Autotaxin–Lysophosphatidic Acid Signaling in Alzheimer’s Disease

 ,
,
Abstract
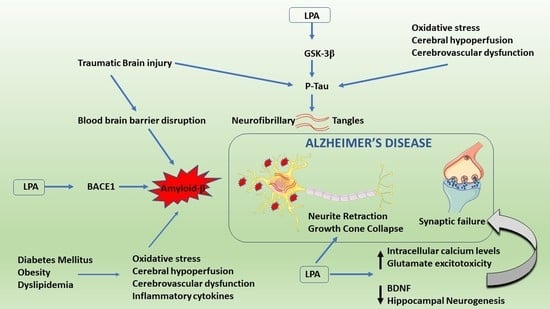
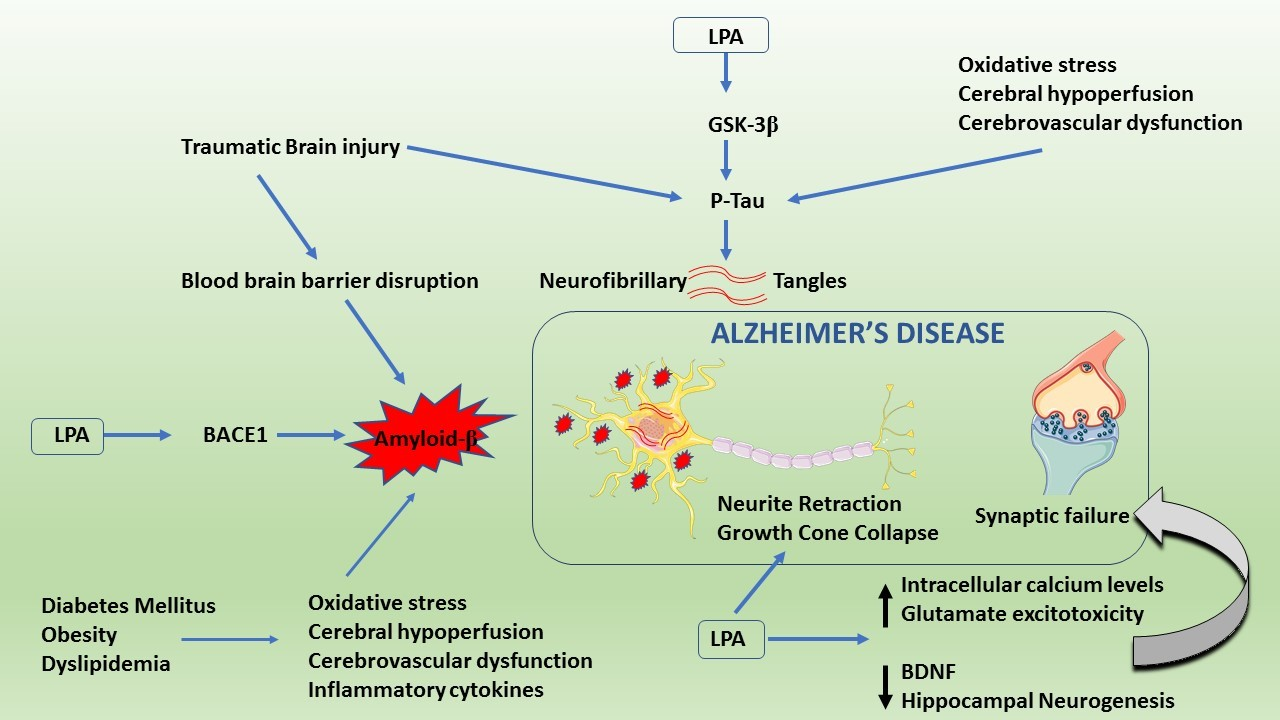
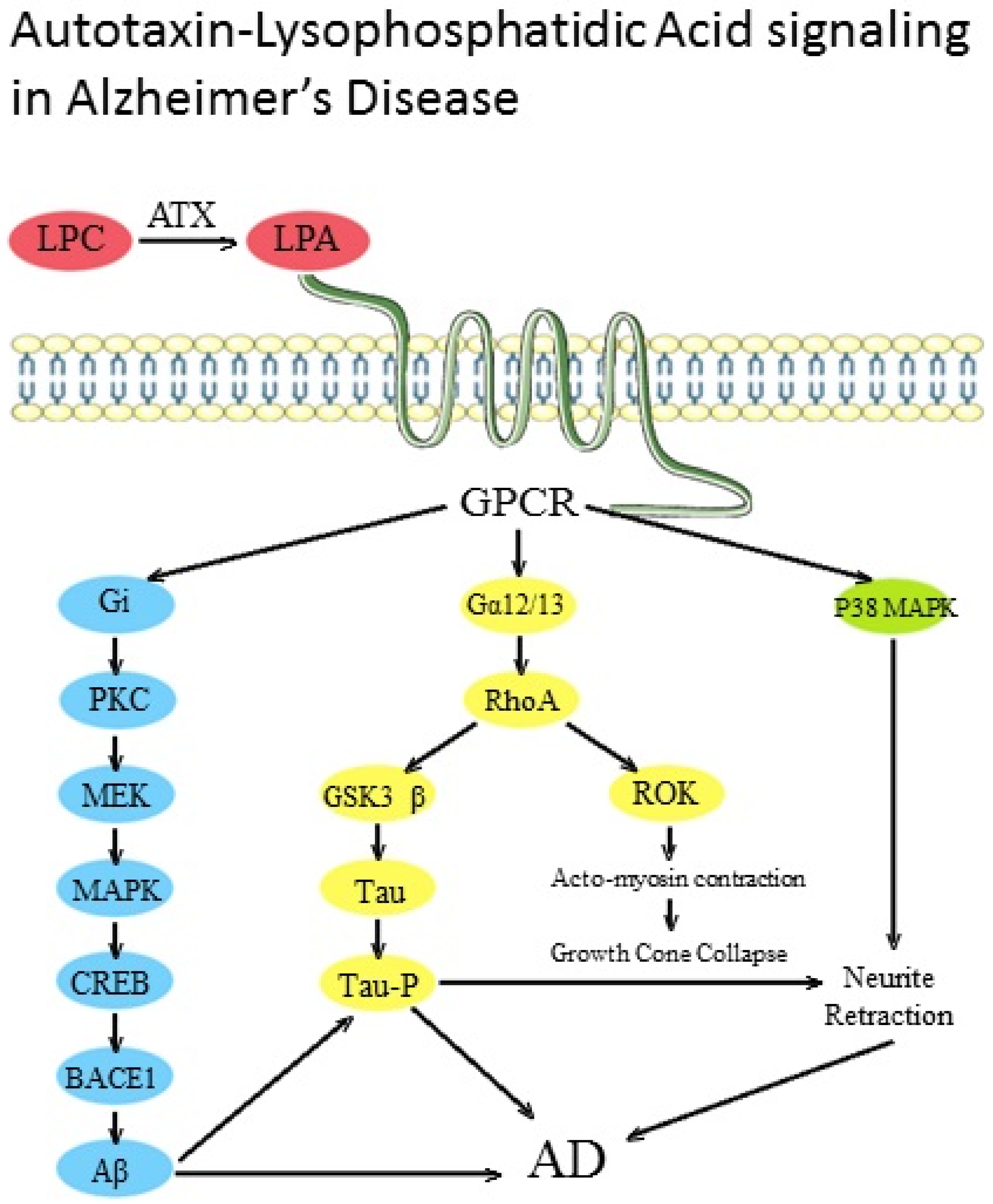
:
1. Introduction
2. Autotaxin (ATX)
3. Lysophosphatidic Acid and its Receptors
LPA Receptors in the Central Nervous System
4. Altered ATX–LPA Signaling and LPARs in Alzheimer’s Disease
4.1. ATX–LPA Signaling and Amyloid β
4.2. ATX–LPA Signaling and Tau Hyperphosphorylation
4.3. Other Effects
5. Cross-Talk between ATX–LPA Signaling and Risk Factors of Alzheimer’s Disease
5.1. Traumatic Brain Injury and AD
5.2. Metabolic Syndrome and AD
5.3. Chronic Hypoperfusion and AD
6. Future Perspective on ATX–LPA Signaling
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ATX | Autotaxin |
| AD | Alzheimer’s disease |
| BBB | Blood–brain barrier |
| BACE | β-site APP-cleaving enzyme |
| ENPP | Ecto-nucleotide pyrophosphatase/phosphodiesterase |
| GPCR | G protein-coupled receptor |
| GSK3β | Glycogen synthase kinase-3β |
| LPA | Lysophosphatidic acid |
| LPC | Lysophosphatidylcholine |
References
- Wortmann, M. Dementia: A global health priority—Highlights from an ADI and World Health Organization report. Alzheimers. Res. Ther. 2012, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Moolenaar, W.H.; van Meeteren, L.A.; Giepmans, B.N.G. The ins and outs of lysophosphatidic acid signaling. BioEssays 2004, 26, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Stefan, C.; Jansen, S.; Bollen, M. NPP-type ectophosphodiesterases: Unity in diversity. Trends Biochem. Sci. 2005, 30, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of Human Plasma Lysophospholipase D, a Lysophosphatidic Acid-producing Enzyme, as Autotaxin, a Multifunctional Phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meeteren, L.A.; Moolenaar, W.H. Regulation and biological activities of the autotaxin–LPA axis. Prog. Lipid Res. 2007, 46, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Aoki, J.; Inoue, A.; Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2008, 1781, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Bolen, A.L.; Naren, A.P.; Yarlagadda, S.; Beranova-Giorgianni, S.; Chen, L.; Norman, D.; Baker, D.L.; Rowland, M.M.; Best, M.D.; Sano, T.; et al. The phospholipase A1 activity of lysophospholipase A-I links platelet activation to LPA production during blood coagulation. J. Lipid Res. 2011, 52, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Cai, L.; Umemoto, E.; Takeda, A.; Tohya, K.; Komai, Y.; Veeraveedu, P.T.; Hata, E.; Sugiura, Y.; Kubo, A.; et al. Constitutive Lymphocyte Transmigration across the Basal Lamina of High Endothelial Venules Is Regulated by the Autotaxin/Lysophosphatidic Acid Axis. J. Immunol. 2013, 190, 2036–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, H.; Newton, R.; Klein, R.; Morita, Y.; Gunn, M.D.; Rosen, S.D. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat. Immunol. 2008, 9, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemoto, E.; Hayasaka, H.; Bai, Z.; Cai, L.; Yonekura, S.; Peng, X.; Takeda, A.; Tohya, K.; Miyasaka, M. Novel regulators of lymphocyte trafficking across high endothelial venules. Crit. Rev. Immunol. 2011, 31, 147–169. [Google Scholar] [CrossRef] [PubMed]
- Dusaulcy, R.; Rancoule, C.; Grès, S.; Wanecq, E.; Colom, A.; Guigné, C.; van Meeteren, L.A.; Moolenaar, W.H.; Valet, P.; Saulnier-Blache, J.S. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J. Lipid Res. 2011, 52, 1247–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.H.G.; van Meeteren, L.A.; Houben, A.J.S.; van Zeijl, L.; et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimasu, H.; Okudaira, S.; Hama, K.; Mihara, E.; Dohmae, N.; Inoue, A.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 2011, 18, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Stracke, M.L.; Krutzsch, H.C.; Unsworth, E.J.; Arestad, A.; Cioce, V.; Schiffmann, E.; Liotta, L.A. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992, 267, 2524–2529. [Google Scholar] [PubMed]
- Jansen, S.; Stefan, C.; Creemers, J.W.M.; Waelkens, E.; Van Eynde, A.; Stalmans, W.; Bollen, M. Proteolytic maturation and activation of autotaxin (NPP2), a secreted metastasis-enhancing lysophospholipase D. J. Cell Sci. 2005, 118, 3081–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, S.; Keino-Masu, K.; Ohto, T.; Masu, M. The N-terminal hydrophobic sequence of autotaxin (ENPP2) functions as a signal peptide. Genes Cells 2006, 11, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrakis, A.; Moolenaar, W.H. Autotaxin: Structure-function and signaling. J. Lipid Res. 2014, 55, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Andries, M.; Vekemans, K.; Vanbilloen, H.; Verbruggen, A.; Bollen, M. Rapid clearance of the circulating metastatic factor autotaxin by the scavenger receptors of liver sinusoidal endothelial cells. Cancer Lett. 2009, 284, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, S.; Hashimoto, T.; Kano, K.; Aoki, J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 2015, 157, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A. A Family of Phospholipid Autacoids: Occurrence, Metabolism and Bioactions. Pergamon Prog. Lipid Res. 1995, 34, 151–184. [Google Scholar] [CrossRef]
- Aoki, J. Mechanisms of lysophosphatidic acid production. Semin. Cell Dev. Biol. 2004, 15, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Nakane, S.; Kishimoto, S.; Waku, K.; Yoshioka, Y.; Tokumura, A.; Hanahan, D.J. Occurrence of lysophosphatidic acid and its alkyl ether-linked analog in rat brain and comparison of their biological activities toward cultured neural cells. Biochim. Biophys. Acta 1999, 1440, 194–204. [Google Scholar] [CrossRef]
- Sano, T.; Baker, D.; Virag, T.; Wada, A.; Yatomi, Y.; Kobayashi, T.; Igarashi, Y.; Tigyi, G. Multiple Mechanisms Linked to Platelet Activation Result in Lysophosphatidic Acid and Sphingosine 1-Phosphate Generation in Blood. J. Biol. Chem. 2002, 277, 21197–21206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anliker, B.; Choi, J.W.; Lin, M.-E.; Gardell, S.E.; Rivera, R.R.; Kennedy, G.; Chun, J. Lysophosphatidic acid (LPA) and its receptor, LPA1, influence embryonic schwann cell migration, myelination, and cell-to-axon segregation. Glia 2013, 61, 2009–2022. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Weiner, J.A.; Kaushal, D.; Contos, J.J.A.; Rehen, S.K.; Kingsbury, M.A.; Kim, K.Y.; Chun, J. Lysophosphatidic acid influences the morphology and motility of young, postmitotic cortical neurons. Mol. Cell. Neurosci. 2002, 20, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Jesionowska, A.; Cecerska, E.; Dolegowska, B. Methods for quantifying lysophosphatidic acid in body fluids: A review. Anal. Biochem. 2014, 453, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Pagès, C.; Simon, M.F.; Valet, P.; Saulnier-Blache, J.S. Lysophosphatidic acid synthesis and release. Prostaglandins Other Lipid Mediat. 2001, 64, 1–10. [Google Scholar] [CrossRef]
- Tokumura, A.; Fukuzawa, K.; Tsukatani, H. Effects of synthetic and natural lysophosphatidic acids on the arterial blood pressure of different animal species. Lipids 1978, 13, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Van Corven, E.J.; Groenink, A.; Jalink, K.; Eichholtz, T.; Moolenaar, W.H. Lysophosphatidate-induced cell proliferation: Identification and dissection of signaling pathways mediated by G proteins. Cell 1989, 59, 45–54. [Google Scholar] [CrossRef]
- Ishii, I.; Fukushima, N.; Ye, X.; Chun, J. Lysophospholipid Receptors: Signaling and Biology. Annu. Rev. Biochem. 2004, 73, 321–354. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Lee, C.-W.; Chun, J. Biological roles of lysophospholipid receptors revealed by genetic null mice: An update. Biochim. Biophys. Acta 2008. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Chun, J. Lysophospholipids and their receptors in the central nervous system. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 20–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, S.T.; Yung, Y.C.; Herr, D.R.; Chun, J. Lysophosphatidic acid in vascular development and disease. IUBMB Life 2009, 61, 791–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, Y.C.; Stoddard, N.C.; Mirendil, H.; Chun, J. Lysophosphatidic Acid Signaling in the Nervous System. Neuron 2015, 85, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Knowlden, S.; Georas, S.N. The autotaxin-LPA axis emerges as a novel regulator of lymphocyte homing and inflammation. J. Immunol. 2014, 192, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Chun, J. Lysophosphatidic acid (LPA) signaling in vertebrate reproduction. Trends Endocrinol. Metab. 2010, 21, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, S.M.; Panupinthu, N.; Lapierre, D.M.; Pereverzev, A.; Dixon, S.J. Lysophosphatidic acid: A potential mediator of osteoblast–osteoclast signaling in bone. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Hartung, H.-P. Mechanism of Action of Oral Fingolimod (FTY720) in Multiple Sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisca, F.; Sabbadini, R.A.; Goldshmit, Y.; Pébay, A. Biological Effects of Lysophosphatidic Acid in the Nervous System. Int. Rev. Cell Mol. Biol. 2012, 296, 273–322. [Google Scholar] [PubMed]
- Lin, M.-E.; Herr, D.R.; Chun, J. Lysophosphatidic acid (LPA) receptors: Signaling properties and disease relevance. Prostaglandins Other Lipid Mediat. 2010, 91, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiropoulos, A.; Gineitis, D.; Copeland, J.; Treisman, R. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell 1999, 98, 159–169. [Google Scholar] [CrossRef]
- Kim, J.H.; Adelstein, R.S. LPA1-induced migration requires nonmuscle myosin II light chain phosphorylation in breast cancer cells. J. Cell. Physiol. 2011, 226, 2881–2893. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.J.; Park, S.Y.; Cho, K.H.; Sohn, J.S.; Lee, J.; Kim, Y.K.; Kang, J.; Park, C.G.; Han, J.W.; Lee, H.Y. The Rho/ROCK pathway for lysophosphatidic acid-induced proteolytic enzyme expression and ovarian cancer cell invasion. Oncogene 2012, 31, 4279–4289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.-Y.; Xiong, Z.-G.; Lei, S.; Orser, B.A.; Dudek, E.; Browning, M.D.; MacDonald, J.F. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat. Neurosci. 1999, 2, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Jongsma, M.; Matas-Rico, E.; Rzadkowski, A.; Jalink, K.; Moolenaar, W.H. LPA Is a Chemorepellent for B16 Melanoma Cells: Action through the cAMP-Elevating LPA5 Receptor. PLoS ONE 2011, 6, e29260. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, O.; Moolenaar, W.H. Ras-MAP kinase signaling by lysophosphatidic acid and other G protein-coupled receptor agonists. Oncogene 2001, 20, 1540–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez, C.; Portela, R.A.; Mellado, M.; Rodríguez-Frade, J.M.; Collard, J.; Serrano, A.; Martínez-A, C.; Avila, J.; Carrera, A.C. Role of the PI3K regulatory subunit in the control of actin organization and cell migration. J. Cell Biol. 2000, 151, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-C.; Kim, K.-M.; Lee, K.-S.; Namkoong, S.; Lee, S.-J.; Han, J.-A.; Jeoung, D.; Ha, K.-S.; Kwon, Y.-G.; Kim, Y.-M. Serum bioactive lysophospholipids prevent TRAIL-induced apoptosis via PI3K/Akt-dependent cFLIP expression and Bad phosphorylation. Cell Death Differ. 2004, 11, 1287–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Fukushima, N.; Kingsbury, M.A.; Chun, J. Lysophosphatidic acid in neural signaling. Neuroreport 2002, 13, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Ishii, I.; Contos, J.J.; Weiner, J.A.; Chun, J. L Ysophospholipid R Eceptors. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 507–534. [Google Scholar] [CrossRef] [PubMed]
- Contos, J.J.A.; Ishii, I.; Fukushima, N.; Kingsbury, M.A.; Ye, X.; Kawamura, S.; Brown, J.H.; Chun, J. Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: Signaling deficits without obvious phenotypic abnormality attributable to lpa(2). Mol. Cell. Biol. 2002, 22, 6921–6929. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Hama, K.; Contos, J.J.A.; Anliker, B.; Inoue, A.; Skinner, M.K.; Suzuki, H.; Amano, T.; Kennedy, G.; Arai, H.; et al. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature 2005, 435, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, J. Lysophospholipid Receptors: Signaling and Biochemistry; Wiley: Hoboken, NJ, USA, 2013; ISBN 9780470569054. [Google Scholar]
- Lee, Z.; Cheng, C.-T.; Zhang, H.; Subler, M.A.; Wu, J.; Mukherjee, A.; Windle, J.J.; Chen, C.-K.; Fang, X. Role of LPA4/p2y9/GPR23 in Negative Regulation of Cell Motility. Mol. Biol. Cell 2008, 19, 5435–5445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundequist, A.; Boyce, J.A. LPA5 Is Abundantly Expressed by Human Mast Cells and Important for Lysophosphatidic Acid Induced MIP-1β Release. PLoS ONE 2011, 6, e18192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azeem, Z.; Jelani, M.; Naz, G.; Tariq, M.; Wasif, N.; Kamran-ul-Hassan Naqvi, S.; Ayub, M.; Yasinzai, M.; Amin-ud-din, M.; Wali, A.; et al. Novel mutations in G protein-coupled receptor gene (P2RY5) in families with autosomal recessive hypotrichosis (LAH3). Hum. Genet. 2008, 123, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Pasternack, S.M.; von Kügelgen, I.; Aboud, K.A.; Lee, Y.-A.; Rüschendorf, F.; Voss, K.; Hillmer, A.M.; Molderings, G.J.; Franz, T.; Ramirez, A.; Nürnberg, P.; et al. G protein–coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat. Genet. 2008, 40, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Petukhova, L.; Sousa, E.C.; Martinez-Mir, A.; Vitebsky, A.; dos Santos, L.G.; Shapiro, L.; Haynes, C.; Gordon, D.; Shimomura, Y.; Christiano, A.M. Genome-wide linkage analysis of an autosomal recessive hypotrichosis identifies a novel P2RY5 mutation. Genomics 2008, 92, 273–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubin, A.E.; Herr, D.R.; Chun, J. Diversity of lysophosphatidic acid receptor-mediated intracellular calcium signaling in early cortical neurogenesis. J. Neurosci. 2010, 30, 7300–7309. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Shano, S.; Moriyama, R.; Chun, J. Lysophosphatidic acid stimulates neuronal differentiation of cortical neuroblasts through the LPA 1–G i/o pathway. Neurochem. Int. 2007, 50, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Kingsbury, M.A.; Rehen, S.K.; Contos, J.J.A.; Higgins, C.M.; Chun, J. Non-proliferative effects of lysophosphatidic acid enhance cortical growth and folding. Nat. Neurosci. 2003, 6, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Dubin, A.E.; Bahnson, T.; Weiner, J.A.; Fukushima, N.; Chun, J. Lysophosphatidic acid stimulates neurotransmitter-like conductance changes that precede GABA and l-glutamate in early, presumptive cortical neuroblasts. J. Neurosci. 1999, 19, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Ishii, I.; Habara, Y.; Allen, C.B.; Chun, J. Dual Regulation of Actin Rearrangement through Lysophosphatidic Acid Receptor in Neuroblast Cell Lines: Actin Depolymerization by Ca2+-alpha-Actinin and Polymerization by Rho. Mol. Biol. Cell 2002, 13, 2692–2705. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.H.; Weiner, J.A.; Post, S.R.; Chun, J. Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J. Cell Biol. 1996, 135, 1071–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, D.; Yamane, M.; Tsujiuchi, T.; Moriyama, R.; Fukushima, N. Lysophosphatidic acid induces neurite branch formation through LPA3. Mol. Cell. Neurosci. 2012, 50, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Yamane, M.; Furuta, D.; Fukushima, N. Lysophosphatidic acid influences initial neuronal polarity establishment. Neurosci. Lett. 2010, 480, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.-W.; Mutoh, T.; Lin, M.-E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA Receptors: Subtypes and Biological Actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Herr, D.R.; Chun, J. Effects of LPA and S1P on the nervous system and implications for their involvement in disease. Curr. Drug Targets 2007, 8, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Herr, D.; Mutoh, T.; Chun, J. Lysophosphatidic acid (LPA) and its receptors. Curr. Opin. Pharmacol. 2009, 9, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Pilpel, Y.; Segal, M. The role of LPA1 in formation of synapses among cultured hippocampal neurons. J. Neurochem. 2006, 97, 1379–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, H. Recent advances in understanding of various chronic pain mechanisms through lysophosphatidic acid (LPA) receptor signaling. Arthritis Res. Ther. 2012, 14, O6. [Google Scholar] [CrossRef]
- Contos, J.J.A.; Fukushima, N.; Weiner, J.A.; Kaushal, D.; Chun, J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc. Natl. Acad. Sci. USA 2000, 97, 13384–13389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldin, M.; Segal, M. Protein kinase C and ERK involvement in dendritic spine plasticity in cultured rodent hippocampal neurons. Eur. J. Neurosci. 2003, 17, 2529–2539. [Google Scholar] [CrossRef] [PubMed]
- Pilpel, Y.; Segal, M. Activation of PKC induces rapid morphological plasticity in dendrites of hippocampal neurons via Rac and Rho-dependent mechanisms. Eur. J. Neurosci. 2004, 19, 3151–3164. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.R.; Holtsberg, F.W.; Keller, J.N.; Mattson, M.P.; Steiner, S.M. Lysophosphatidic acid induction of neuronal apoptosis and necrosis. Ann. N. Y. Acad. Sci. 2000, 905, 132–141. [Google Scholar] [CrossRef] [PubMed]
- García-Morales, V.; Montero, F.; González-Forero, D.; Rodríguez-Bey, G.; Gómez-Pérez, L.; Medialdea-Wandossell, M.J.; Domínguez-Vías, G.; García-Verdugo, J.M.; Moreno-López, B. Membrane-Derived Phospholipids Control Synaptic Neurotransmission and Plasticity. PLOS Biol. 2015, 13, e1002153. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R. One Lipid, Two Synaptic Plasticity Pathways. PLOS Biol. 2015, 13, e1002154. [Google Scholar] [CrossRef] [PubMed]
- Shiono, S.; Kawamoto, K.; Yoshida, N.; Kondo, T.; Inagami, T. Neurotransmitter Release from Lysophosphatidic Acid-Stimulated PC12 Cells: Involvement of Lysophosphatidic Acid Receptors. Biochem. Biophys. Res. Commun. 1993, 193, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, S.; Kume, K.; Aihara, M.; Shimizu, T. Expression of Lysophosphatidic Acid Receptor in Rat Astrocytes: Mitogenic Effect and Expression of Neurotrophic Genes. Neurochem. Res. 2000, 25, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Weiner, J.A.; Fukushima, N.; Contos, J.J.; Scherer, S.S.; Chun, J. Regulation of Schwann cell morphology and adhesion by receptor-mediated lysophosphatidic acid signaling. J. Neurosci. 2001, 21, 7069–7078. [Google Scholar] [CrossRef] [PubMed]
- Cervera, P.; Tirard, M.; Barron, S.; Allard, J.; Trottier, S.; Lacombe, J.; Daumas-Duport, C.; Sokoloff, P. Immunohistological localization of the myelinating cell-specific receptor LPA1. Glia 2002, 38, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Shano, S.; Moriyama, R.; Chun, J.; Fukushima, N. Lysophosphatidic acid stimulates astrocyte proliferation through LPA1. Neurochem. Int. 2008, 52, 216–220. [Google Scholar] [CrossRef] [PubMed]
- De Sampaio e Spohr, T.C.L.; Dezonne, R.S.; Rehen, S.K.; Gomes, F.C.A. Astrocytes treated by lysophosphatidic acid induce axonal outgrowth of cortical progenitors through extracellular matrix protein and epidermal growth factor signaling pathway. J. Neurochem. 2011, 119, 113–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spohr, T.C.; Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rehen, S.K.; Gomes, F.C.A.; Chun, J. Lysophosphatidic Acid Receptor-dependent Secondary Effects via Astrocytes Promote Neuronal Differentiation. J. Biol. Chem. 2008, 283, 7470–7479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, T.S.; Lariosa-Willingham, K.D.; Lin, F.-F.; Palfreyman, E.L.; Yu, N.; Chun, J.; Webb, M. Pharmacological characterization of lysophospholipid receptor signal transduction pathways in rat cerebrocortical astrocytes. Brain Res. 2003, 990, 182–194. [Google Scholar] [CrossRef]
- Sorensen, S.D.; Nicole, O.; Peavy, R.D.; Montoya, L.M.; Lee, C.J.; Murphy, T.J.; Traynelis, S.F.; Hepler, J.R. Common Signaling Pathways Link Activation of Murine PAR-1, LPA, and S1P Receptors to Proliferation of Astrocytes. Mol. Pharmacol. 2003, 64, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Steiner, M.R.; Holtsberg, F.W.; Mattson, M.P.; Steiner, S.M. Lysophosphatidic acid-induced proliferation-related signals in astrocytes. J. Neurochem. 1997, 69, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Steiner, M.R.; Mattson, M.P.; Steiner, S.M. Lysophosphatidic acid decreases glutamate and glucose uptake by astrocytes. J. Neurochem. 1996, 67, 2300–2305. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Horiuchi, Y.; Jin, Y.; Malchinkhuu, E.; Komachi, M.; Kondo, T.; Okajima, F. Unmasking of LPA1 receptor-mediated migration response to lysophosphatidic acid by interleukin-1β-induced attenuation of Rho signaling pathways in rat astrocytes. J. Neurochem. 2011, 117, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New Roles for the Synaptic Stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science (80-.). 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhang, Y.; Chen, Y.; Zhu, J.; Yang, Y.; Zhang, H.-L. Role of Microglia in Neurological Disorders and Their Potentials as a Therapeutic Target. Mol. Neurobiol. 2017, 54, 7567–7584. [Google Scholar] [CrossRef] [PubMed]
- Lucin, K.M.; Wyss-Coray, T. Immune Activation in Brain Aging and Neurodegeneration: Too Much or Too Little? Neuron 2009, 64, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Lariosa-Willingham, K.D.; Lin, F.-F.; Webb, M.; Rao, T.S. Characterization of lysophosphatidic acid and sphingosine-1-phosphate-mediated signal transduction in rat cortical oligodendrocytes. Glia 2004, 45, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Pébay, A.; Torrens, Y.; Toutant, M.; Cordier, J.; Glowinski, J.; Tencé, M. Pleiotropic effects of lysophosphatidic acid on striatal astrocytes. Glia 1999, 28, 25–33. [Google Scholar] [CrossRef]
- Möller, T.; Contos, J.J.; Musante, D.B.; Chun, J.; Ransom, B.R. Expression and Function of Lysophosphatidic Acid Receptors in Cultured Rodent Microglial Cells. J. Biol. Chem. 2001, 276, 25946–25952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhart, E.; Kollroser, M.; Rechberger, G.; Reicher, H.; Heinemann, A.; Schratl, P.; Hallström, S.; Wintersperger, A.; Nusshold, C.; DeVaney, T.; et al. Lysophosphatidic acid receptor activation affects the C13NJ microglia cell line proteome leading to alterations in glycolysis, motility, and cytoskeletal architecture. Proteomics 2010, 10, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Repp, H.; Richter, H.; Koschinski, A.; Heinemann, U.; Dreyer, F.; Eder, C. Lysophospholipids induce membrane hyperpolarization in microglia by activation of IKCa1 Ca(2+)-dependent K(+) channels. Neuroscience 2002, 109, 827–835. [Google Scholar] [CrossRef]
- Schilling, T.; Stock, C.; Schwab, A.; Eder, C. Functional importance of Ca2+-activated K+ channels for lysophosphatidic acid-induced microglial migration. Eur. J. Neurosci. 2004, 19, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Awada, R.; Rondeau, P.; Grès, S.; Saulnier-Blache, J.S.; Lefebvre d’Hellencourt, C.; Bourdon, E. Autotaxin protects microglial cells against oxidative stress. Free Radic. Biol. Med. 2012, 52, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Awada, R.; Saulnier-Blache, J.S.; Grès, S.; Bourdon, E.; Rondeau, P.; Parimisetty, A.; Orihuela, R.; Harry, G.J.; d’Hellencourt, C.L. Autotaxin downregulates LPS-induced microglia activation and pro-inflammatory cytokines production. J. Cell. Biochem. 2014, 115, 2123–2132. [Google Scholar] [CrossRef] [PubMed]
- Wille, C.; Seufferlein, T.; Eiseler, T. Protein Kinase D family kinases. Bioarchitecture 2014, 4, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozengurt, E. Protein Kinase D Signaling: Multiple Biological Functions in Health and Disease. Physiology 2011, 26, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, T.T.; Leung, W.Y.; Moyer, M.P.; Strieter, R.M.; Rozengurt, E. Protein kinase D2 mediates lysophosphatidic acid-induced interleukin 8 production in nontransformed human colonic epithelial cells through NF-κB. Am. J. Physiol. Physiol. 2007, 292, C767–C777. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Döppler, H.; Toker, A. Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol. Cell. Biol. 2004, 24, 2614–2626. [Google Scholar] [CrossRef] [PubMed]
- Popiolek-Barczyk, K.; Mika, J. Targeting the Microglial Signaling Pathways: New Insights in the Modulation of Neuropathic Pain. Curr. Med. Chem. 2016, 23, 2908–2928. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Xie, L.; Chung, C.Y. Signaling Pathways Controlling Microglia Chemotaxis. Mol. Cells 2017, 40, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Weiner, J.A.; Hecht, J.H.; Chun, J. Lysophosphatidic Acid Receptor Gene vzg-1/lp A1 /edg-2 Is Expressed by Mature Oligodendrocytes During Myelination in the Postnatal Murine Brain. J. Comp. Neurol 1998, 398, 587–598. [Google Scholar] [CrossRef]
- Möller, T.; Musante, D.B.; Ransom, B.R. Lysophosphatidic acid-induced calcium signals in cultured rat oligodendrocytes. Neuroreport 1999, 10, 2929–2932. [Google Scholar] [CrossRef] [PubMed]
- Nogaroli, L.; Yuelling, L.M.; Dennis, J.; Gorse, K.; Payne, S.G.; Fuss, B. Lysophosphatidic acid can support the formation of membranous structures and an increase in MBP mRNA levels in differentiating oligodendrocytes. Neurochem. Res. 2009, 34, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, D.A.; Jin, K. From angiogenesis to neuropathology. Nature 2005, 438, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, A.; Thomas, J.-L. Molecular Parallels between Neural and Vascular Development. Cold Spring Harb. Perspect. Med. 2013, 3, a006551. [Google Scholar] [CrossRef] [PubMed]
- Ruhrberg, C.; Bautch, V.L. Neurovascular development and links to disease. Cell. Mol. Life Sci. 2013, 70, 1675–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-I.; Chen, C.-N.; Huang, M.-T.; Lee, S.-J.; Lin, C.-H.; Chang, C.-C.; Lee, H. Lysophosphatidic acid upregulates vascular endothelial growth factor-C and tube formation in human endothelial cells through LPA1/3, COX-2, and NF-κB activation- and EGFR transactivation-dependent mechanisms. Cell. Signal. 2008, 20, 1804–1814. [Google Scholar] [CrossRef] [PubMed]
- Ptaszynska, M.M.; Pendrak, M.L.; Stracke, M.L.; Roberts, D.D. Autotaxin Signaling via Lysophosphatidic Acid Receptors Contributes to Vascular Endothelial Growth Factor-Induced Endothelial Cell Migration. Mol. Cancer Res. 2010, 8, 309–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, S.; Wang, F.; Wu, H.; Mukherjee, T.J.; Fishman, D.A. The NF-κB pathway mediates lysophosphatidic acid (LPA)-induced VEGF signaling and cell invasion in epithelial ovarian cancer (EOC). Gynecol. Oncol. 2011, 123, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Tigyi, G.; Hong, L.; Yakubu, M.; Parfenova, H.; Shibata, M.; Leffler, C.W. Lysophosphatidic acid alters cerebrovascular reactivity in piglets. Am. J. Physiol. Circ. Physiol. 1995, 268, H2048–H2055. [Google Scholar] [CrossRef] [PubMed]
- Ruisanchez, É.; Dancs, P.; Kerék, M.; Németh, T.; Faragó, B.; Balogh, A.; Patil, R.; Jennings, B.L.; Liliom, K.; Malik, K.U.; et al. Lysophosphatidic acid induces vasodilation mediated by LPA1 receptors, phospholipase C, and endothelial nitric oxide synthase. FASEB J. 2014, 28, 880–890. [Google Scholar] [CrossRef] [PubMed]
- On, N.H.; Savant, S.; Toews, M.; Miller, D.W. Rapid and reversible enhancement of blood-brain barrier permeability using lysophosphatidic acid. J. Cereb. Blood Flow Metab. 2013, 33, 1944–1954. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Qin, J.; Liu, M.; Ruan, Q.; Li, Y.; Zhang, Z. Role of Rho kinase in lysophosphatidic acid-induced altering of blood-brain barrier permeability. Int. J. Mol. Med. 2014, 33, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.A.; Thompson, R.C.; Bunney, W.E.; Schatzberg, A.F.; Barchas, J.D.; Myers, R.M.; Akil, H.; Watson, S.J. Altered choroid plexus gene expression in major depressive disorder. Front. Hum. Neurosci. 2014, 8, 238. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, H.; Hamada, A.; Matsuda, H.; Takagi, A.; Tanaka, M.; Aoki, J.; Arai, H.; Noji, S. Expression patterns of the lysophospholipid receptor genes during mouse early development. Dev. Dyn. 2008, 237, 3280–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bächner, D.; Ahrens, M.; Betat, N.; Schröder, D.; Gross, G. Developmental expression analysis of murine autotaxin (ATX). Mech. Dev. 1999, 84, 121–125. [Google Scholar] [CrossRef]
- Savaskan, N.E.; Rocha, L.; Kotter, M.R.; Baer, A.; Lubec, G.; van Meeteren, L.A.; Kishi, Y.; Aoki, J.; Moolenaar, W.H.; Nitsch, R.; et al. Autotaxin (NPP-2) in the brain: Cell type-specific expression and regulation during development and after neurotrauma. Cell. Mol. Life Sci. 2007, 64, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Rowan, M.J.; Selkoe, D.J. Amyloid-beta oligomers: Their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans. 2002, 30, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science (80-.). 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science (80-.). 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Bienkowski, M.J.; Shuck, M.E.; Miao, H.; Tory, M.C.; Pauley, A.M.; Brashler, J.R.; Stratman, N.C.; Mathews, W.R.; Buhl, A.E.; et al. Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activity. Nature 1999, 402, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Powell, D.; Howlett, D.R.; Tew, D.G.; Meek, T.D.; Chapman, C.; Gloger, I.S.; Murphy, K.E.; Southan, C.D.; Ryan, D.M.; et al. Identification of a Novel Aspartic Protease (Asp 2) as β-Secretase. Mol. Cell. Neurosci. 1999, 14, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Xu, X. γ-Secretase Catalyzes Sequential Cleavages of the AβPP Transmembrane Domain. J. Alzheimer Dis. 2009, 16, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Casado, Á.; Encarnación López-Fernández, M.; Concepción Casado, M.; de La Torre, R. Lipid Peroxidation and Antioxidant Enzyme Activities in Vascular and Alzheimer Dementias. Neurochem. Res. 2008, 33, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Ferna´ndez-Novoa, L.; Lombardi, V.; Corzo, L.; Pichel, V.; Kubota, Y. Cerebrovascular risk factors in Alzheimer’s disease: Brain hemodynamics and pharmacogenomic implications. Neurol. Res. 2003, 25, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; D’Introno, A.; Colacicco, A.M.; Basile, A.M.; Capurso, C.; Kehoe, P.G.; Capurso, A.; Solfrizzi, V. Vascular risk and genetics of sporadic late-onset Alzheimer’s disease. J. Neural Transm. 2004, 111, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Aldred, S.; Bennett, S.; Mecocci, P. Increased low-density lipoprotein oxidation, but not total plasma protein oxidation, in Alzheimer’s disease. Clin. Biochem. 2010, 43, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Draczynska-Lusiak, B.; Doung, A.; Sun, A.Y. Oxidized lipoproteins may play a role in neuronal cell death in Alzheimer disease. Mol. Chem. Neuropathol. 1998, 33, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Ischiropoulos, H.; Lee, V.M.-Y.; Trojanowski, J.Q. The relationship between oxidative/nitrative stress and pathological inclusions in Alzheimer’s and Parkinson’s diseases. Free Radic. Biol. Med. 2002, 32, 1264–1275. [Google Scholar] [CrossRef]
- Bassett, C.N.; Neely, M.D.; Sidell, K.R.; Markesbery, W.R.; Switt, L.L.; Montine, T.J. Cerebrospinal fluid lipoproteins are more vulnerable to oxidation in Alzheimer’s disease and are neurotoxic when oxidized ex vivo. Lipids 1999, 34, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-X.; Minthon, L.; Wallmark, A.; Warkentin, S.; Blennow, K.; Janciauskiene, S. Inflammatory Markers in Matched Plasma and Cerebrospinal Fluid from Patients with Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2003, 16, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Frisardi, V.; Panza, F.; Seripa, D.; Farooqui, T.; Farooqui, A.A. Glycerophospholipids and glycerophospholipid-derived lipid mediators: A complex meshwork in Alzheimer’s disease pathology. Prog. Lipid Res. 2011, 50, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Dong, Y.; Cui, M.-Z.; Xu, X. Lysophosphatidic acid induces increased BACE1 expression and Aβ formation. Biochim. Biophys. Acta 2013, 1832, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Jolly-Tornetta, C.; Wolf, B.A. Regulation of amyloid precursor protein (APP) secretion by protein kinase calpha in human ntera 2 neurons (NT2N). Biochemistry 2000, 39, 7428–7435. [Google Scholar] [CrossRef] [PubMed]
- Rossner, S.; Mendla, K.; Schliebs, R.; Bigl, V. Protein kinase Calpha and beta1 isoforms are regulators of alpha-secretory proteolytic processing of amyloid precursor protein in vivo. Eur. J. Neurosci. 2001, 13, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, D.; Lin, Y.-H.; McMahon, T.; Koo, E.H.; Messing, R.O. Protein Kinase C ϵ Suppresses Aβ Production and Promotes Activation of α-Secretase. Biochem. Biophys. Res. Commun. 2001, 285, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Kinouchi, T.; Sorimachi, H.; Maruyama, K.; Mizuno, K.; Ohno, S.; Ishiura, S.; Suzuki, K. Conventional protein kinase C (PKC)-alpha and novel PKC epsilon, but not -delta, increase the secretion of an N-terminal fragment of Alzheimer’s disease amyloid precursor protein from PKC cDNA transfected 3Y1 fibroblasts. FEBS Lett. 1995, 364, 203–206. [Google Scholar] [PubMed]
- Blois, J.T.; Mataraza, J.M.; Mecklenbraüker, I.; Tarakhovsky, A.; Chiles, T.C. B Cell Receptor-induced cAMP-response Element-binding Protein Activation in B Lymphocytes Requires Novel Protein Kinase Cδ. J. Biol. Chem. 2004, 279, 30123–30132. [Google Scholar] [CrossRef] [PubMed]
- Cordy, J.M.; Cordy, J.M.; Hooper, N.M.; Turner, A.J. The involvement of lipid rafts in Alzheimer’s disease (Review). Mol. Membr. Biol. 2006, 23, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.A.; Nalivaeva, N.N.; Turner, A.J. Lipid rafts and Alzheimer’s disease: Protein-lipid interactions and perturbation of signaling. Front. Physiol. 2012, 3, 189. [Google Scholar] [CrossRef] [PubMed]
- Kalvodova, L.; Kahya, N.; Schwille, P.; Ehehalt, R.; Verkade, P.; Drechsel, D.; Simons, K. Lipids as modulators of proteolytic activity of BACE: Involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 2005, 280, 36815–36823. [Google Scholar] [CrossRef] [PubMed]
- Dias, I.H.K.; Mistry, J.; Fell, S.; Reis, A.; Spickett, C.M.; Polidori, M.C.; Lip, G.Y.H.; Griffiths, H.R. Oxidized LDL lipids increase β-amyloid production by SH-SY5Y cells through glutathione depletion and lipid raft formation. Free Radic. Biol. Med. 2014, 75, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Mandell, J.W.; Banker, G.A. Microtubule-associated proteins, phosphorylation gradients, and the establishment of neuronal polarity. Perspect. Dev. Neurobiol. 1996, 4, 125–135. [Google Scholar] [PubMed]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Steiner, B.; Mandelkow, E.M.; Biernat, J.; Gustke, N.; Meyer, H.E.; Schmidt, B.; Mieskes, G.; Söling, H.D.; Drechsel, D.; Kirschner, M.W.; et al. Phosphorylation of microtubule-associated protein tau: Identification of the site for Ca2(+)-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J. 1990, 9, 3539–3544. [Google Scholar] [PubMed]
- Evans, D.B.; Rank, K.B.; Bhattacharya, K.; Thomsen, D.R.; Gurney, M.E.; Sharma, S.K. Tau Phosphorylation at Serine 396 and Serine 404 by Human Recombinant Tau Protein Kinase II Inhibits Tau’s Ability to Promote Microtubule Assembly. J. Biol. Chem. 2000, 275, 24977–24983. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Tomizawa, K.; Kato, R.; Sato, K.; Uchida, T.; Fujita, S.C.; Imahori, K. Localization and developmental changes of tau protein kinase I/glycogen synthase kinase-3 beta in rat brain. J. Neurochem. 1994, 63, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.V.; Shanley, J.; Correll, M.P.; Fieles, W.E.; Keith, R.A.; Scott, C.W.; Lee, C.-M. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. USA 2000, 97, 11074–11079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Li, M.; Vrana, J.; Schaller, M.D. Glycogen synthase kinase 3- and extracellular signal-regulated kinase-dependent phosphorylation of paxillin regulates cytoskeletal rearrangement. Mol. Cell. Biol. 2006, 26, 2857–2868. [Google Scholar] [CrossRef] [PubMed]
- Goold, R.G.; Owen, R.; Gordon-Weeks, P.R. Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J. Cell Sci. 1999, 112. [Google Scholar]
- Sánchez, C.; Pérez, M.; Avila, J. GSK3beta-mediated phosphorylation of the microtubule-associated protein 2C (MAP2C) prevents microtubule bundling. Eur. J. Cell Biol. 2000, 79, 252–260. [Google Scholar] [CrossRef]
- Sperber, B.R.; Leight, S.; Goedert, M.; Lee, V.M. Glycogen synthase kinase-3 beta phosphorylates tau protein at multiple sites in intact cells. Neurosci. Lett. 1995, 197, 149–153. [Google Scholar] [CrossRef]
- Sayas, C.L.; Moreno-Flores, M.T.; Avila, J.; Wandosell, F. The neurite retraction induced by lysophosphatidic acid increases Alzheimer’s disease-like Tau phosphorylation. J. Biol. Chem. 1999, 274, 37046–370452. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L.; Avila, J.; Wandosell, F. Regulation of neuronal cytoskeleton by lysophosphatidic acid: Role of GSK-3. Biochim. Biophys. Acta 2002, 1582, 144–153. [Google Scholar] [CrossRef]
- Amano, M.; Kaneko, T.; Maeda, A.; Nakayama, M.; Ito, M.; Yamauchi, T.; Goto, H.; Fukata, Y.; Oshiro, N.; Shinohara, A.; et al. Identification of Tau and MAP2 as novel substrates of Rho-kinase and myosin phosphatase. J. Neurochem. 2003, 87, 780–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayas, C.L.; Avila, J.; Wandosell, F. Glycogen synthase kinase-3 is activated in neuronal cells by Galpha12 and Galpha13 by Rho-independent and Rho-dependent mechanisms. J. Neurosci. 2002, 22, 6863–6875. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Kim, N.-H.; Yang, H.; Kim, S.-H.; Huh, S.-O. Lysophosphatidic acid induces neurite retraction in differentiated neuroblastoma cells via GSK-3β activation. Mol. Cells 2011, 31, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, Y.; Lee, M.H.; Lee, J.; Jung, J.; Lee, S.H.; Yang, D.-J.; Kim, B.W.; Son, H.; Lee, B.; Chang, S.; et al. TRPM2 mediates the lysophosphatidic acid-induced neurite retraction in the developing brain. Pflügers Arch. Eur. J. Physiol. 2014, 466, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Crowder, R.J.; Freeman, R.S. Glycogen Synthase Kinase-3β Activity Is Critical for Neuronal Death Caused by Inhibiting Phosphatidylinositol 3-Kinase or Akt but Not for Death Caused by Nerve Growth Factor Withdrawal. J. Biol. Chem. 2000, 275, 34266–34271. [Google Scholar] [CrossRef] [PubMed]
- Tigyi, G.; Fischer, D.J.; Sebök, A.; Yang, C.; Dyer, D.L.; Miledi, R. Lysophosphatidic acid-induced neurite retraction in PC12 cells: Control by phosphoinositide-Ca2+ signaling and Rho. J. Neurochem. 1996, 66, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Holtsberg, F.W.; Steiner, M.R.; Furukawa, K.; Keller, J.N.; Mattson, M.P.; Steiner, S.M. Lysophosphatidic acid induces a sustained elevation of neuronal intracellular calcium. J. Neurochem. 1997, 69, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Schulze, C.; Smales, C.; Rubin, L.L.; Staddon, J.M. Lysophosphatidic Acid Increases Tight Junction Permeability in Cultured Brain Endothelial Cells. J. Neurochem. 2002, 68, 991–1000. [Google Scholar] [CrossRef] [Green Version]
- Estivill-Torrús, G.; Llebrez-Zayas, P.; Matas-Rico, E.; Santín, L.; Pedraza, C.; De Diego, I.; Del Arco, I.; Fernández-Llebrez, P.; Chun, J.; De Fonseca, F.R. Absence of LPA1 Signaling Results in Defective Cortical Development. Cereb. Cortex 2008, 18, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Sebök, A.; Meakin, S.; Kobayashi, T.; Murakami-Murofushi, K.; Tigyi, G. Cyclic phosphatidic acid elicits neurotrophin-like actions in embryonic hippocampal neurons. J. Neurochem. 2003, 87, 1272–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin Rhee, H.; Nam, J.-S.; Sun, Y.; Kim, M.-J.; Choi, H.-K.; Han, D.-H.; Kim, N.-H.; Huh, S.-O. Lysophosphatidic acid stimulates cAMP accumulation and cAMP response element-binding protein phosphorylation in immortalized hippocampal progenitor cells. Neuroreport 2006, 17, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Castilla-Ortega, E.; Sánchez-López, J.; Hoyo-Becerra, C.; Matas-Rico, E.; Zambrana-Infantes, E.; Chun, J.; De Fonseca, F.R.; Pedraza, C.; Estivill-Torrús, G.; Santin, L.J. Exploratory, anxiety and spatial memory impairments are dissociated in mice lacking the LPA1 receptor. Neurobiol. Learn. Mem. 2010, 94, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matas-Rico, E.; García-Diaz, B.; Llebrez-Zayas, P.; López-Barroso, D.; Santín, L.; Pedraza, C.; Smith-Fernández, A.; Fernández-Llebrez, P.; Tellez, T.; Redondo, M.; et al. Deletion of lysophosphatidic acid receptor LPA1 reduces neurogenesis in the mouse dentate gyrus. Mol. Cell. Neurosci. 2008, 39, 342–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, S.M.; Reavill, C.; Brown, G.; Brown, J.T.; Cluderay, J.E.; Crook, B.; Davies, C.H.; Dawson, L.A.; Grau, E.; Heidbreder, C.; et al. LPA1 receptor-deficient mice have phenotypic changes observed in psychiatric disease. Mol. Cell. Neurosci. 2003, 24, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.; Winter, P.; Shilliam, C.S.; Hughes, Z.A.; Langmead, C.; Maycox, P.R.; Dawson, L.A. Neurochemical Changes in LPA1 Receptor Deficient Mice – A Putative Model of Schizophrenia. Neurochem. Res. 2005, 30, 371–377. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Stein, T.D.; Nowinski, C.J.; Stern, R.A.; Daneshvar, D.H.; Alvarez, V.E.; Lee, H.-S.; Hall, G.; Wojtowicz, S.M.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Washington, P.M.; Morffy, N.; Parsadanian, M.; Zapple, D.N.; Burns, M.P. Experimental Traumatic Brain Injury Induces Rapid Aggregation and Oligomerization of Amyloid-Beta in an Alzheimer’s Disease Mouse Model. J. Neurotrauma 2014, 31, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Abisambra, J.F.; Scheff, S. Brain injury in the context of tauopathies. J. Alzheimers. Dis. 2014, 40, 495–518. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.C.; Sturm, V.E.; Peterson, M.J.; Pieper, C.F.; Bullock, T.; Boeve, B.F.; Miller, B.L.; Guskiewicz, K.M.; Berger, M.S.; Kramer, J.H.; et al. Association of traumatic brain injury with subsequent neurological and psychiatric disease: A meta-analysis. J. Neurosurg. 2016, 124, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-β pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The Pathobiology of Vascular Dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Furmaga-Jabłońska, W.; Maciejewski, R.; Ułamek-Kozioł, M.; Jabłoński, M. Brain ischemia activates β- and γ-secretase cleavage of amyloid precursor protein: Significance in sporadic Alzheimer’s disease. Mol. Neurobiol. 2013, 47, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Wolters, F.J.; Zonneveld, H.I.; Hofman, A.; van der Lugt, A.; Koudstaal, P.J.; Vernooij, M.W.; Ikram, M.A. Heart-Brain Connection Collaborative Research Group Cerebral Perfusion and the Risk of Dementia: A Population-Based Study. Circulation 2017, 136, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Srivastava, K.R.; Nagarajan, S.; Lapidus, L.J. Monomer Dynamics of Alzheimer Peptides and Kinetic Control of Early Aggregation in Alzheimer’s Disease. ChemPhysChem 2016, 17, 3470–3479. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldshmit, Y.; Munro, K.; Leong, S.Y.; Pébay, A.; Turnley, A.M. LPA receptor expression in the central nervous system in health and following injury. Cell Tissue Res. 2010, 341, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Frugier, T.; Crombie, D.; Conquest, A.; Tjhong, F.; Taylor, C.; Kulkarni, T.; McLean, C.; Pébay, A. Modulation of LPA Receptor Expression in the Human Brain Following Neurotrauma. Cell. Mol. Neurobiol. 2011, 31, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Crack, P.J.; Zhang, M.; Morganti-Kossmann, M.; Morris, A.J.; Wojciak, J.M.; Fleming, J.K.; Karve, I.; Wright, D.; Sashindranath, M.; Goldshmit, Y.; et al. Anti-lysophosphatidic acid antibodies improve traumatic brain injury outcomes. J. Neuroinflammation 2014, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Johnson, S.C.; Birdsill, A.C.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P.; et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimer Dement. 2015, 11, 504–510.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willette, A.A.; Modanlo, N.; Kapogiannis, D. Alzheimer’s Disease Neuroimaging Initiative Insulin Resistance Predicts Medial Temporal Hypermetabolism in Mild Cognitive Impairment Conversion to Alzheimer Disease. Diabetes 2015, 64, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Pannacciulli, N.; Del Parigi, A.; Chen, K.; Le, D.S.N.T.; Reiman, E.M.; Tataranni, P.A. Brain abnormalities in human obesity: A voxel-based morphometric study. Neuroimage 2006, 31, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.C.; Cada, A.; Nelson, N.; Davis, G.; Sutherland, R.J.; Gonzalez-Lima, F. Reduced cytochrome oxidase and memory dysfunction after chronic brain ischemia in aged rats. Neurosci. Lett. 1997, 223, 165–168. [Google Scholar] [CrossRef]
- Willette, A.A.; Xu, G.; Johnson, S.C.; Birdsill, A.C.; Jonaitis, E.M.; Sager, M.A.; Hermann, B.P.; La Rue, A.; Asthana, S.; Bendlin, B.B. Insulin Resistance, Brain Atrophy, and Cognitive Performance in Late Middle-Aged Adults. Diabetes Care 2013, 36, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Obesity in Aging Exacerbates Blood-Brain Barrier Disruption, Neuroinflammation, and Oxidative Stress in the Mouse Hippocampus: Effects on Expression of Genes Involved in Beta-Amyloid Generation and Alzheimer’s Disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Castanon, N.; Luheshi, G.; Layé, S. Role of neuroinflammation in the emotional and cognitive alterations displayed by animal models of obesity. Front. Neurosci. 2015, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Rancoule, C.; Dusaulcy, R.; Tréguer, K.; Grès, S.; Attané, C.; Saulnier-Blache, J.S. Involvement of autotaxin/lysophosphatidic acid signaling in obesity and impaired glucose homeostasis. Biochimie 2014, 96, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Ferry, G.; Tellier, E.; Try, A.; Grés, S.; Naime, I.; Simon, M.F.; Rodriguez, M.; Boucher, J.; Tack, I.; Gesta, S.; et al. Autotaxin Is Released from Adipocytes, Catalyzes Lysophosphatidic Acid Synthesis, and Activates Preadipocyte Proliferation. J. Biol. Chem. 2003, 278, 18162–18169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, J.; Quilliot, D.; Praderes, J.P.; Simon, M.F.; Gres, S.; Guigne, C.; Prevot, D.; Ferry, G.; Boutin, J.A.; Carpene, C.; et al. Potential involvement of adipocyte insulin resistance in obesity-associated up-regulation of adipocyte lysophospholipase D/autotaxin expression. Diabetologia 2005, 48, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Umemura, K.; Yamashita, N.; Yu, X.; Arima, K.; Asada, T.; Makifuchi, T.; Murayama, S.; Saito, Y.; Kanamaru, K.; Goto, Y.; et al. Autotaxin expression is enhanced in frontal cortex of Alzheimer-type dementia patients. Neurosci. Lett. 2006, 400, 97–100. [Google Scholar] [CrossRef] [PubMed]
- McLimans, K.E.; Willette, A.A. Alzheimer’s Disease Neuroimaging Initiative, for the A. D. N. Autotaxin is Related to Metabolic Dysfunction and Predicts Alzheimer’s Disease Outcomes. J. Alzheimers. Dis. 2017, 56, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Luiten, P.G. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef]
- Thal, D.R.; Griffin, W.S.T.; de Vos, R.A.I.; Ghebremedhin, E. Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol. 2008, 115, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Jabłoński, M.; Ułamek-Kozioł, M.; Kocki, J.; Brzozowska, J.; Januszewski, S.; Furmaga-Jabłońska, W.; Bogucka-Kocka, A.; Maciejewski, R.; Czuczwar, S.J. Sporadic Alzheimer’s Disease Begins as Episodes of Brain Ischemia and Ischemically Dysregulated Alzheimer’s Disease Genes. Mol. Neurobiol. 2013, 48, 500–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierenga, C.E.; Hays, C.C.; Zlatar, Z.Z. Cerebral blood flow measured by arterial spin labeling MRI as a preclinical marker of Alzheimer’s disease. J. Alzheimers. Dis. 2014, 42 (Suppl. 4), S411–S419. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, L.Y.; Venneri, A.; Farkas, E.; Evans, P.C.; Marzo, A.; Frangi, A.F. Vascular dysfunction in the pathogenesis of Alzheimer’s disease—A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol. Dis. 2015, 82, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, A.; O’Connor, J.J. Hypoxia-inducible factor signalling mechanisms in the central nervous system. Acta Physiol. 2013, 208, 298–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Park, S.Y.; Lee, E.K.; Park, C.G.; Chung, H.C.; Rha, S.Y.; Kim, Y.K.; Bae, G.-U.; Kim, B.K.; Han, J.-W.; et al. Activation of Hypoxia-Inducible Factor-1 Is Necessary for Lysophosphatidic Acid-Induced Vascular Endothelial Growth Factor Expression. Clin. Cancer Res. 2006, 12, 6351–6358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-J.; No, Y.R.; Dang, D.T.; Dang, L.H.; Yang, V.W.; Shim, H.; Yun, C.C. Regulation of Hypoxia-inducible Factor 1α (HIF-1α) by Lysophosphatidic Acid Is Dependent on Interplay between p53 and Krüppel-like Factor 5. J. Biol. Chem. 2013, 288, 25244–25253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fotopoulou, S.; Oikonomou, N.; Grigorieva, E.; Nikitopoulou, I.; Paparountas, T.; Thanassopoulou, A.; Zhao, Z.; Xu, Y.; Kontoyiannis, D.L.; Remboutsika, E.; et al. ATX expression and LPA signalling are vital for the development of the nervous system. Dev. Biol. 2010, 339, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Herr, K.J.; Herr, D.R.; Lee, C.-W.; Noguchi, K.; Chun, J. Stereotyped fetal brain disorganization is induced by hypoxia and requires lysophosphatidic acid receptor 1 (LPA1) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 15444–15449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef] [PubMed]
- Villa, J.C.; Chiu, D.; Brandes, A.H.; Escorcia, F.E.; Villa, C.H.; Maguire, W.F.; Hu, C.-J.; de Stanchina, E.; Simon, M.C.; Sisodia, S.S.; et al. Nontranscriptional Role of Hif-1α in Activation of γ-Secretase and Notch Signaling in Breast Cancer. Cell Rep. 2014, 8, 1077–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, K.; Mizuno, A.; Ueda, M.; Li, S.; Suzuki, Y.; Hida, Y.; Hayakawa-Yano, Y.; Itoh, M.; Ohta, E.; Kobori, M.; et al. Autophagy impairment stimulates PS1 expression and γ-secretase activity. Autophagy 2010, 3, 345–352. [Google Scholar] [CrossRef]
- Cai, Z.; Zhou, Y.; Liu, Z.; Ke, Z.; Zhao, B. Autophagy dysfunction upregulates beta-amyloid peptides via enhancing the activity of γ-secretase complex. Neuropsychiatr. Dis. Treat. 2015, 11, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- De Gasperi, R.; Sosa, M.; Dracheva, S.; Elder, G.A. Presenilin-1 regulates induction of hypoxia inducible factor-1α: Altered activation by a mutation associated with familial Alzheimer’s disease. Mol. Neurodegener. 2010, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, M.R.; Barth, S.; Konietzko, U.; Wu, B.; Egger, S.; Kunze, R.; Marti, H.H.; Hick, M.; Muller, U.; Camenisch, G.; et al. Dysregulation of Hypoxia-Inducible Factor by Presenilin/ -Secretase Loss-of-Function Mutations. J. Neurosci. 2013, 33, 1915–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savitz, S.I.; Dhallu, M.S.; Malhotra, S.; Mammis, A.; Ocava, L.C.; Rosenbaum, P.S.; Rosenbaum, D.M. EDG receptors as a potential therapeutic target in retinal ischemia–reperfusion injury. Brain Res. 2006, 1118, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Lafleur, J.; Mwaikambo, B.R.; Zhu, T.; Gagnon, C.; Chemtob, S.; Di Polo, A.; Hardy, P. The Role of Lysophosphatidic Acid Receptor (LPA1) in the Oxygen-Induced Retinal Ganglion Cell Degeneration. Investig. Opthalmol. Vis. Sci. 2009, 50, 1290. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; Tang, X.; Venkatraman, G.; Bekele, R.T.; Brindley, D.N. Recent advances in targeting the autotaxin-lysophosphatidate-lipid phosphate phosphatase axis in vivo. J. Biomed. Res. 2016, 30, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.; MacIntyre, I.; McMullen, T.; Brindley, D. Coming of Age for Autotaxin and Lysophosphatidate Signaling: Clinical Applications for Preventing, Detecting and Targeting Tumor-Promoting Inflammation. Cancers 2018, 10, 73. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Gα Subunit | Signaling Pathway | Cellular Effects | References |
|---|---|---|---|
| Gα12/13 | Rho/ROCK and Rho/SRF pathways | Cell motility Invasion Cytoskeletal changes Vasodilation | [44,45,46] |
| Gαq/11 | IP3-DAG pathway | Cell growth Immunity Learning and memory | [44,45,46,47] |
| Gαs | Adenylyl cyclase | Inhibits cell migration | [48] |
| Gαi/O | Ras/MAPK pathway PI3K/Rac pathway PI3K/Akt pathway | Reorganization of the actin cytoskeleton Cytoskeletal changes and cell migration Cell survival and apoptosis | [49,50,51,52] |
| LPAR Subtypes | Biological Functions | References |
|---|---|---|
| LPA1 | Cell survival, proliferation, adhesion, migration, immune function, and myelination | [53] |
| LPA2 | Similar to and complementary to LPA1-mediated effects | [33,54] |
| LPA3 | Mainly involved in reproductive functions—fertility, implantation of the embryo | [55] |
| LPA4 | Cell adhesion and aggregation, vascular development, regulation of osteogenesis, offset LPA1- and LPA2-mediated chemokine release | [56,57] |
| LPA5 | Inhibits cell motility Involved in chemokine release | [48,58] |
| LPA6 | Not fully elucidated Mutations linked to hair loss and hypotrichosis | [59,60,61] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramesh, S.; Govindarajulu, M.; Suppiramaniam, V.; Moore, T.; Dhanasekaran, M. Autotaxin–Lysophosphatidic Acid Signaling in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1827. https://doi.org/10.3390/ijms19071827
Ramesh S, Govindarajulu M, Suppiramaniam V, Moore T, Dhanasekaran M. Autotaxin–Lysophosphatidic Acid Signaling in Alzheimer’s Disease. International Journal of Molecular Sciences. 2018; 19(7):1827. https://doi.org/10.3390/ijms19071827
Chicago/Turabian StyleRamesh, Sindhu, Manoj Govindarajulu, Vishnu Suppiramaniam, Timothy Moore, and Muralikrishnan Dhanasekaran. 2018. "Autotaxin–Lysophosphatidic Acid Signaling in Alzheimer’s Disease" International Journal of Molecular Sciences 19, no. 7: 1827. https://doi.org/10.3390/ijms19071827
APA StyleRamesh, S., Govindarajulu, M., Suppiramaniam, V., Moore, T., & Dhanasekaran, M. (2018). Autotaxin–Lysophosphatidic Acid Signaling in Alzheimer’s Disease. International Journal of Molecular Sciences, 19(7), 1827. https://doi.org/10.3390/ijms19071827