar] [CrossRef] [PubMed]
- German, C.L.; Hanson, G.R.; Fleckenstein, A.E. Amphetamine and methamphetamine reduce striatal dopamine transporter function without concurrent dopamine transporter relocalization. J. Neurochem. 2012, 123, 288–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Li, W.; Huang, S.; Song, J.; Kim, J.Y.; Tian, X.; Kang, E.; Sano, Y.; Liu, C.; Balaji, J.; et al. mTOR Inhibition ameliorates cognitive and affective deficits caused by Disc1 knockdown in adult-born dentate granule neurons. Neuron 2013, 77, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, H.; Elgersma, Y.; Bahn, S. A brain proteomic investigation of rapamycin effects in the Tsc1+/− mouse model. Mol. Autism 2017, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Sotnikova, T.D.; Yao, W.D.; Kockeritz, L.; Woodgett, J.R.; Gainetdinov, R.R.; Caron, M.G. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. USA 2004, 101, 5099–5104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrián, C.; Mosharov, E.V.; Tang, G.; Cheng, H.C.; Kholodilov, N.; Yarygina, O.; Burke, R.E.; et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron 2012, 74, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.A.; Staal, R.G.; Sonsalla, P.K. Analysis of VMAT2 binding after methamphetamine or MPTP treatment: Disparity between homogenates and vesicle preparations. J. Neurochem. 2000, 74, 2217–2220. [Google Scholar] [CrossRef] [PubMed]
- Guillot, T.S.; Shepherd, K.R.; Richardson, J.R.; Wang, M.Z.; Li, Y.; Emson, P.C.; Miller, G.W. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J. Neurochem. 2008, 106, 2205–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubells, J.F.; Rayport, S.; Rajendran, G.; Sulzer, D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J. Neurosci. 1994, 14, 2260–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornai, F.; Chen, K.; Giorgi, F.S.; Gesi, M.; Alessandri, M.G.; Shih, J.C. Striatal dopamine metabolism in monoamine oxidase B-deficient mice: A brain dialysis study. J. Neurochem. 1999, 73, 2434–2440. [Google Scholar] [CrossRef] [PubMed]
- Gesi, M.; Santinami, A.; Ruffoli, R.; Conti, G.; Fornai, F. Novel aspects of dopamine oxidative metabolism (confounding outcomes take place of certainties). Pharmacol. Toxicol. 2001, 89, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, G.; Lenzi, P.; Busceti, C.L.; Ferrucci, M.; Falleni, A.; Bruno, V.; Paparelli, A.; Fornai, F. Mechanisms involved in the formation of dopamine-induced intracellular bodies within striatal neurons. J. Neurochem. 2007, 101, 1414–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornai, F.; Lenzi, P.; Frenzilli, G.; Gesi, M.; Ferrucci, M.; Lazzeri, G.; Biagioni, F.; Nigro, M.; Falleni, A.; Giusiani, M.; et al. DNA damage and ubiquitinated neuronal inclusions in the substantia nigra and striatum of mice following MDMA (ecstasy). Psychopharmacology 2004, 173, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Butler, B.; Gamble-George, J.; Prins, P.; North, A.; Clarke, J.T.; Khoshbouei, H. Chronic Methamphetamine Increases Alpha-Synuclein Protein Levels in the Striatum and Hippocampus but not in the Cortex of Juvenile Mice. J. Addict. Prev. 2014, 2, 6. [Google Scholar] [PubMed]
- Ferrucci, M.; Ryskalin, L.; Biagioni, F.; Gambardella, S.; Busceti, C.L.; Falleni, A.; Lazzeri, G.; Fornai, F. Methamphetamine increases Prion Protein and induces dopamine-dependent expression of protease resistant PrPsc. Arch. Ital. Biol. 2017, 155, 81–97. [Google Scholar] [PubMed]
- Kobayashi, H.; Ide, S.; Hasegawa, J.; Ujike, H.; Sekine, Y.; Ozaki, N.; Inada, T.; Harano, M.; Komiyama, T.; Yamada, M.; et al. Study of association between α-synuclein gene polymorphism and methamphetamine psychosis/dependence. Ann. N. Y. Acad. Sci. 2004, 1025, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, J.; Nilsson, C.; Kachergus, J.; Munz, M.; Larsson, E.M.; Schüle, B.; Langston, J.W.; Middleton, F.A.; Ross, O.A.; Hulihan, M.; et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 2007, 68, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Kolomeets, N.S.; Uranova, N.A. Synaptic contacts in schizophrenia: Study with immunocytochemical identification of dopaminergic neurons. Zhurnal Nevrol. Psikhiatrii Imeni S. S. Korsakova 1997, 97, 39–43. [Google Scholar] [CrossRef]
- Uranova, N.A.; Levité, O.I. Ultrastructure of the substantia nigra in schizophrenia. Zhurnal Nevrol. Psikhiatrii Imeni S. S. Korsakova 1987, 87, 1017–1024. [Google Scholar]
- Arnold, S.E.; Lee, V.M.; Gur, R.E.; Trojanowski, J.Q. Abnormal expression of two microtubule-associated proteins (MAP2 and MAP5) in specific subfields of the hippocampal formation in schizophrenia. Proc. Natl. Acad. Sci. USA 1991, 88, 10850–10854. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.; Kerwin, R.; Doshi, B.; Martin, C.S.; Everall, I.P. Alterations in hippocampal non-phosphorylated MAP2 protein expression in schizophrenia. Brain Res. 1997, 765, 238–246. [Google Scholar] [CrossRef]
- Rosoklija, G.; Keilp, J.G.; Toomayan, G.; Mancevski, B.; Haroutunian, V.; Liu, D.; Malespina, D.; Hays, A.P.; Sadiq, S.; Latov, N.; et al. Altered subicular MAP2 immunoreactivity in schizophrenia. Prilozi 2005, 26, 13–34. [Google Scholar] [PubMed]
- Somenarain, L.; Jones, L.B. A comparative study of MAP2 immunostaining in areas 9 and 17 in schizophrenia and Huntington chorea. J. Psychiatr. Res. 2010, 44, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.A.; Newman, J.T.; Gu, H.; Sampson, A.R.; Fish, K.N.; MacDonald, M.L.; Moyer, C.E.; DiBitetto, J.V.; Dorph-Petersen, K.A.; Penzes, P.; et al. Loss of Microtubule-Associated Protein 2 Immunoreactivity Linked to Dendritic Spine Loss in Schizophrenia. Biol. Psychiatry 2015, 78, 374–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soustek, Z. Ultrastructure of cortical synapses in the brain of schizophrenics. Zentralbl. Allg. Pathol. 1989, 135, 25–32. [Google Scholar] [PubMed]
- Prohovnik, I.; Dwork, A.J.; Kaufman, M.A.; Willson, N. Alzheimer-type neuropathology in elderly schizophrenia patients. Schizophr. Bull. 1993, 19, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Constantinidis, J.; Wegiel, J.; Bobinski, M.; Tarnawski, M. Neurofibrillary pathology in brains of elderly schizophrenics treated with neuroleptics. Alzheimer Dis. Assoc. Disord. 1994, 8, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Trojanowski, J.Q.; Gur, R.E.; Blackwell, P.; Han, L.Y.; Choi, C. Absence of neurodegeneration and neural injury in the cerebral cortex in a sample of elderly patients with schizophrenia. Arch. Gen. Psychiatry 1998, 55, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Purohit, D.P.; Perl, D.P.; Haroutunian, V.; Powchik, P.; Davidson, M.; Davis, K.L. Alzheimer disease and related neurodegenerative diseases in elderly patients with schizophrenia: A postmortem neuropathologic study of 100 cases. Arch. Gen. Psychiatry 1998, 55, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Falke, E.; Han, L.Y.; Arnold, S.E. Absence of neurodegeneration in the thalamus and caudate of elderly patients with schizophrenia. Psychiatry Res. 2000, 93, 103–110. [Google Scholar] [CrossRef]
- Bozikas, V.P.; Kövari, E.; Bouras, C.; Karavatos, A. Neurofibrillary tangles in elderly patients with late onset schizophrenia. Neurosci. Lett. 2002, 324, 109–112. [Google Scholar] [CrossRef]
- Momeni, P.; Wickremaratchi, M.M.; Bell, J.; Arnold, R.; Beer, R.; Hardy, J.; Revesz, T.; Neal, J.W.; Morris, H.R. Familial early onset frontotemporal dementia caused by a novel S356T MAPT mutation, initially diagnosed as schizophrenia. Clin. Neurol. Neurosurg. 2010, 112, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Uranova, N.A.; Vikhreva, O.V.; Rachmanova, V.I.; Orlovskaya, D.D. Ultrastructural alterations of myelinated fibers and oligodendrocytes in the prefrontal cortex in schizophrenia: A postmortem morphometric study. Schizophr. Res. Treat. 2011, 2011, 325789. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.K.; Roche, J.K.; Sinha, V.; Roberts, R.C. Substantia nigra ultrastructural pathology in schizophrenia. Schizophr. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Leliveld, S.R.; Bader, V.; Hendriks, P.; Prikulis, I.; Sajnani, G.; Requena, J.R.; Korth, C. Insolubility of disrupted-in-schizophrenia 1 disrupts oligomer-dependent interactions with nuclear distribution element 1 and is associated with sporadic mental disease. J. Neurosci. 2008, 28, 3839–3845. [Google Scholar] [CrossRef] [PubMed]
- Ottis, P.; Bader, V.; Trossbach, S.V.; Kretzschmar, H.; Michel, M.; Leliveld, S.R.; Korth, C. Convergence of two independent mental disease genes on the protein level: Recruitment of dysbindin to cell-invasive disrupted-in-schizophrenia 1 aggresomes. Biol. Psychiatry 2011, 70, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Atkin, T.; Kittler, J. DISC1 and the aggresome: A disruption to cellular function? Autophagy 2012, 8, 851–852. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sun, Y.; Ye, H.; Zhu, L.; Liu, J.; Wu, X.; Wang, L.; He, T.; Shen, Y.; Wu, J.Y.; et al. Increased dysbindin-1B isoform expression in schizophrenia and its propensity in aggresome formation. Cell Discov. 2015, 1, 15032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camargo, L.M.; Collura, V.; Rain, J.C.; Mizuguchi, K.; Hermjakob, H.; Kerrien, S.; Bonnert, T.P.; Whiting, P.J.; Brandon, N.J. Disrupted in Schizophrenia 1 Interactome: Evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol. Psychiatry 2007, 12, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Martins-de-Souza, D.; Cassoli, J.S.; Nascimento, J.M.; Hensley, K.; Guest, P.C.; Pinzon-Velasco, A.M.; Turck, C.W. The protein interactome of collapsin response mediator protein-2 (CRMP2/DPYSL2) reveals novel partner proteins in brain tissue. Proteom. Clin. Appl. 2015, 9, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Edgar, P.F.; Douglas, J.E.; Cooper, G.J.; Dean, B.; Kydd, R.; Faull, R.L. Comparative proteome analysis of the hippocampus implicates chromosome 6q in schizophrenia. Mol. Psychiatry 2000, 5, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston-Wilson, N.L.; Sims, C.D.; Hofmann, J.P.; Anderson, L.; Shore, A.D.; Torrey, E.F.; Yolken, R.H. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol. Psychiatry 2000, 5, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Beasley, C.L.; Pennington, K.; Behan, A.; Wait, R.; Dunn, M.J.; Cotter, D. Proteomic analysis of the anterior cingulate cortex in the major psychiatric disorders: Evidence for disease-associated changes. Proteomics 2006, 6, 3414–3425. [Google Scholar] [CrossRef] [PubMed]
- Martins-de-Souza, D.; Gattaz, W.F.; Schmitt, A.; Maccarrone, G.; Hunyadi-Gulyás, E.; Eberlin, M.N.; Souza, G.H.; Marangoni, S.; Novello, J.C.; Turck, C.W.; et al. Proteomic analysis of dorsolateral prefrontal cortex indicates the involvement of cytoskeleton, oligodendrocyte, energy metabolism and new potential markers in schizophrenia. J. Psychiatr. Res. 2009, 43, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.; Verstreken, P. Autophagy in the presynaptic compartment in health and disease. J. Cell Biol. 2017, 216, 1895–1906. [Google Scholar] [CrossRef] [PubMed]
- Merenlender-Wagner, A.; Shemer, Z.; Touloumi, O.; Lagoudaki, R.; Giladi, E.; Andrieux, A.; Grigoriadis, N.C.; Gozes, I. New horizons in schizophrenia treatment: Autophagy protection is coupled with behavioral improvements in a mouse model of schizophrenia. Autophagy 2014, 10, 2324–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merenlender-Wagner, A.; Malishkevich, A.; Shemer, Z.; Udawela, M.; Gibbons, A.; Scarr, E.; Dean, B.; Levine, J.; Agam, G.; Gozes, I. Autophagy has a key role in the pathophysiology of schizophrenia. Mol. Psychiatry 2015, 20, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown, R.H., Jr.; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef] [PubMed]
- Daoust, A.; Bohic, S.; Saoudi, Y.; Debacker, C.; Gory-Fauré, S.; Andrieux, A.; Barbier, E.L.; Deloulme, J.C. Neuronal transport defects of the MAP6 KO mouse—A model of schizophrenia- and alleviation by Epothilone D treatment, as observed using MEMRI. Neuroimage 2014, 96, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Riva, M.A.; Molteni, R. Synaptic alterations associated with depression and schizophrenia: Potential as a therapeutic target. Expert Opin. Ther. Targets 2016, 20, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Bridi, J.C.; Hirth, F. Mechanisms of α-Synuclein Induced Synaptopathy in Parkinson’s Disease. Front. Neurosci. 2018, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- St Clair, D.; Blackwood, D.; Muir, W.; Carothers, A.; Walker, M.; Spowart, G.; Gosden, C.; Evans, H.J. Association within a family of a balanced autosomal translocation with major mental illness. Lancet 1990, 336, 13–16. [Google Scholar] [CrossRef]
- Schurov, I.L.; Handford, E.J.; Brandon, N.J.; Whiting, B.J. Expression of disrupted in schizophrenia 1 (DISC1) protein in the adult and developing mouse brain indicates its role in neurodevelopment. Mol. Psychiatry 2004, 9, 1100–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandon, N.J.; Sawa, A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat. Rev. Neurosci. 2011, 12, 707–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hikida, T.; Gamo, N.J.; Sawa, A. DISC1 as a therapeutic target for mental illnesses. Expert Opin. Ther. Targets 2012, 16, 1151–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipska, B.K.; Mitkus, S.N.; Mathew, S.V.; Fatula, R.; Hyde, T.M.; Weinberger, D.R.; Kleinman, J.E. Functional genomics in postmortem human brain: Abnormalities in a DISC1 molecular pathway in schizophrenia. Dialogues Clin. Neurosci. 2006, 8, 353–357. [Google Scholar] [PubMed]
- Dahoun, T.; Trossbach, S.V.; Brandon, N.J.; Korth, C.; Howes, O.D. The impact of Disrupted-in-Schizophrenia 1 (DISC1) on the dopaminergic system: A systematic review. Transl. Psychiatry 2017, 7, e1015. [Google Scholar] [CrossRef] [PubMed]
- Nakai, T.; Nagai, T.; Wang, R.; Yamada, S.; Kuroda, K.; Kaibuchi, K.; Yamada, K. Alterations of GABAergic and dopaminergic systems in mutant mice with disruption of exons 2 and 3 of the Disc1 gene. Neurochem. Int. 2014, 74, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogorelov, V.M.; Nomura, J.; Kim, J.; Kannan, G.; Ayhan, Y.; Yang, C.; Taniguchi, Y.; Abazyan, B.; Valentine, H.; Krasnova, I.N.; et al. Mutant DISC1 affects methamphetamine-induced sensitization and conditioned place preference: A comorbidity model. Neuropharmacology 2012, 62, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Niwa, M.; Lee, R.S.; Tanaka, T.; Okada, K.; Kano, S.; Sawa, A. A critical period of vulnerability to adolescent stress: Epigenetic mediators in mesocortical dopaminergic neurons. Hum. Mol. Genet. 2016, 25, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Duan, X.; Liu, C.Y.; Jang, M.H.; Guo, J.U.; Pow-anpongkul, N.; Kang, E.; Song, H.; Ming, G.L. DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron 2009, 63, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, D.H.; Fordyce, A.; Walker, M.T.; St Clair, D.M.; Porteous, D.J.; Muir, W.J. Schizophrenia and affective disorders—Cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: Clinical and P300 findings in a family. Am. J. Hum. Genet. 2001, 69, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Clapcote, S.J.; Lipina, T.V.; Millar, J.K.; Mackie, S.; Christie, S.; Ogawa, F.; Lerch, J.P.; Trimble, K.; Uchiyama, M.; Sakuraba, Y.; et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 2007, 54, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Chubb, J.E.; Bradshaw, N.J.; Soares, D.C.; Porteous, D.J.; Millar, J.K. The DISC locus in psychiatric illness. Mol. Psychiatry 2008, 13, 36–64. [Google Scholar] [CrossRef] [PubMed]
- Gangarossa, G.; Ceolin, L.; Paucard, A.; Lerner-Natoli, M.; Perroy, J.; Fagni, L.; Valjent, E. Repeated stimulation of dopamine D1-like receptor and hyperactivation of mTOR signaling lead to generalized seizures, altered dentate gyrus plasticity, and memory deficits. Hippocampus 2014, 24, 1466–1481. [Google Scholar] [CrossRef] [PubMed]
- Siuta, M.A.; Robertson, S.D.; Kocalis, H.; Saunders, C.; Gresch, P.J.; Khatri, V.; Shiota, C.; Kennedy, J.P.; Lindsley, C.W.; Daws, L.C.; et al. Dysregulation of the norepinephrine transporter sustains cortical hypodopaminergia and schizophrenia-like behaviors in neuronal rictor null mice. PLoS Biol. 2010, 8, e1000393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emamian, E.S.; Hall, D.; Birnbaum, M.J.; Karayiorgou, M.; Gogos, J.A. Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nat. Genet. 2004, 36, 131–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, M.; Iwata, N.; Suzuki, T.; Kitajima, T.; Yamanouchi, Y.; Kinoshita, Y.; Inada, T.; Ozaki, N. Association of AKT1 with schizophrenia confirmed in a Japanese population. Biol. Psychiatry 2004, 56, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Schwab, S.G.; Hoefgen, B.; Hanses, C.; Hassenbach, M.B.; Albus, M.; Lerer, B.; Trixler, M.; Maier, W.; Wildenauer, D.B. Further evidence for association of variants in the AKT1 gene with schizophrenia in a sample of European sib-pair families. Biol. Psychiatry 2005, 58, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Bajestan, S.N.; Sabouri, A.H.; Nakamura, M.; Takashima, H.; Keikhaee, M.R.; Behdani, F.; Fayyazi, M.R.; Sargolzaee, M.R.; Bajestan, M.N.; Sabouri, Z.; et al. Association of AKT1 haplotype with the risk of schizophrenia in Iranian population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Q.; Xing, Q.H.; Zheng, Y.L.; Li, S.; Gao, J.J.; He, G.; Guo, T.W.; Feng, G.Y.; Xu, F.; He, L. Association of AKT1 gene polymorphisms with risk of schizophrenia and with response to antipsychotics in the Chinese population. J. Clin. Psychiatry 2007, 68, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Thiselton, D.L.; Vladimirov, V.I.; Kuo, P.H.; McClay, J.; Wormley, B.; Fanous, A.; O’Neill, F.A.; Walsh, D.; Van den Oord, E.J.; Kendler, K.S.; et al. AKT1 is associated with schizophrenia across multiple symptom dimensions in the Irish study of high density schizophrenia families. Biol. Psychiatry 2008, 63, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Karege, F.; Méary, A.; Perroud, N.; Jamain, S.; Leboyer, M.; Ballmann, E.; Fernandez, R.; Malafosse, A.; Schürhoff, F. Genetic overlap between schizophrenia and bipolar disorder: A study with AKT1 gene variants and clinical phenotypes. Schizophr. Res. 2012, 135, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Nohesara, S.; Ghadirivasfi, M.; Barati, M.; Ghasemzadeh, M.R.; Narimani, S.; Mousavi-Behbahani, Z.; Joghataei, M.; Soleimani, M.; Taban, M.; Mehrabi, S.; et al. Methamphetamine-induced psychosis is associated with DNA hypomethylation and increased expression of AKT1 and key dopaminergic genes. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Petryshen, T.L.; Middleton, F.A.; Kirby, A.; Aldinger, K.A.; Purcell, S.; Tahl, A.R.; Morley, C.P.; McGann, L.; Gentile, K.L.; Rockwell, G.N.; et al. Support for involvement of neuregulin 1 in schizophrenia pathophysiology. Mol. Psychiatry 2005, 10, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Law, A.J.; Kleinman, J.E.; Weinberger, D.R.; Weickert, C.S. Disease-associated intronic variants in the ErbB4 gene are related to altered ErbB4 splice-variant expression in the brain in schizophrenia. Hum. Mol. Genet. 2007, 16, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.G.; Wang, H.Y.; Cho, D.S.; Talbot, K.; Gur, R.E.; Berrettini, W.H.; Bakshi, K.; Kamins, J.; Borgmann-Winter, K.E.; Siegel, S.J.; et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat. Med. 2006, 12, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Xiong, W.C. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. 2008, 9, 437–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshadri, S.; Kamiya, A.; Yokota, Y.; Prikulis, I.; Kano, S.; Hayashi-Takagi, A.; Stanco, A.; Eom, T.Y.; Rao, S.; Ishizuka, K.; et al. Disrupted-in-Schizophrenia-1 expression is regulated by beta-site amyloid precursor protein cleaving enzyme-1-neuregulin cascade. Proc. Natl. Acad. Sci. USA 2010, 107, 5622–5627. [Google Scholar] [CrossRef] [PubMed]
- Skirzewski, M.; Karavanova, I.; Shamir, A.; Erben, L.; Garcia-Olivares, J.; Shin, J.H.; Vullhorst, D.; Alvarez, V.A.; Amara, S.G.; Buonanno, A. ErbB4 Signaling in Dopaminergic Axonal Projections Increases Extracellular Dopamine Levels and Regulates Spatial/Working Memory Behaviors. Mol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Murtie, J.C.; El-Khodor, B.F.; Edgar, N.; Sardi, S.P.; Hooks, B.M.; Benoit-Marand, M.; Chen, C.; Moore, H.; O’Donnell, P.; et al. Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 8131–8136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Tang, R.; Xiao, Z.; Shi, Y.; Feng, G.; Gu, N.; Shi, J.; Xing, Y.; Yan, L.; Sang, H.; et al. An investigation of the dihydropyrimidinase-like 2 (DPYSL2) gene in schizophrenia: Genetic association study and expression analysis. Int. J. Neuropsychopharmacol. 2006, 9, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Ujike, H.; Sakai, A.; Takaki, M.; Imamura, T.; Tanaka, Y.; Kuroda, S. The human dihydropyrimidinase-related protein 2 gene on chromosome 8p21 is associated with paranoid-type schizophrenia. Biol. Psychiatry 2003, 53, 571–576. [Google Scholar] [CrossRef]
- Fallin, M.D.; Lasseter, V.K.; Liu, Y.; Avramopoulos, D.; McGrath, J.; Wolyniec, P.S.; Nestadt, G.; Liang, K.Y.; Chen, P.L.; Valle, D.; et al. Linkage and association on 8p21.2-p21.1 in schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pham, X.; Zhang, L.; Chen, P.L.; Burzynski, G.; McGaughey, D.M.; He, S.; McGrath, J.A.; Wolyniec, P.; Fallin, M.D.; et al. Functional variants in DPYSL2 sequence increase risk of schizophrenia and suggest a link to mTOR signaling. G3 2014, 5, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Chong, S.A.; Keefe, R. Psychosocial factors in the neurobiology of schizophrenia: A selective review. Ann. Acad. Med. Singap. 2009, 38, 402–406. [Google Scholar] [PubMed]
- Lee, H.; Joo, J.; Nah, S.S.; Kim, J.W.; Kim, H.K.; Kwon, J.T.; Lee, H.Y.; Kim, Y.O.; Kim, H.J. Changes in Dpysl2 expression are associated with prenatally stressed rat offspring and susceptibility to schizophrenia in humans. Int. J. Mol. Med. 2015, 35, 1574–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobeissy, F.H.; Warren, M.W.; Ottens, A.K.; Sadasivan, S.; Zhang, Z.; Gold, M.S.; Wang, K.K. Psychoproteomic analysis of rat cortex following acute methamphetamine exposure. J. Proteome Res. 2008, 7, 1971–1983. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Yamashita, N.; Kimura, A.; Kimura, Y.; Hirano, H.; Makihara, H.; Kawamoto, Y.; Jitsuki-Takahashi, A.; Yonezaki, K.; Takase, K.; et al. Comprehensive behavioral study and proteomic analyses of CRMP2-deficient mice. Genes Cells 2016, 21, 1059–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Sasamoto, K.; Nagai, J.; Yamazaki, Y.; Saito, K.; Goshima, Y.; Inoue, T.; Ohshima, T. Phosphorylation of CRMP2 by Cdk5 Regulates Dendritic Spine Development of Cortical Neuron in the Mouse Hippocampus. Neural Plast. 2016, 2016, 6790743. [Google Scholar] [CrossRef] [PubMed]
- Benavides, D.R.; Bibb, J.A. Role of Cdk5 in drug abuse and plasticity. Ann. N. Y. Acad. Sci. 2004, 1025, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Mlewski, E.C.; Arias, C.; Paglini, G. Association between the expression of amphetamine-induced behavioral sensitization and Cdk5/p35 activity in dorsal striatum. Behav. Neurosci. 2016, 130, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Lebel, M.; Patenaude, C.; Allyson, J.; Massicotte, G.; Cyr, M. Dopamine D1 receptor activation induces tau phosphorylation via Cdk5 and GSK3 signaling pathways. Neuropharmacology 2009, 57, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Cantrup, R.; Sathanantham, K.; Rushlow, W.J.; Rajakumar, N. Chronic hyperdopaminergic activity of schizophrenia is associated with increased ΔFosB levels and cdk-5 signaling in the nucleus accumbens. Neuroscience 2012, 222, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Ferreras, S.; Fernández, G.; Danelon, V.; Pisano, M.V.; Masseroni, L.; Chapleau, C.A.; Krapacher, F.A.; Mlewski, E.C.; Mascó, D.H.; Arias, C.; et al. Cdk5 Is Essential for Amphetamine to Increase Dendritic Spine Density in Hippocampal Pyramidal Neurons. Front. Cell. Neurosci. 2017, 11, 372. [Google Scholar] [CrossRef] [PubMed]
- Engmann, O.; Hortobágyi, T.; Pidsley, R.; Troakes, C.; Bernstein, H.G.; Kreutz, M.R.; Mill, J.; Nikolic, M.; Giese, K.P. Schizophrenia is associated with dysregulation of a Cdk5 activator that regulates synaptic protein expression and cognition. Brain 2011, 134, 2408–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Eissa, M.M.; Fiorentino, A.; Sharp, S.I.; O’Brien, N.L.; Wolfe, K.; Giaroli, G.; Curtis, D.; Bass, N.J.; McQuillin, A. Exome sequence analysis and follow up genotyping implicates rare ULK1 variants to be involved in susceptibility to schizophrenia. Ann. Hum. Genet. 2018, 82, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Horesh, Y.; Katsel, P.; Haroutunian, V.; Domany, E. Gene expression signature is shared by patients with Alzheimer’s disease and schizophrenia at the superior temporal gyrus. Eur. J. Neurol. 2011, 18, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, P.W.; Wright, I.C.; Bullmore, E.T.; Brammer, M.; Howard, R.J.; Williams, S.C.; Shapleske, J.; Rossell, S.; David, A.S.; McGuire, P.K.; et al. Auditory hallucinations and the temporal cortical response to speech in schizophrenia: A functional magnetic resonance imaging study. Am. J. Psychiatry 1997, 154, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.; Larøi, F.; McGuire, P.K.; Aleman, A. The hallucinating brain: A review of structural and functional neuroimaging studies of hallucinations. Neurosci. Biobehav. Rev. 2008, 32, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.R.; Huxley-Jones, J.; Maycox, P.R.; Lennon, M.; Thornber, A.; Kelly, F.; Bates, S.; Taylor, A.; Reid, J.; Jones, N.; et al. Transcription and pathway analysis of the superior temporal cortex and anterior prefrontal cortex in schizophrenia. J. Neurosci. Res. 2011, 89, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Vinayagam, A.; Stelzl, U.; Foulle, R.; Plassmann, S.; Zenkner, M.; Timm, J.; Assmus, H.E.; Andrade-Navarro, M.A.; Wanker, E.E. A directed protein interaction network for investigating intracellular signal transduction. Sci. Signal. 2011, 4, rs8. [Google Scholar] [CrossRef] [PubMed]
- Dresner, E.; Agam, G.; Gozes, I. Activity-dependent neuroprotective protein (ADNP) expression level is correlated with the expression of the sister protein ADNP2: Deregulation in schizophrenia. Eur. Neuropsychopharmacol. 2011, 21, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C.; Buchanan, R.W.; Keefe, R.S.; Kern, R.; McMahon, R.P.; Green, M.F.; Lieberman, J.; Goff, D.C.; Csernansky, J.G.; McEvoy, J.P.; et al. Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr. Res. 2012, 136, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Krishna, G.; Imarisio, S.; Saili, S.; O’Kane, C.J.; Rubinsztein, D.C. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum. Mol. Genet. 2008, 17, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.C.; Lin, C.C.; Chen, T.T.; Chen, J.Y.; Tsai, H.J.; Wang, C.Y.; Chen, S.Y. Clozapine induces autophagic cell death in non-small cell lung cancer cells. Cell. Physiol. Biochem. 2015, 35, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Sade, Y.; Toker, L.; Kara, N.Z.; Einat, H.; Rapoport, S.; Moechars, D.; Berry, G.T.; Bersudsky, Y.; Agam, G. IP3 accumulation and/or inositol depletion: Two downstream lithium’s effects that may mediate its behavioral and cellular changes. Transl. Psychiatry 2016, 6, e968. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, S.; Yu, H.S.; Ko, K.H.; Park, H.G.; Kim, Y.S. The antipsychotic agent clozapine induces autophagy via the AMPK-ULK1-Beclin1 signaling pathway in the rat frontal cortex. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 81, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.D.; Jeong, E.I.; Nah, J.; Yoo, S.M.; Lee, W.J.; Kim, Y.; Moon, S.; Hong, S.H.; Jung, Y.K. Pimozide reduces toxic forms of tau in TauC3 mice via 5′ adenosine monophosphate-activated protein kinase-mediated autophagy. J. Neurochem. 2017, 142, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Park, S.J.; Kim, E.S.; Jo, Y.K.; Hong, J.; Cho, D.H. Sertindole, a potent antagonist at dopamine D₂ receptors, induces autophagy by increasing reactive oxygen species in SH-SY5Y neuroblastoma cells. Biol. Pharm. Bull. 2012, 35, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Charvin, D.; Roze, E.; Perrin, V.; Deyts, C.; Betuing, S.; Pagès, C.; Régulier, E.; Luthi-Carter, R.; Brouillet, E.; Déglon, N.; et al. Haloperidol protects striatal neurons from dysfunction induced by mutated huntingtin in vivo. Neurobiol. Dis. 2008, 29, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Höllerhage, M.; Goebel, J.N.; de Andrade, A.; Hildebrandt, T.; Dolga, A.; Culmsee, C.; Oertel, W.H.; Hengerer, B.; Höglinger, G.U. Trifluoperazine rescues human dopaminergic cells from wild-type α-synuclein-induced toxicity. Neurobiol. Aging 2014, 35, 1700–1711. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Jeong, H.J.; Liu, Q.F.; Oh, S.T.; Koo, B.S.; Kim, Y.; Chung, I.W.; Kim, Y.S.; Jeon, S. Clozapine Improves Memory Impairment and Reduces Aβ Level in the Tg-APPswe/PS1dE9 Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chung, S.; An, H.; Kim, J.; Seo, J.; Kim, D.H.; Yoon, S.Y. Haloperidol and clozapine block formation of autophagolysosomes in rat primary neurons. Neuroscience 2012, 209, 64–73. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The mammalian target of rapamycin (mTOR) pathway. The cartoon summarizes the main up- and down-stream components of the mTOR pathway. Growth factors, glucose, and amino acids activate mTOR, which in turn promotes protein synthesis, lipid metabolism, and mitochondrial biogenesis, while autophagy is under the negative control mTOR. GPCRs—G-protein coupled receptors. TSC—tuberous sclerosis complex; TSC1—hamartin; TSC2—tuberin; RTKs—receptor tyrosine kinase receptors; Akt—protein kinase B; PTEN—Phosphatase and Tensin Homolog; BDNF—Brain-derived neurotrophic factor; IGF—insulin-like growth factor; NGF—nerve growth factor.
Figure 1.
The mammalian target of rapamycin (mTOR) pathway. The cartoon summarizes the main up- and down-stream components of the mTOR pathway. Growth factors, glucose, and amino acids activate mTOR, which in turn promotes protein synthesis, lipid metabolism, and mitochondrial biogenesis, while autophagy is under the negative control mTOR. GPCRs—G-protein coupled receptors. TSC—tuberous sclerosis complex; TSC1—hamartin; TSC2—tuberin; RTKs—receptor tyrosine kinase receptors; Akt—protein kinase B; PTEN—Phosphatase and Tensin Homolog; BDNF—Brain-derived neurotrophic factor; IGF—insulin-like growth factor; NGF—nerve growth factor.

Figure 2.
Overlap of dopamine-dependent molecular mechanisms underlying methamphetamine (METH) and schizophrenia. In normal conditions (A), the amount of intra-cytosolic dopamine is determined by the rate limiting enzyme tyrosine hydroxylase (TH), which converts tyrosine into L-dihydroxyphenylalanine (L-DOPA) and eventually dopamine (DA). DA is selectively taken-up into synaptic vesicles by the vesicular monoamine transporter type-2 (VMAT-2), which is key to surveil the physiological storage of vesicular DA. DA-containing synaptic vesicles are coated with soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein receptor (SNARE) proteins co-chaperoned by alpha-synuclein, which mediate docking, priming, and release of DA-synaptic vesicles via exocytosis. Once exocytosis has occurred, synaptic vesicles and their associated proteins are endocytosed and sorted for autophagy (ATG) degradation. In this way, ATG monitors the amount of releasable DA synaptic vesicles, thus playing a key role in restraining DA release and in the turnover of synaptic proteins. In the synaptic cleft, the dopamine transporter (DAT) is key to take-up extracellular DA in order to guarantee a physiological stimulation of post-synaptic DA receptors. On the other hand, METH addicted and schizophrenic brains (B) feature alterations of DA metabolism and handling, which consist of the following: (i) increased levels of TH, which produces high levels of intra-cytosolic DA; (ii) a decrease in VMAT-2, which leads to a loss of DA vesicular storage and increases the amount freely diffusible intra-cytosolic DA; (iii) free cytosolic DA is highly prone to auto-oxidation into reactive DA-quinones, which produce structural modifications of presynaptic proteins such as alpha synuclein; (iv) a rapid and massive release of DA occurs via either exocytosis or efflux from the axoplasm; (v) extracellular DA rapidly accumulates as DAT is inhibited or downregulated, thus leading to abnormal stimulation of post-synaptic DA receptors, mainly D1-like receptors; (vi) dysfunctions in the ATG machinery, which cannot restrain DA release, are likely to play a key role in such a mechanism. In addition, impaired ATG cannot handle the oxidatively modified alpha-synuclein, thus leading to a progressive accumulation of alpha-synuclein aggregates fueling synaptic pathology.
Figure 2.
Overlap of dopamine-dependent molecular mechanisms underlying methamphetamine (METH) and schizophrenia. In normal conditions (A), the amount of intra-cytosolic dopamine is determined by the rate limiting enzyme tyrosine hydroxylase (TH), which converts tyrosine into L-dihydroxyphenylalanine (L-DOPA) and eventually dopamine (DA). DA is selectively taken-up into synaptic vesicles by the vesicular monoamine transporter type-2 (VMAT-2), which is key to surveil the physiological storage of vesicular DA. DA-containing synaptic vesicles are coated with soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein receptor (SNARE) proteins co-chaperoned by alpha-synuclein, which mediate docking, priming, and release of DA-synaptic vesicles via exocytosis. Once exocytosis has occurred, synaptic vesicles and their associated proteins are endocytosed and sorted for autophagy (ATG) degradation. In this way, ATG monitors the amount of releasable DA synaptic vesicles, thus playing a key role in restraining DA release and in the turnover of synaptic proteins. In the synaptic cleft, the dopamine transporter (DAT) is key to take-up extracellular DA in order to guarantee a physiological stimulation of post-synaptic DA receptors. On the other hand, METH addicted and schizophrenic brains (B) feature alterations of DA metabolism and handling, which consist of the following: (i) increased levels of TH, which produces high levels of intra-cytosolic DA; (ii) a decrease in VMAT-2, which leads to a loss of DA vesicular storage and increases the amount freely diffusible intra-cytosolic DA; (iii) free cytosolic DA is highly prone to auto-oxidation into reactive DA-quinones, which produce structural modifications of presynaptic proteins such as alpha synuclein; (iv) a rapid and massive release of DA occurs via either exocytosis or efflux from the axoplasm; (v) extracellular DA rapidly accumulates as DAT is inhibited or downregulated, thus leading to abnormal stimulation of post-synaptic DA receptors, mainly D1-like receptors; (vi) dysfunctions in the ATG machinery, which cannot restrain DA release, are likely to play a key role in such a mechanism. In addition, impaired ATG cannot handle the oxidatively modified alpha-synuclein, thus leading to a progressive accumulation of alpha-synuclein aggregates fueling synaptic pathology.

Figure 3.
The Akt/mTOR pathway in schizophrenia. The cartoon summarizes key proteins involved in schizophrenia (lightning bolts), which converge on the overactivation of the Akt/mTOR pathway. These include disrupted in schizophrenia 1 (DISC1), neuregulin-1 (NRG1)/avian erythroblastosis oncogene B4-like protein (ErbB4), and collapsin response mediator protein 2 (CRMP2), as well as dopamine D1 receptors (D1R), which in turn are modulated by DISC1 and NRG1/ErbB4.
Figure 3.
The Akt/mTOR pathway in schizophrenia. The cartoon summarizes key proteins involved in schizophrenia (lightning bolts), which converge on the overactivation of the Akt/mTOR pathway. These include disrupted in schizophrenia 1 (DISC1), neuregulin-1 (NRG1)/avian erythroblastosis oncogene B4-like protein (ErbB4), and collapsin response mediator protein 2 (CRMP2), as well as dopamine D1 receptors (D1R), which in turn are modulated by DISC1 and NRG1/ErbB4.


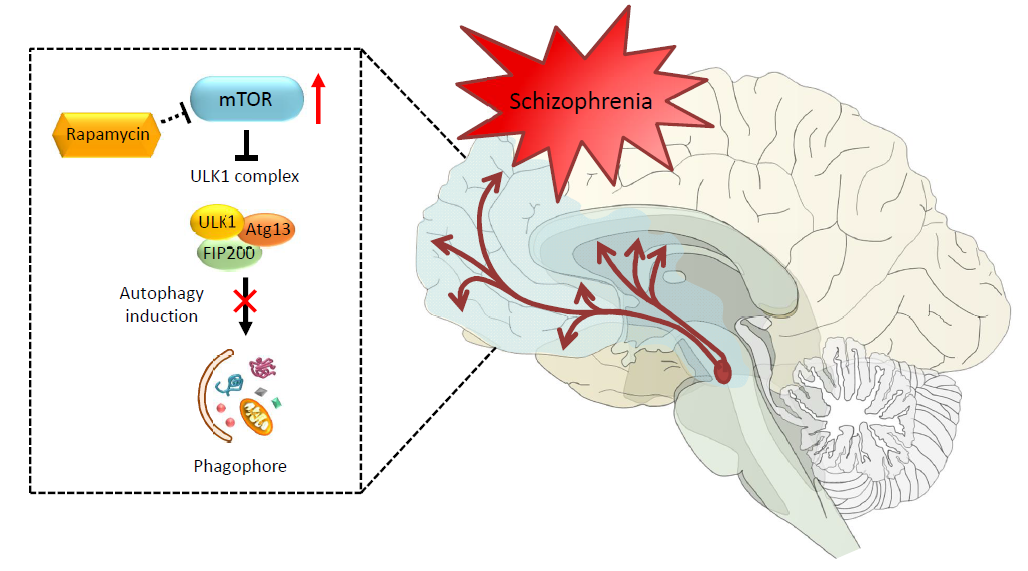{kind=link}
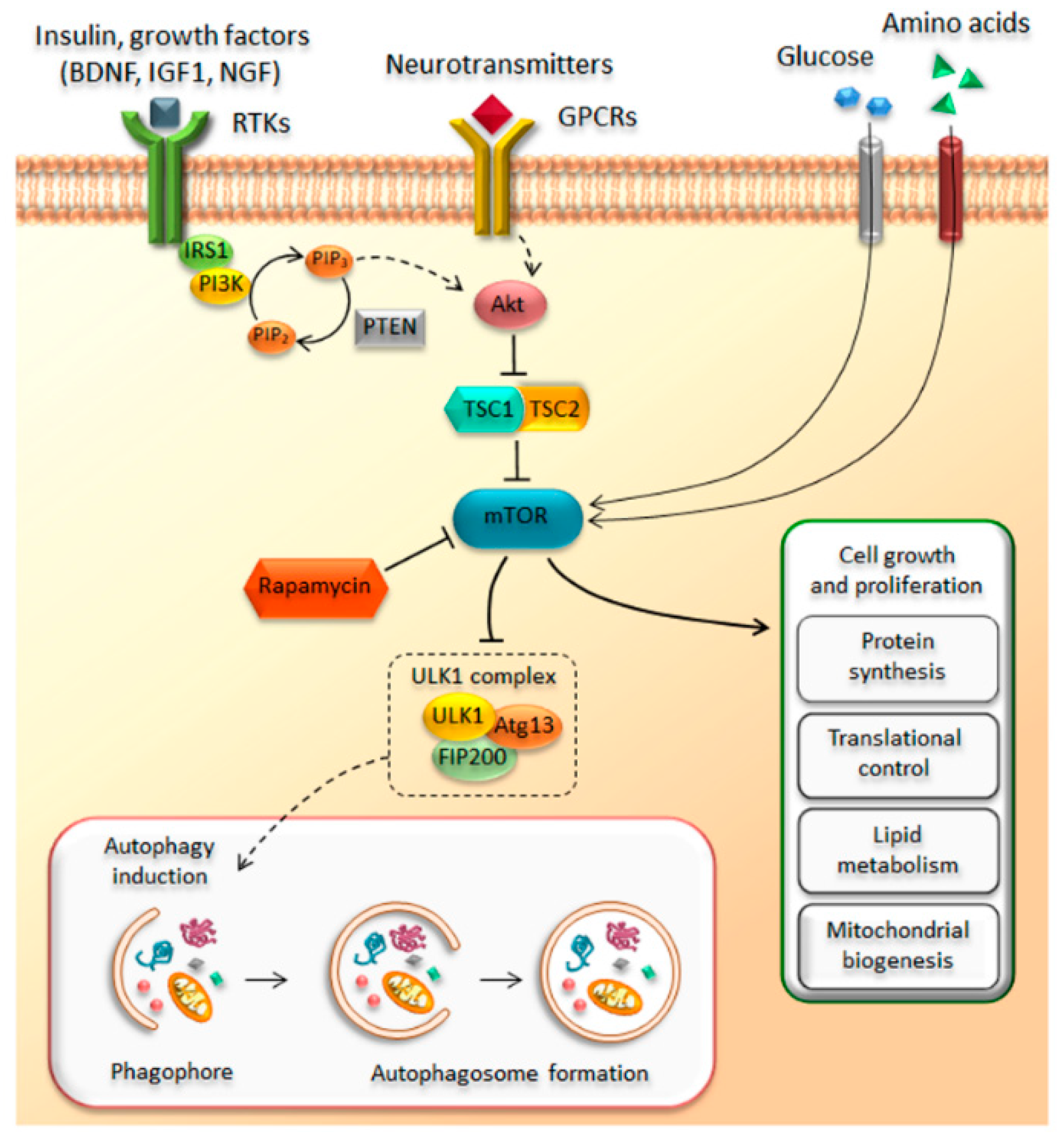{kind=link}
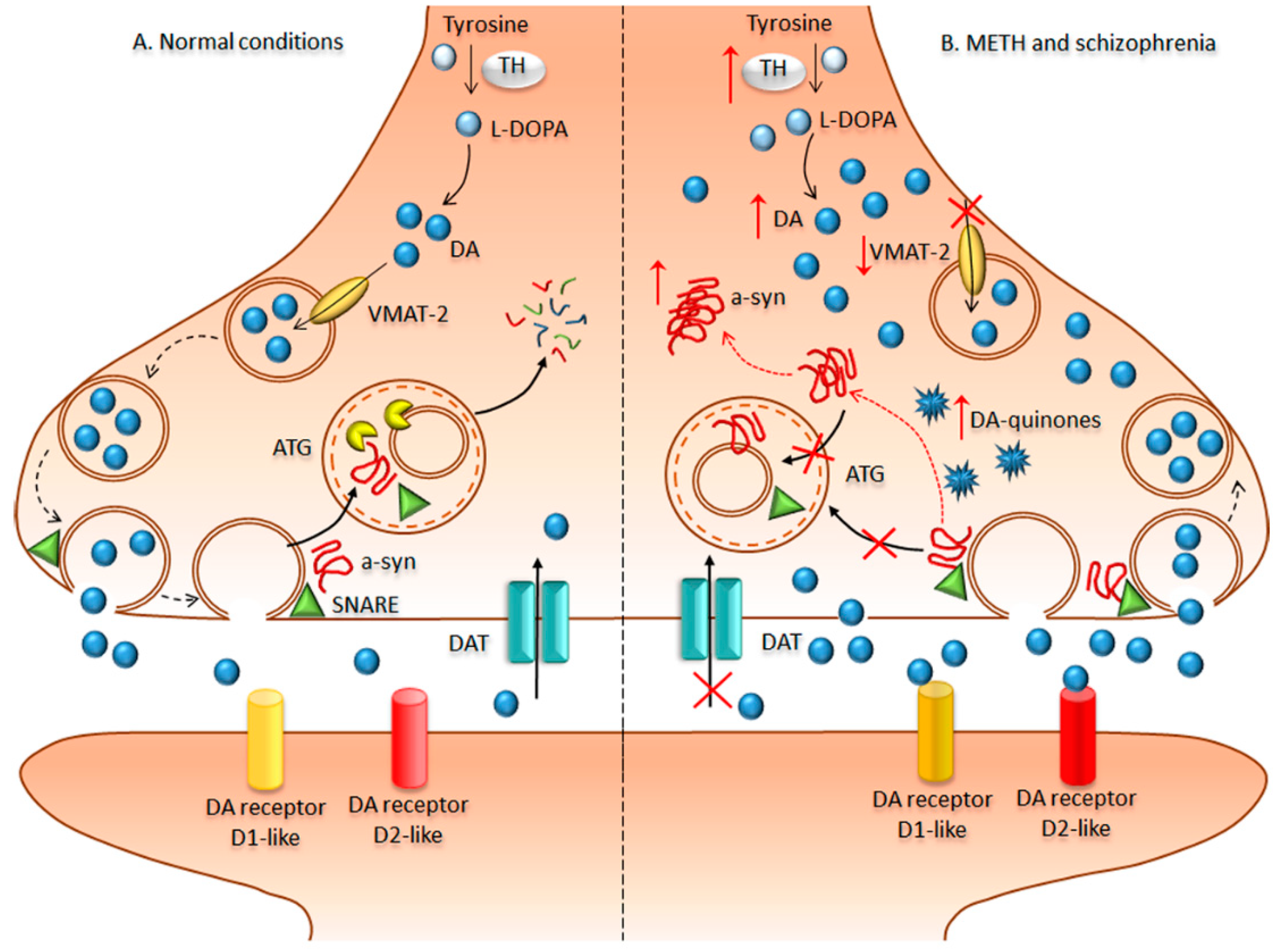{kind=link}
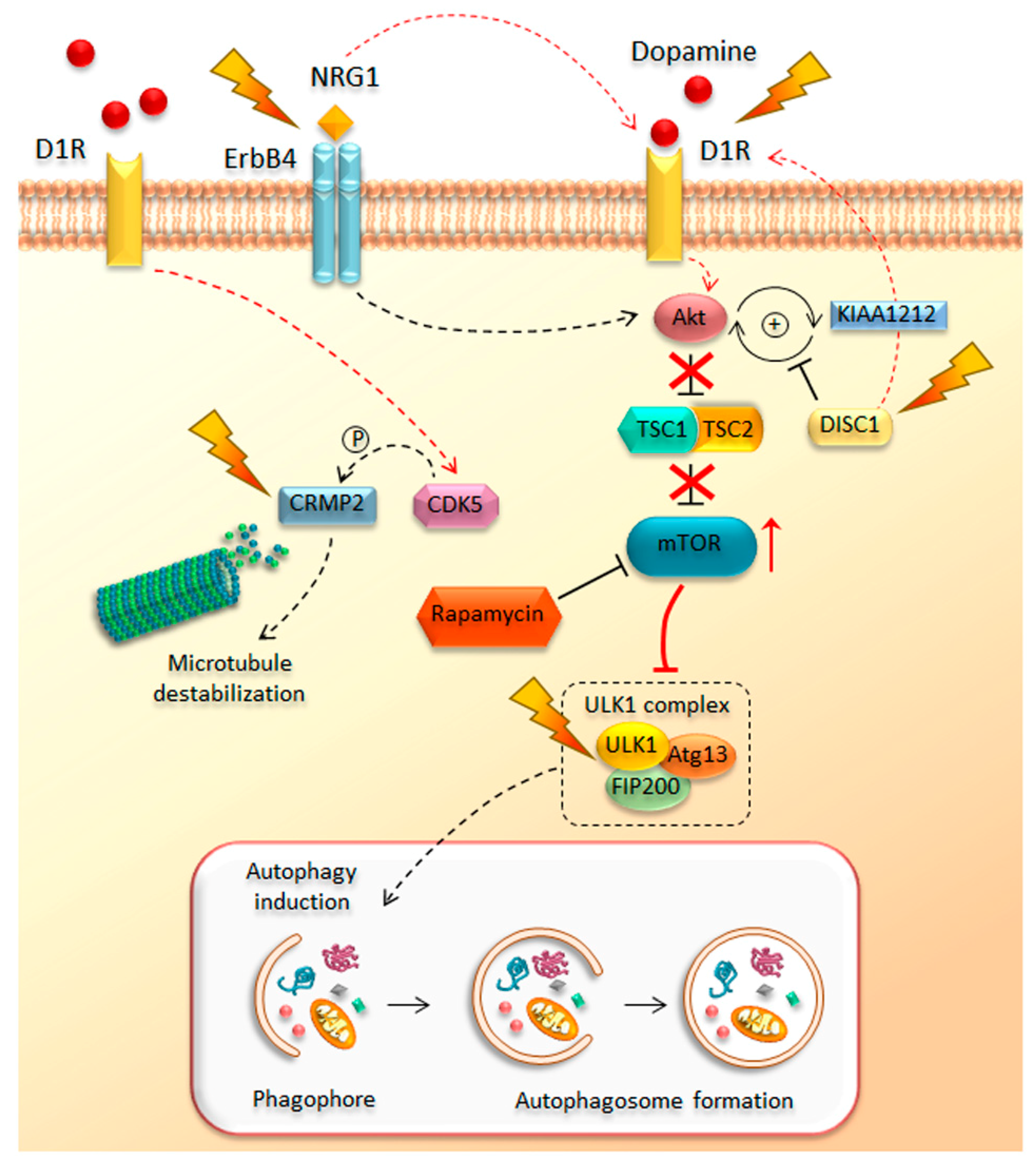{kind=link}
Table 1.
Altered proteins converging on the mammalian target of rapamycin (mTOR) pathway during schizophrenia and methamphetamine addiction. DISC1—disrupted in schizophrenia 1; Akt—protein kinase B; NGR1—neuregulin-1; ErbB4—avian erythroblastosis oncogene B4-like protein; CRMP2—collapsin response mediator protein 2; CDK5—cyclin-dependent kinase 5.
Table 1.
Altered proteins converging on the mammalian target of rapamycin (mTOR) pathway during schizophrenia and methamphetamine addiction. DISC1—disrupted in schizophrenia 1; Akt—protein kinase B; NGR1—neuregulin-1; ErbB4—avian erythroblastosis oncogene B4-like protein; CRMP2—collapsin response mediator protein 2; CDK5—cyclin-dependent kinase 5.
| Protein | Schizophrenia | Methamphetamine |
|---|---|---|
| DISC1 | [153,212,213,214,215] | [208,209] |
| Akt | [218,219,220,221,222,223,224] | [225] |
| NRG1/ErbB4 | [228,230,232] | [231] |
| CRMP2 | [236,238] | [239,240] |
| CDK5/p35 | [247] | [130,242,243] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ryskalin, L.; Limanaqi, F.; Frati, A.; Busceti, C.L.; Fornai, F. mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders. Int. J. Mol. Sci. 2018, 19, 2226. https://doi.org/10.3390/ijms19082226
AMA Style
Ryskalin L, Limanaqi F, Frati A, Busceti CL, Fornai F. mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders. International Journal of Molecular Sciences. 2018; 19(8):2226. https://doi.org/10.3390/ijms19082226
Chicago/Turabian StyleRyskalin, Larisa, Fiona Limanaqi, Alessandro Frati, Carla L. Busceti, and Francesco Fornai. 2018. "mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders" International Journal of Molecular Sciences 19, no. 8: 2226. https://doi.org/10.3390/ijms19082226
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.