Application of Nanoparticles for Targeting G Protein-Coupled Receptors


Abstract
:1. GPCR Activation and GPCRs in Cancer
2. Nanoparticles
2.1. Passive Targeting and Positive Targeting
2.2. Drug Design
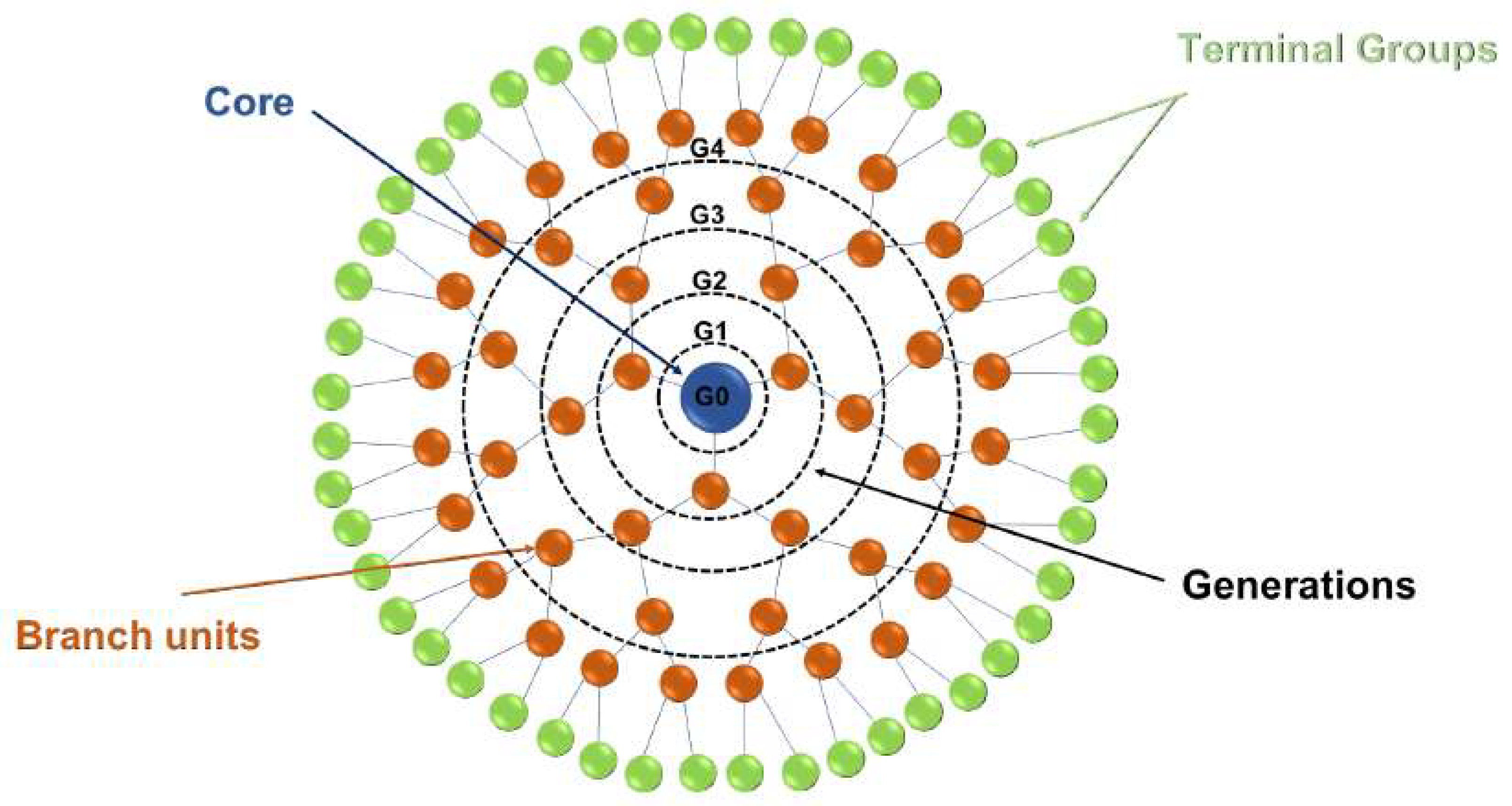
2.3. Dendrimers
2.4. Quantum Dots
2.5. Gold Nanoparticles
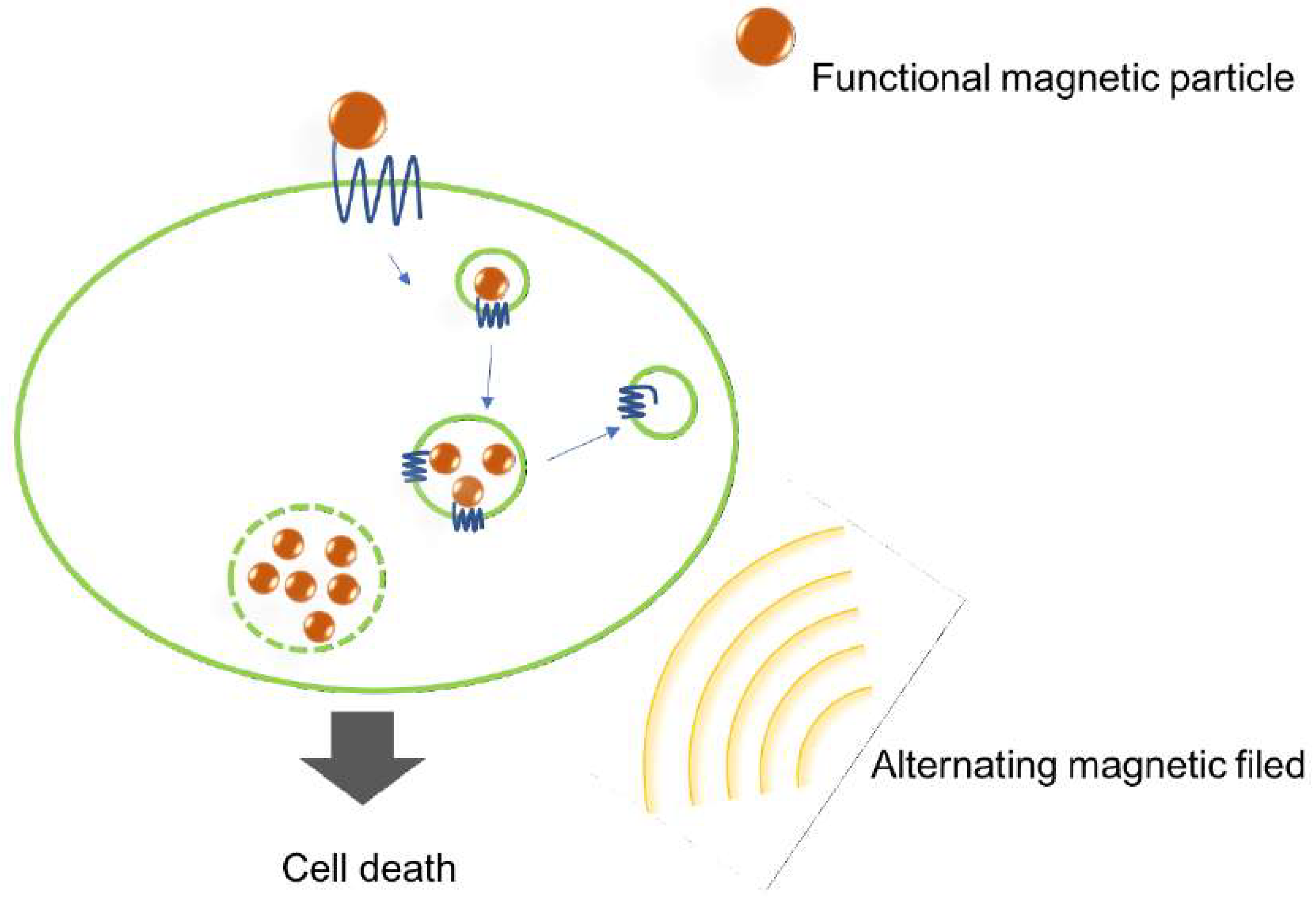
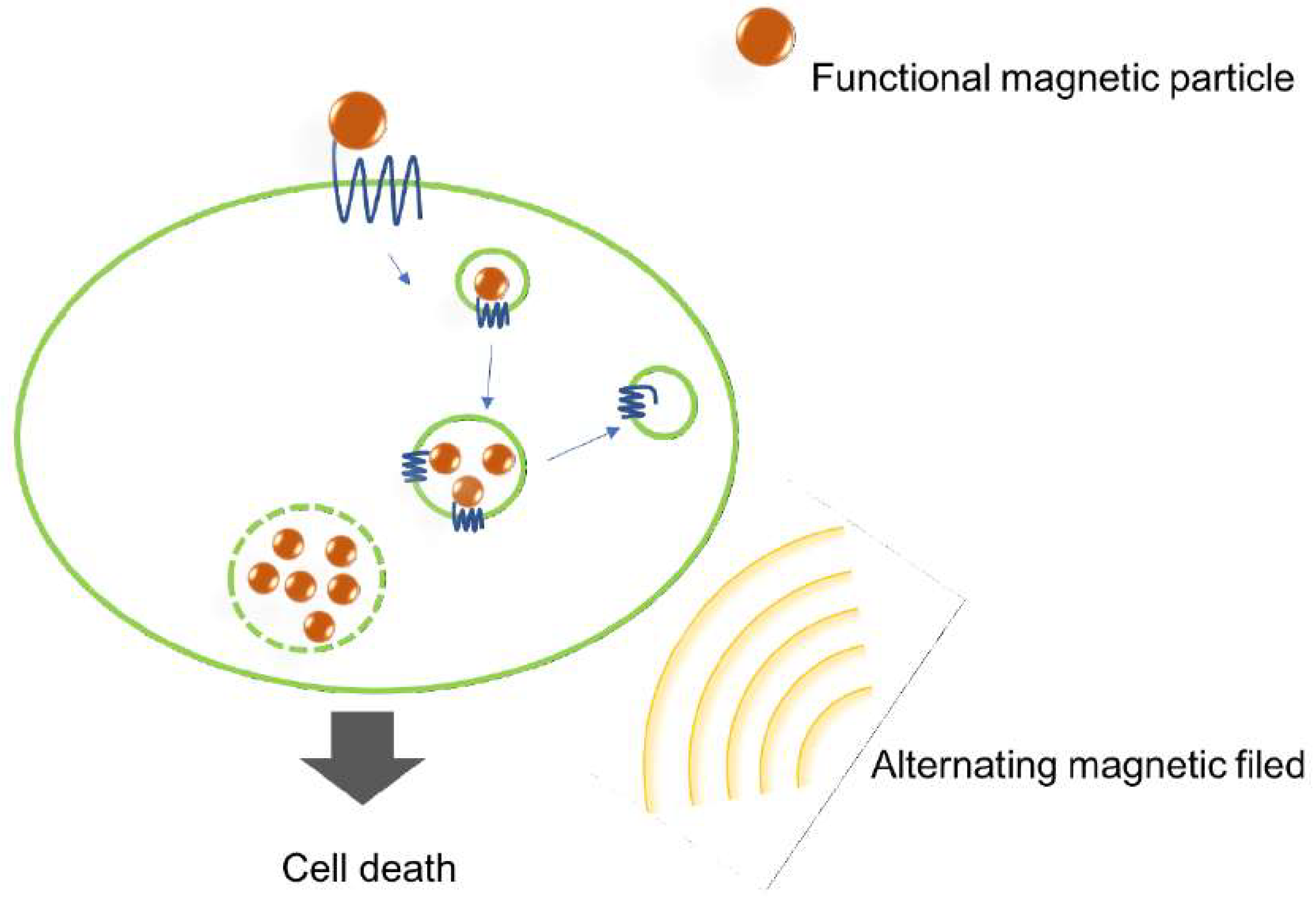
2.6. Magnetic Nanoparticles
2.7. Others
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANGII | Angiotensin II |
| AR | Adenosine Receptor |
| AuNP | Gold Nanoparticle |
| CCK2R | Cholecystokinin-2 Receptor |
| CXCR4 | Chemokine Receptor 4 |
| EGFR | Epidermal Growth Factor Receptor |
| ELP | Elastin-Like Polypeptide |
| EPR | Enhanced Permeation and Retention Effect |
| ETA | Endothelin Receptor |
| GLiDe | GPCR Ligand-Dendrimer |
| GNR | Gold Nanorod |
| GPCR | G Protein-Coupled Receptor |
| GRK | G protein-Coupled Receptor Kinase |
| GRPR | Gastrin-Releasing Peptide Receptor |
| GTP | Guanosine Triphosphate |
| 5-HT1A | Serotonin Receptor Subtype 1A |
| IONP | Iron Oxide Nanoparticle |
| LPAR | Lysophosphatidic Acid Receptor |
| PDGFR | Platelet-Derived Growth Factor Receptor |
| RTK | Receptor Tyrosine Kinases |
References
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G Protein-Coupled Receptors in Cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef] [PubMed]
- Munk, C.; Isberg, V.; Mordalski, S.; Harpsoe, K.; Rataj, K.; Hauser, A.S.; Kolb, P.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E. GPCRdb: The G protein-coupled receptor database—An introduction. Br. J. Pharmacol. 2016, 173, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Kobilka, B.K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta (BBA) Biomembr. 2007, 1768, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Maudsley, S.; Bohn, L.M. Fulfilling the Promise of “Biased” G Protein-Coupled Receptor Agonism. Mol. Pharmacol. 2015, 88, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.-P.; Bünemann, M.; Feinstein, T.N.; Lambert, N.; Nikolaev, V.O.; Engelhardt, S.; Lohse, M.J.; Hoffmann, C. Minireview: GPCR and G Proteins: Drug Efficacy and Activation in Live Cells. Mol. Endocrinol. 2009, 23, 590–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bologna, Z.; Teoh, J.-P.; Bayoumi, A.S.; Tang, Y.; Kim, I.-M. Biased G Protein-Coupled Receptor Signaling: New Player in Modulating Physiology and Pathology. Biomol. Ther. 2017, 25, 12–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcourt, N.; Bockaert, J.; Marin, P. GPCR-jacking: From a new route in RTK signalling to a new concept in GPCR activation. Trends Pharmacol. Sci. 2007, 28, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Receptor tyrosine kinase-G-protein-coupled receptor signalling platforms: Out of the shadow? Trends Pharmacol. Sci. 2011, 32, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Waters, C.M.; Long, J.S.; Moughal, N.A.; Tigyi, G.; Pyne, S. Receptor tyrosine kinase-G-protein coupled receptor complex signaling in mammalian cells. Adv. Enzyme Regul. 2007, 47, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onfroy, L.; Galandrin, S.; Pontier, S.M.; Seguelas, M.H.; N’Guyen, D.; Senard, J.M.; Gales, C. G protein stoichiometry dictates biased agonism through distinct receptor-G protein partitioning. Sci. Rep. 2017, 7, 7885. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Tang, C.-M.; Hahntow, I.; Michel, M.C. Impact of GPCRs in clinical medicine: Genetic variants and drug targets. Biochim. Biophys. Acta 2007, 1768, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; An, S.; Ward, R.; Yang, Y.; Guo, X.-X.; Li, W.; Xu, T.-R. G protein-coupled receptors as promising cancer targets. Cancer Lett. 2016, 376, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.; Proia, R.L. The role of G protein-coupled receptors in lymphoid malignancies. Cell. Signal. 2017, 39, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, V.; Sokolov, E.; Swet, J.H.; Ahrens, W.A.; Showlater, V.; Iannitti, D.A.; McKillop, I.H. Expression and function of lysophosphatidic acid receptors (LPARs) 1 and 3 in human hepatic cancer progenitor cells. Oncotarget 2016, 7, 2951–2967. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Murph, M.M.; Lu, Y.; Liu, S.; Hall, H.S.; Liu, J.; Stephens, C.; Fang, X.; Mills, G.B. Lysophosphatidic Acid Receptors Determine Tumorigenicity and Aggressiveness of Ovarian Cancer Cells. J. Natl. Cancer Inst. 2008, 100, 1630–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, M.T.; Luster, A.D. Chemokines in Cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brault, L.; Menter, T.; Obermann, E.C.; Knapp, S.; Thommen, S.; Schwaller, J.; Tzankov, A. PIM kinases are progression markers and emerging therapeutic targets in diffuse large B-cell lymphoma. Br. J. Cancer 2012, 107, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgat, C.; MacGrogan, G.; Brouste, V.; Velasco, V.; Sevenet, N.; Bonnefoi, H.; Fernandez, P.; Debled, M.; Hindie, E. Expression of Gastrin-Releasing Peptide Receptor in Breast Cancer and Its Association with Pathologic, Biologic, and Clinical Parameters: A Study of 1,432 Primary Tumors. J. Nucl. Med. 2017, 58, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Laukkanen, M.O.; Castellone, M.D. Gastrin-Releasing Peptide Receptor Targeting in Cancer Treatment: Emerging Signaling Networks and Therapeutic Applications. Curr. Drug Targets 2016, 17, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Schwartsmann, G.; DiLeone, L.P.; Horowitz, M.; Schunemann, D.; Cancella, A.; Pereira, A.S.; Richter, M.; Souza, F.; da Rocha, A.B.; Souza, F.H.; et al. A phase I trial of the bombesin/gastrin-releasing peptide (BN/GRP) antagonist RC3095 in patients with advanced solid malignancies. Investig. New Drugs 2006, 24, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Cheng, S.-Y. Angiopoietin-2: Development of Inhibitors for Cancer Therapy. Curr. Oncol. Rep. 2009, 11, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Arafat, H.A.; Gong, Q.; Chipitsyna, G.; Rizvi, A.; Saa, C.T.; Yeo, C.J. Antihypertensives as novel antineoplastics: Angiotensin-I-converting enzyme inhibitors and angiotensin II type 1 receptor blockers in pancreatic ductal adenocarcinoma. J. Am. Coll. Surg. 2007, 204, 996–1005, discussion 1005–1006. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Guevara, P.; Escobar, E.; García-Navarrete, R.; Pineda, B.; Sotelo, J. Blockage of angiotensin II type I receptor decreases the synthesis of growth factors and induces apoptosis in C6 cultured cells and C6 rat glioma. Br. J. Cancer 2005, 92, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Isayama, H.; Ijichi, H.; Sasaki, T.; Takahara, N.; Ito, Y.; Matsubara, S.; Uchino, R.; Yagioka, H.; Arizumi, T.; et al. A multicenter phase II trial of gemcitabine and candesartan combination therapy in patients with advanced pancreatic cancer: GECA2. Investig. New Drugs 2013, 31, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Haley, B.; Frenkel, E. Nanoparticles for drug delivery in cancer treatment. Urol. Oncol.-Semin. Orig. Investig. 2008, 26, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Yameen, B.; Choi, W.I.; Vilos, C.; Swami, A.; Shi, J.J.; Farokhzad, O.C. Insight into nanoparticle cellular uptake and intracellular targeting. J. Control. Release 2014, 190, 485–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulbrich, K.; Holá, K.; Šubr, V.; Bakandritsos, A.; Tuček, J.; Zbořil, R. Targeted Drug Delivery with Polymers and Magnetic Nanoparticles: Covalent and Noncovalent Approaches, Release Control, and Clinical Studies. Chem. Rev. 2016, 116, 5338–5431. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.J.; Labgaa, I.; Villacorta-Martin, C.; Ningarhari, M.; Villanueva, A. Tumor Heterogeneity and Resistance to Targeted Therapies in Hepatocellular Carcinoma. In Resistance to Molecular Therapies for Hepatocellular Carcinoma; Villanueva, A., Ed.; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Richard, S.; Boucher, M.; Saric, A.; Herbet, A.; Lalatonne, Y.; Petit, P.X.; Meriaux, S.; Boquet, D.; Motte, L. Optimization of pegylated iron oxide nanoplatforms for antibody coupling and bio-targeting. J. Mater. Chem. B 2017, 5, 2896–2907. [Google Scholar] [CrossRef]
- Palankar, R.; Pinchasik, B.-E.; Khlebtsov, B.N.; Kolesnikova, T.A.; Möhwald, H.; Winterhalter, M.; Skirtach, A.G. Nanoplasmonically-Induced Defects in Lipid Membrane Monitored by Ion Current: Transient Nanopores versus Membrane Rupture. Nano Lett. 2014, 14, 4273–4279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Gao, H.J.; Bao, G. Physical Principles of Nanoparticle Cellular Endocytosis. ACS Nano 2015, 9, 8655–8671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.J.; Trase, I.; Ren, M.Q.; Duval, K.; Guo, X.; Chen, Z. Design of Nanoparticle-Based Carriers for Targeted Drug Delivery. J. Nanomater. 2016, 2016, 1087250. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-T. Modeling the scaling law of surface plasmon resonance in gold spherical nanoshells. J. Nanophoton 2010, 4, 049507. [Google Scholar] [CrossRef]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, P.; Iyer, A.K. Recent advances in dendrimer-based nanovectors for tumor-targeted drug and gene delivery. Drug Discov. Today 2015, 20, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampathkumar Srinvasa-Gopalan, Y.K.J. Dendrimers in Cancer Treatment and Diagnosis. In Naonomaterials for Cancer Diagnosis; Kumar, C.S., Ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007. [Google Scholar] [Green Version]
- Kurniasih, I.N.; Keilitz, J.; Haag, R. Dendritic nanocarriers based on hyperbranched polymers. Chem. Soc. Rev. 2015, 44, 4145–4164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caliman, A.D.; Miao, Y.L.; McCammon, J.A. Mapping the allosteric sites of the A(2A) adenosine receptor. Chem. Biol. Drug Des. 2018, 91, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S.; Brito, R.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Adenosine Receptors: Expression, Function and Regulation. Int. J. Mol. Sci. 2014, 15, 2024–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A. GPCR ligand-dendrimer (GLiDe) conjugates: Future smart drugs? Trends Pharmacol. Sci. 2010, 31, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Hechler, B.; Klutz, A.M.; Gachet, C.; Jacobson, K.A. Toward Multivalent Signaling across G Protein-Coupled Receptors from Poly(amidoamine) Dendrimers. Bioconj. Chem. 2008, 19, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Klutz, A.M.; Hechler, B.; Gao, Z.-G.; Gachet, C.; Jacobson, K.A. Application of the functionalized congener approach to dendrimer-based signaling agents acting through A(2A) adenosine receptors. Purinergic Signal. 2009, 5, 39–50. [Google Scholar] [CrossRef] [PubMed]
- De Castro, S.; Maruoka, H.; Hong, K.; Kilbey, S.M.; Costanzi, S.; Hechler, B.; Brown, G.G.; Gachet, C.; Harden, T.K.; Jacobson, K.A. Functionalized Congeners of P2Y(1) Receptor Antagonists: 2-Alkynyl (N)-Methanocarba 2′-Deoxyadenosine 3′,5′-Bisphosphate Analogues and Conjugation to a Polyamidoamine (PAMAM) Dendrimer Carrier. Bioconj. Chem. 2010, 21, 1190–1205. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Zhou, Y.; Ivanov, A.A.; Carter, R.L.; Harden, T.K.; Jacobson, K.A. Enhanced Potency of Nucleotide–Dendrimer Conjugates as Agonists of the P2Y(14) Receptor: Multivalent Effect in G Protein-Coupled Receptor Recognition. Bioconj. Chem. 2009, 20, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.K.; Yoo, L.S.; Chinn, M.; Hong, K.; Kilbey, S.M., 2nd; Barrett, M.O.; Fricks, I.P.; Harden, T.K.; Gao, Z.G.; Jacobson, K.A. Polyamidoamine (PAMAM) dendrimer conjugates of “clickable” agonists of the A3 adenosine receptor and coactivation of the P2Y14 receptor by a tethered nucleotide. Bioconj. Chem. 2010, 21, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Leopoldo, M.; Lacivita, E.; Berardi, F.; Perrone, R. Developments in fluorescent probes for receptor research. Drug Discov. Today 2009, 14, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Pisanic, T.R.; Zhang, Y.; Wang, T.H. Quantum dots in diagnostics and detection: Principles and paradigms. Analyst 2014, 139, 2968–2981. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.Q.; Huang, X.Y.; Ren, J.C. Characterization of water-soluble luminescent quantum dots by fluorescence correlation spectroscopy. Ann. N. Y. Acad. Sci. 2008, 1130, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T. Nano-Biomedical Engineering 2012: Proceedings of the Tohoku University Global Centre of Excellence Programme; Imperial College Press: London, UK, 2012. [Google Scholar]
- Young, S.H.; Rozengurt, E. Qdot nanocrystal conjugates conjugated to bombesin or ANG II label the cognate G protein-coupled receptor in living cells. Am. J. Physiol. Cell Physiol. 2006, 290, C728–C732. [Google Scholar] [CrossRef] [PubMed]
- Michalet, X.; Pinaud, F.F.; Bentolila, L.A.; Tsay, J.M.; Doose, S.; Li, J.J.; Sundaresan, G.; Wu, A.M.; Gambhir, S.S.; Weiss, S. Quantum dots for live cells, in vivo imaging, and diagnostics. Science 2005, 307, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Hild, W.; Pollinger, K.; Caporale, A.; Cabrele, C.; Keller, M.; Pluym, N.; Buschauer, A.; Rachel, R.; Tessmar, J.; Breunig, M.; et al. G protein-coupled receptors function as logic gates for nanoparticle binding and cell uptake. Proc. Natl. Acad. Sci. USA 2010, 107, 10667–10672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsuzaki, A.; Ohyanagi, T.; Tsukasaki, Y.; Miyanaga, Y.; Ueda, M.; Jin, T. Compact Halo-Ligand-Conjugated Quantum Dots for Multicolored Single-Molecule Imaging of Overcrowding GPCR Proteins on Cell Membranes. Small 2015, 11, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.J.; Zheng, J.Y.; Xu, J.M.; Rastogi, V.K.; Cheng, T.C.; DeFrank, J.J.; Leblanc, R.M. (CdSe)ZnS quantum dots and organophosphorus hydrolase bioconjugate as biosensors for detection of paraoxon. J. Phys. Chem. B 2005, 109, 3793–3799. [Google Scholar] [CrossRef] [PubMed]
- Hennig, R.; Pollinger, K.; Tessmar, J.; Goepferich, A. Multivalent targeting of AT(1) receptors with angiotensin II-functionalized nanoparticles. J. Drug Target. 2015, 23, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Fichter, K.M.; Flajolet, M.; Greengard, P.; Vu, T.Q. Kinetics of G-protein-coupled receptor endosomal trafficking pathways revealed by single quantum dots. Proc. Natl. Acad. Sci. USA 2010, 107, 18658–18663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreaden, E.C.; Mackey, M.A.; Huang, X.H.; Kang, B.; El-Sayed, M.A. Beating cancer in multiple ways using nanogold. Chem. Soc. Rev. 2011, 40, 3391–3404. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.H.; Peng, X.H.; Wang, Y.Q.; Wang, Y.X.; Shin, D.M.; El-Sayed, M.A.; Nie, S.M. A Reexamination of Active and Passive Tumor Targeting by Using Rod-Shaped Gold Nanocrystals and Covalently Conjugated Peptide Ligands. ACS Nano 2010, 4, 5887–5896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khlebtsov, N.; Dykman, L. Biodistribution and toxicity of engineered gold nanoparticles: A review of in vitro and in vivo studies. Chem. Soc. Rev. 2011, 40, 1647–1671. [Google Scholar] [CrossRef] [PubMed]
- Patra, C.R.; Bhattacharya, R.; Mukhopadhyay, D.; Mukherjee, P. Application of gold nanoparticles for targeted therapy in cancer. J. Biomed. Nanotechnol. 2008, 4, 99–132. [Google Scholar] [CrossRef]
- Shukla, R.; Bansal, V.; Chaudhary, M.; Basu, A.; Bhonde, R.R.; Sastry, M. Biocompatibility of Gold Nanoparticles and Their Endocytotic Fate Inside the Cellular Compartment: A Microscopic Overview. Langmuir 2005, 21, 10644–10654. [Google Scholar] [CrossRef] [PubMed]
- DeLong, R.K.; Reynolds, C.M.; Malcolm, Y.; Schaeffer, A.; Severs, T.; Wanekaya, A. Functionalized gold nanoparticles for the binding, stabilization, and delivery of therapeutic DNA, RNA, and other biological macromolecules. Nanotechnol. Sci. Appl. 2010, 3, 53–63. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, I.H.; Huang, X.; El-Sayed, M.A. Surface plasmon resonance scattering and absorption of anti-EGFR antibody conjugated gold nanoparticles in cancer diagnostics: Applications in oral cancer. Nano Lett. 2005, 5, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Peng, Q. Protein-gold nanoparticle interactions and their possible impact on biomedical applications. Acta Biomater. 2017, 55, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.-C.; Creran, B.; Rotello, V.M. Gold Nanoparticles: Preparation, Properties, and Applications in Bionanotechnology. Nanoscale 2012, 4, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Bischof, J.C. Thermophysical and biological responses of gold nanoparticle laser heating. Chem. Soc. Rev. 2012, 41, 1191–1217. [Google Scholar] [CrossRef] [PubMed]
- Xiong, R.; Raemdonck, K.; Peynshaert, K.; Lentacker, I.; De Cock, I.; Demeester, J.; De Smedt, S.C.; Skirtach, A.G.; Braeckmans, K. Comparison of Gold Nanoparticle Mediated Photoporation: Vapor Nanobubbles Outperform Direct Heating for Delivering Macromolecules in Live Cells. ACS Nano 2014, 8, 6288–6296. [Google Scholar] [CrossRef] [PubMed]
- Delcea, M.; Sternberg, N.; Yashchenok, A.M.; Georgieva, R.; Baumler, H.; Mohwald, H.; Skirtach, A.G. Nanoplasmonics for dual-molecule release through nanopores in the membrane of red blood cells. ACS Nano 2012, 6, 4169–4180. [Google Scholar] [CrossRef] [PubMed]
- Skirtach, A.G.; Dejugnat, C.; Braun, D.; Susha, A.S.; Rogach, A.L.; Parak, W.J.; Mohwald, H.; Sukhorukov, G.B. The role of metal nanoparticles in remote release of encapsulated materials. Nano Lett. 2005, 5, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Jayasekara, P.S.; Phan, K.; Tosh, D.K.; Kumar, T.S.; Moss, S.M.; Zhang, G.F.; Barchi, J.J.; Gao, Z.G.; Jacobson, K.A. Modulation of G protein-coupled adenosine receptors by strategically functionalized agonists and antagonists immobilized on gold nanoparticles. Purinergic Signal. 2013, 9, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Nripen, C.; Shukla, R.; Katti, K.V.; Kannan, R. Gastrin Releasing Protein Receptor—Specific Gold Nanorods: Breast and Prostate Tumor-avid Nanovectors for Molecular Imaging. Nano Lett. 2009, 9, 1798–1805. [Google Scholar] [CrossRef] [Green Version]
- Arruebo, M.; Fernández-Pacheco, R.; Ibarra, M.R.; Santamaría, J. Magnetic nanoparticles for drug delivery. Nano Today 2007, 2, 22–32. [Google Scholar] [CrossRef]
- Bañobre-López, M.; Teijeiro, A.; Rivas, J. Magnetic nanoparticle-based hyperthermia for cancer treatment. Rep. Pract. Oncol. Radiother. 2013, 18, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Gobbo, O.L.; Sjaastad, K.; Radomski, M.W.; Volkov, Y.; Prina-Mello, A. Magnetic Nanoparticles in Cancer Theranostics. Theranostics 2015, 5, 1249–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, C.; El Hajj Diab, D.; Connord, V.; Clerc, P.; Meunier, E.; Pipy, B.; Payré, B.; Tan, R.P.; Gougeon, M.; Carrey, J.; et al. Targeting a G-Protein-Coupled Receptor Overexpressed in Endocrine Tumors by Magnetic Nanoparticles To Induce Cell Death. ACS Nano 2014, 8, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Chen, J.; Zhang, X.; Liu, Y.; Xu, C. Follicle-stimulating hormone polypeptide modified nanoparticle drug delivery system in the treatment of lymphatic metastasis during ovarian carcinoma therapy. Gynecol. Oncol. 2014, 135, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Garg, S.; Eldi, P.; Zhou, F.H.-h.; Johnson, I.R.D.; Brooks, D.A.; Lam, F.; Rychkov, G.; Hayball, J.; Albrecht, H. Targeting prostate cancer cells with genetically engineered polypeptide-based micelles displaying gastrin-releasing peptide. Int. J. Pharm. 2016, 513, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Kecskes, A.; Tosh, D.K.; Wei, Q.; Gao, Z.G.; Jacobson, K.A. GPCR Ligand Dendrimer (GLiDe) Conjugates: Adenosine Receptor Interactions of a Series of Multivalent Xanthine Antagonists. Bioconj. Chem. 2011, 22, 1115–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Hechler, B.; Gao, Z.L.; Gachet, C.; Jacobson, K.A. PEGylated Dendritic Unimolecular Micelles as Versatile Carriers for Ligands of G Protein-Coupled Receptors. Bioconj. Chem. 2009, 20, 1888–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Nakatani, E.; Gronenberg, L.S.; Tokimoto, T.; Wirth, M.J.; Hruby, V.J.; Roberts, A.; Lynch, R.M.; Ghosh, I. Peptide-labeled quantum dots for imaging GPCRs in whole cells and as single molecules. Bioconj. Chem. 2007, 18, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Thurner, P.; Gsandtner, I.; Kudlacek, O.; Choquet, D.; Nanoff, C.; Freissmuth, M.; Zezula, J. A Two-state Model for the Diffusion of the A(2A) Adenosine Receptor in Hippocampal Neurons. J. Biol. Chem. 2014, 289, 9263–9274. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, T.; Kruusmagi, M.; Sourial-Bassillious, N.; Bondar, A.; Svenningsson, P.; Flajolet, M.; Greengard, P.; Scott, L.; Brismar, H.; Aperia, A. A Noncanonical Postsynaptic Transport Route for a GPCR Belonging to the Serotonin Receptor Family. J. Neurosci. 2012, 32, 17998–18008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kwon, Y.J.; Choi, Y.; Kim, H.C.; Kim, K.; Kim, J.; Park, S.; Song, R. Quantum Dot-Based Screening System for Discovery of G Protein-Coupled Receptor Agonists. ChemBioChem 2012, 13, 1503–1508. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Kim, K.; Hong, S.; Kim, H.; Kwon, Y.J.; Song, R. Intracellular Protein Target Detection by Quantum Dots Optimized for Live Cell Imaging. Bioconj. Chem. 2011, 22, 1576–1586. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasan, V.K.A.; Wan Razali, W.A.; Zhang, K.; Pillai, R.R.; Saini, A.; Denkova, D.; Santiago, M.; Brown, H.; Thompson, J.; Connor, M.; et al. Development of Bright and Biocompatible Nanoruby and Its Application to Background-Free Time-Gated Imaging of G-Protein-Coupled Receptors. ACS Appl. Mater. Interfaces 2017, 9, 39197–39208. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Nanoparticles | Ligand/Antibody | GPCR | Function | Reference |
|---|---|---|---|---|
| Dendrimer | MRS2500 (antagonist) | P2Y(1) | Affinity to receptor (Ki 23 nM); Inhibiting ADP-promoted human platelet aggregation; Antithrombotic drug | [46] |
| XAC (antagonist) | Adenosine receptor (A2AAR) | Affinity to receptor (Ki 3.7 nM); Treating Parkinson’s disease and asthma | [81] | |
| CGS21680 (agonist) | Adenosine receptor (A2AAR) | Affinity to receptor (Ki 12 ± 6 nM); Activating adenylate cyclase and accumulating cAMP; Inhibiting ADP-promoted human platelet aggregation; Using short PEG chains in nanocarriers targeting ligand-receptor interactions | [82] | |
| UDPGA-A3AR 3a-G4 PAMAM (agonist) | Both Adenosine receptor (A3AR) and P2Y14 | Affinity to receptor (Ki 39.5 nM to A3AR); Inhibiting cAMP cumulation; Antithrombotic drug | [46] | |
| DITC-APEC (agonist) | Adenosine receptor (A2AAR) | Affinity to receptor (Ki 70 ± 3 nM); Inhibiting ADP-promoted human platelet aggregation; Antithrombotic drug | [45] | |
| Quantum dots (QDs) | α-melanocyte-simulating hormone NDP and MT-II | Human melanocortin receptor (hMCR) | Specific marking of surface receptors; Single molecule imaging of GPCR; Multiplexing; Investigating GPCR localization and trafficking in live cells and in single molecule studies | [83] |
| Deltorphin-II | Human δ-opioid receptor (hDOR) | |||
| ANGII | ANGIIReceptor | Monitoring ligand-receptor binding in live cells; Monitoring GPCR internalization; Detecting and labeling several GPCRs in living cells | [53] | |
| BBN | BBN receptor | |||
| HaloTag ligand | Cyclic AMP receptor 1 (cAR1) | Single molecule imaging of GPCR; Using multi-colored, single molecule imaging to study membrane protein dynamics | [56] | |
| Antibody against flag tag (HA) | Serotonin receptor subtype 1A (5-HT1A) | Monitoring internalization and endosomal trafficking; Identifying two distinct GPCR recycling pathways | [59] | |
| ANGII | AGTR1 | Higher affinity to receptor than native ANGII; Multivalent ANGII-targeted NP-AGTR1 binding; Targeting tissues that overexpress AGTR1 | [58] | |
| Adenosine | Adenosine receptor (A2AAR) | Studying the mobility of the receptor in neurons | [84] | |
| Antibody against flag tag (GFP) | Serotonin receptor subtype 1B (5-HT1B) | Monitoring receptor trafficking in rat hippocampal neurons; Suggesting alternative mechanism of mood regulation via GPCR | [85] | |
| Antibody against flag tag (HA) | κ-opioid receptors (κ-ORs) | Displaying receptor affinity and specificity; Monitoring receptor trafficking; Detecting two receptors in one cell; Application in drug screening | [86] | |
| Antibody against flag tag (GFP) | Adenosine receptor (A2AAR) | |||
| Antibody against flag tag (GFP) | Endothelin A receptor (ET(A)R) | Monitoring receptor intracellular translocation; Detecting intracellular targets (when conjugated with a cell penetrating agent) | [87] | |
| Antibody against flag tag (HA) | Serotonin receptor (5-HT1A) | Single molecule imaging to monitor receptor trafficking; Understanding the mechanism of therapies targeting GPCR | [59] | |
| Gold nanoparticle (AuNP) | Several AR agonists and antagonists | Adenosine receptor (AR) | Affinity to receptor (Ki 37 nM to A3AAR); Cancer diagnosing and treatment | [73] |
| BBN | Gastrin-releasing peptide receptor (GRPR) | Monitoring receptor internalization; Cancer therapy and molecular imaging in vivo | [74] | |
| Magnetic nanoparticle (MNP) | Anti-ETA antibody | Endothelin receptor (ETA) | Efficient receptor targeting; Bimodal contrast agent and imaging agent; Targeting tumor pathologies; Early Cancer diagnosing | [32] |
| MG | Cholecystokinin-2 receptor (CCK2R) | Inducing cell death in cancer | [59] | |
| Elastin-like polypeptide (ELP) micelles | GRP | Gastrin-releasing peptide receptor (GRPR) | Specific receptor targeting; Delivering cytotoxic drugs to cancer | [80] |
| Unmodified NPs | FSH | Follicle-Stimulating hormone receptor (FSHR) | Successfully delivering drug to lymph nodes; Reducing metastasis; Ovarian cancer therapy | [79] |
| Nanorubies | Antibody against flag tag (HA) | μ-opioid receptor | Imaging functionality; Achieving real-time single molecule imaging on biological samples | [88] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Xiong, Y.; Lee, L.T.O. Application of Nanoparticles for Targeting G Protein-Coupled Receptors. Int. J. Mol. Sci. 2018, 19, 2006. https://doi.org/10.3390/ijms19072006
Ma X, Xiong Y, Lee LTO. Application of Nanoparticles for Targeting G Protein-Coupled Receptors. International Journal of Molecular Sciences. 2018; 19(7):2006. https://doi.org/10.3390/ijms19072006
Chicago/Turabian StyleMa, Xin, Yunfang Xiong, and Leo Tsz On Lee. 2018. "Application of Nanoparticles for Targeting G Protein-Coupled Receptors" International Journal of Molecular Sciences 19, no. 7: 2006. https://doi.org/10.3390/ijms19072006
APA StyleMa, X., Xiong, Y., & Lee, L. T. O. (2018). Application of Nanoparticles for Targeting G Protein-Coupled Receptors. International Journal of Molecular Sciences, 19(7), 2006. https://doi.org/10.3390/ijms19072006