Cx43 Channel Gating and Permeation: Multiple Phosphorylation-Dependent Roles of the Carboxyl Terminus

Abstract
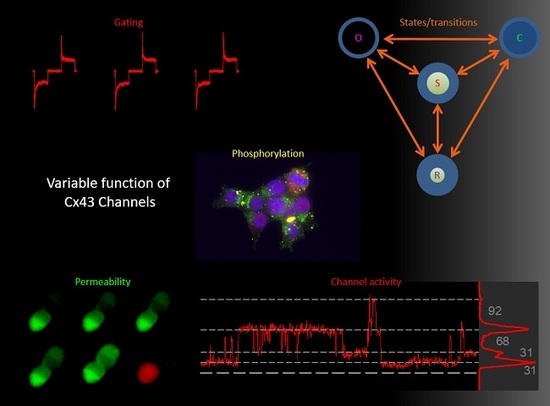
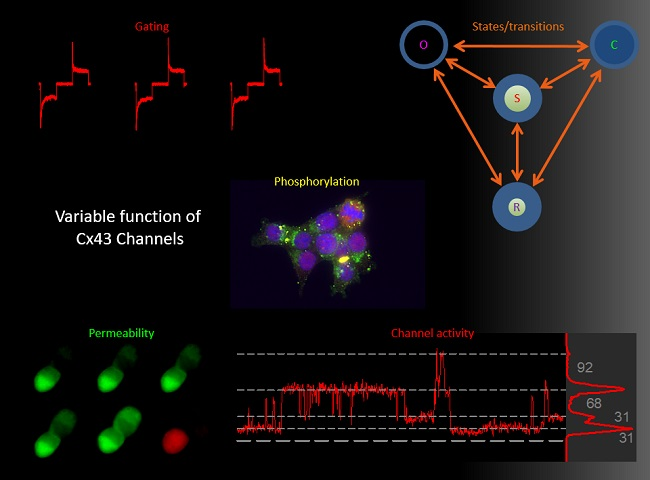
:
1. Introduction
2. Results
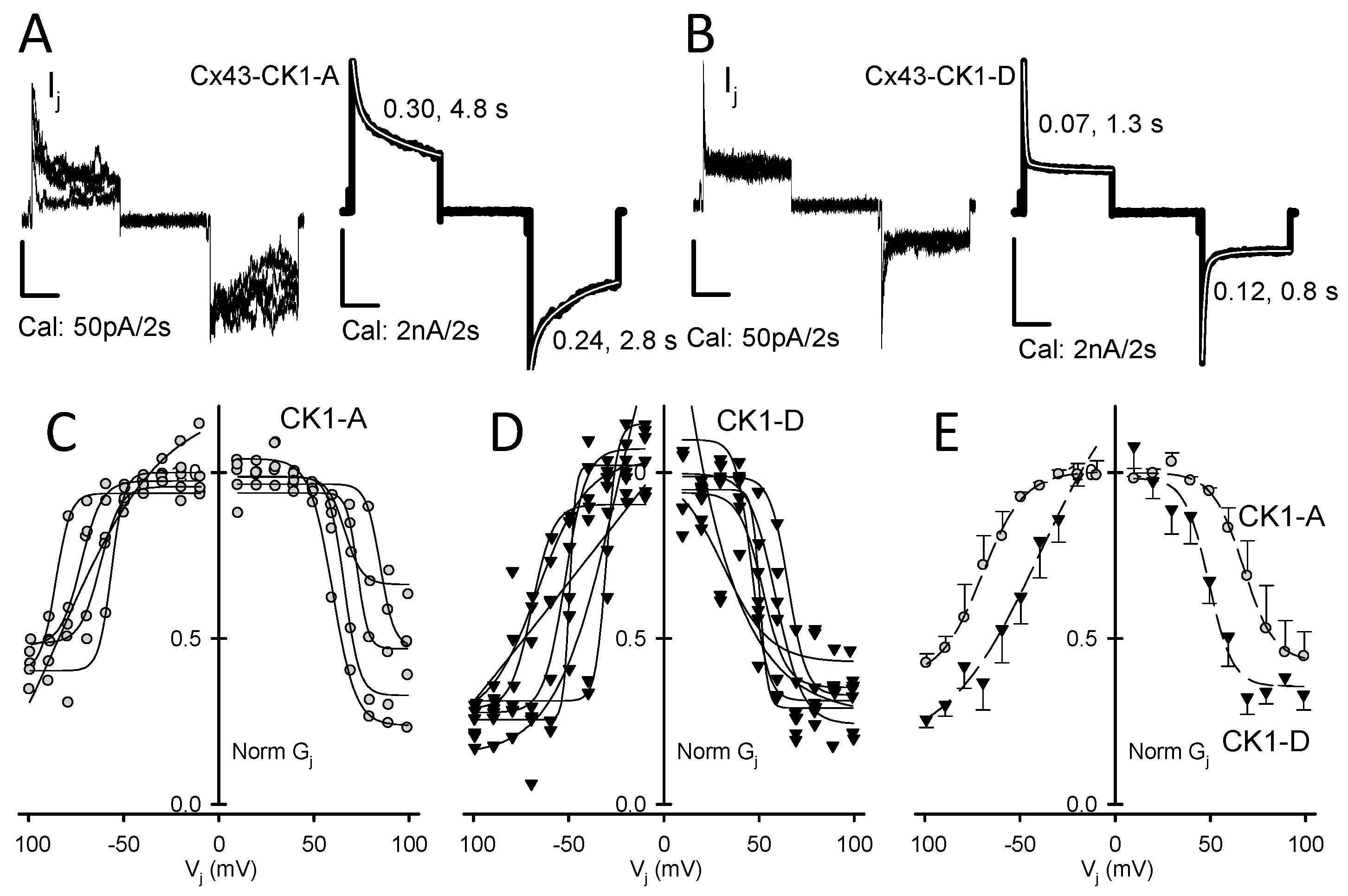
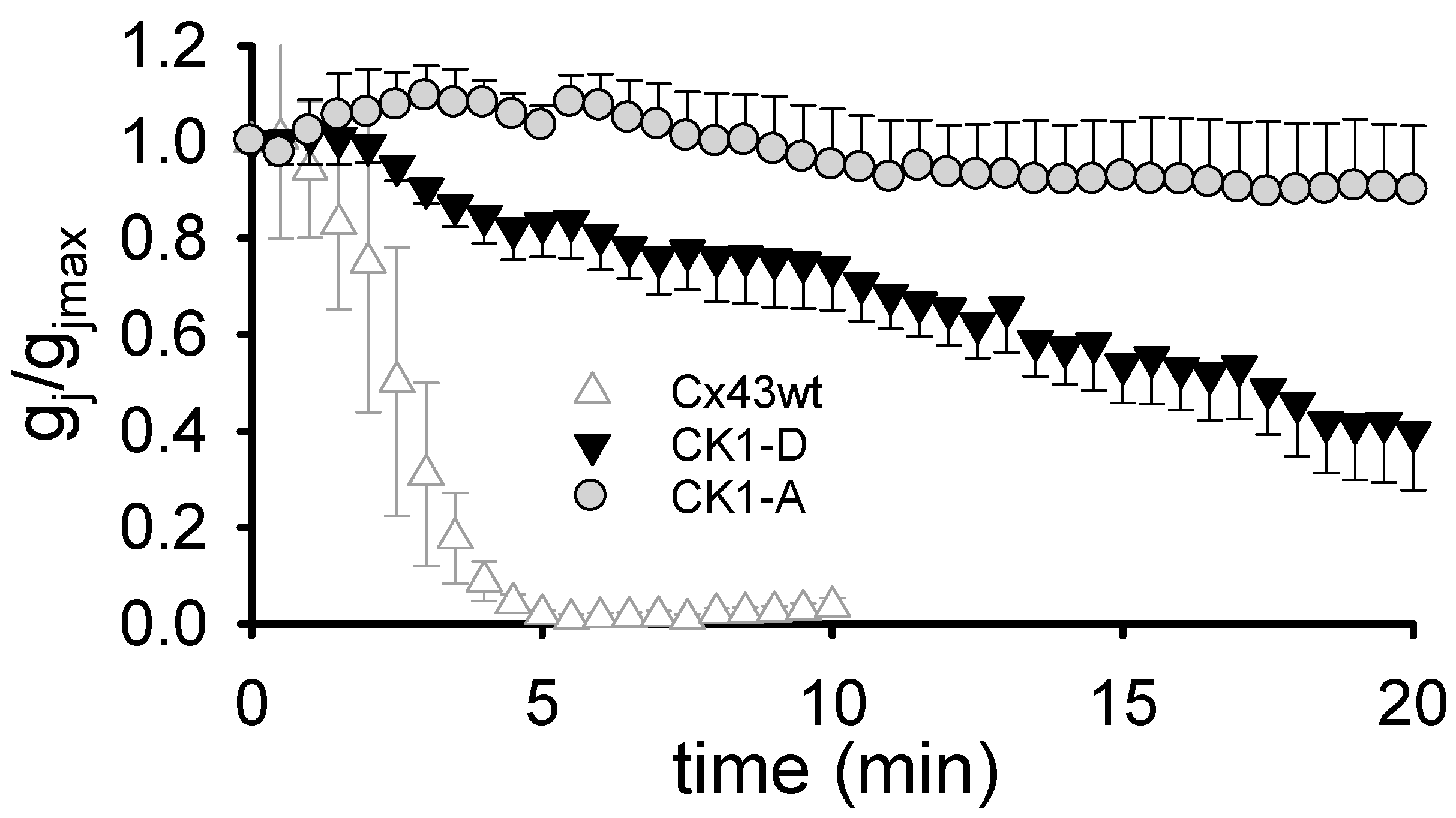
2.1. Cx43-CK1-D Displays Stronger Vj-Sensitivity than Cx43-CK1-A
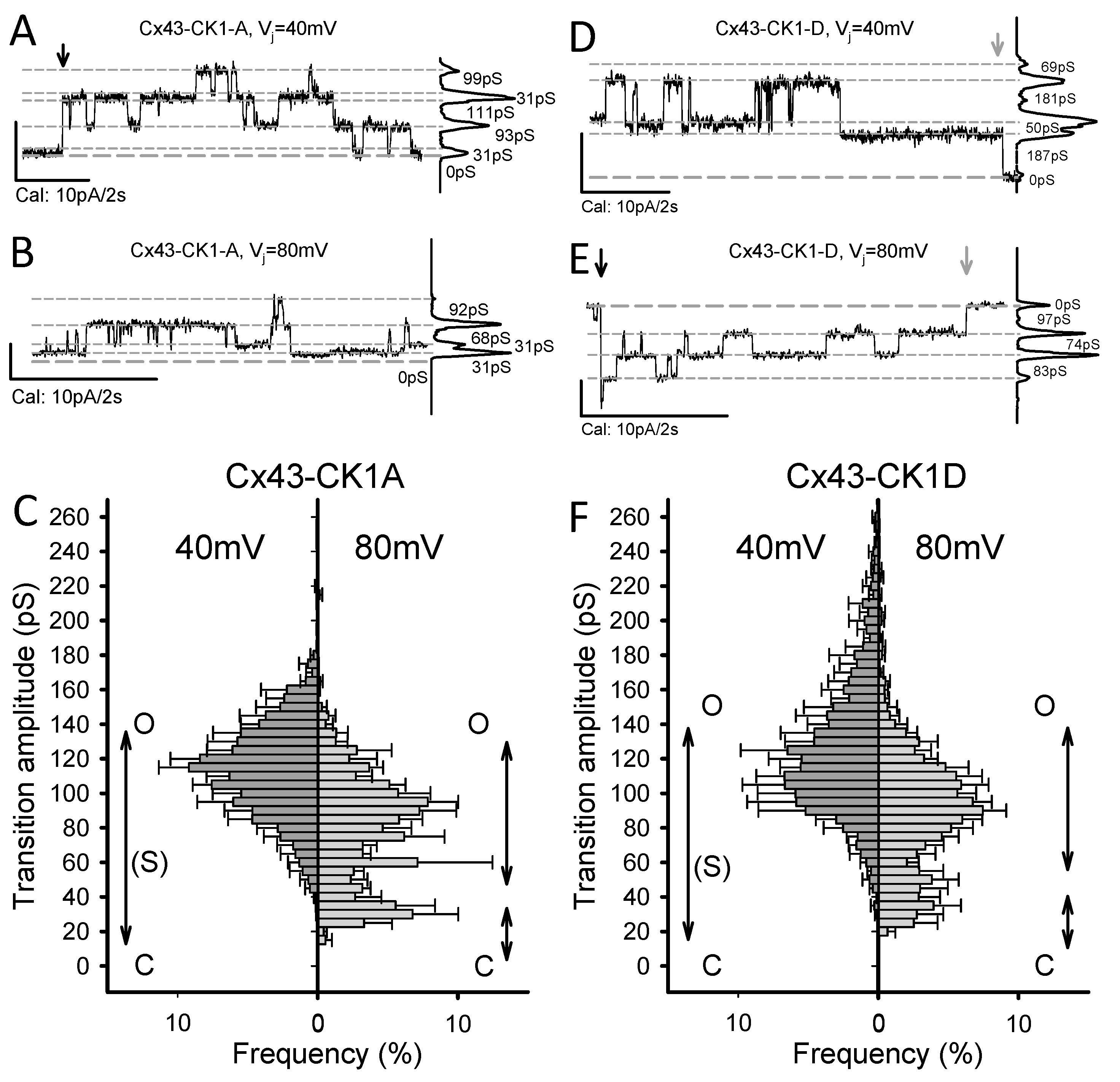
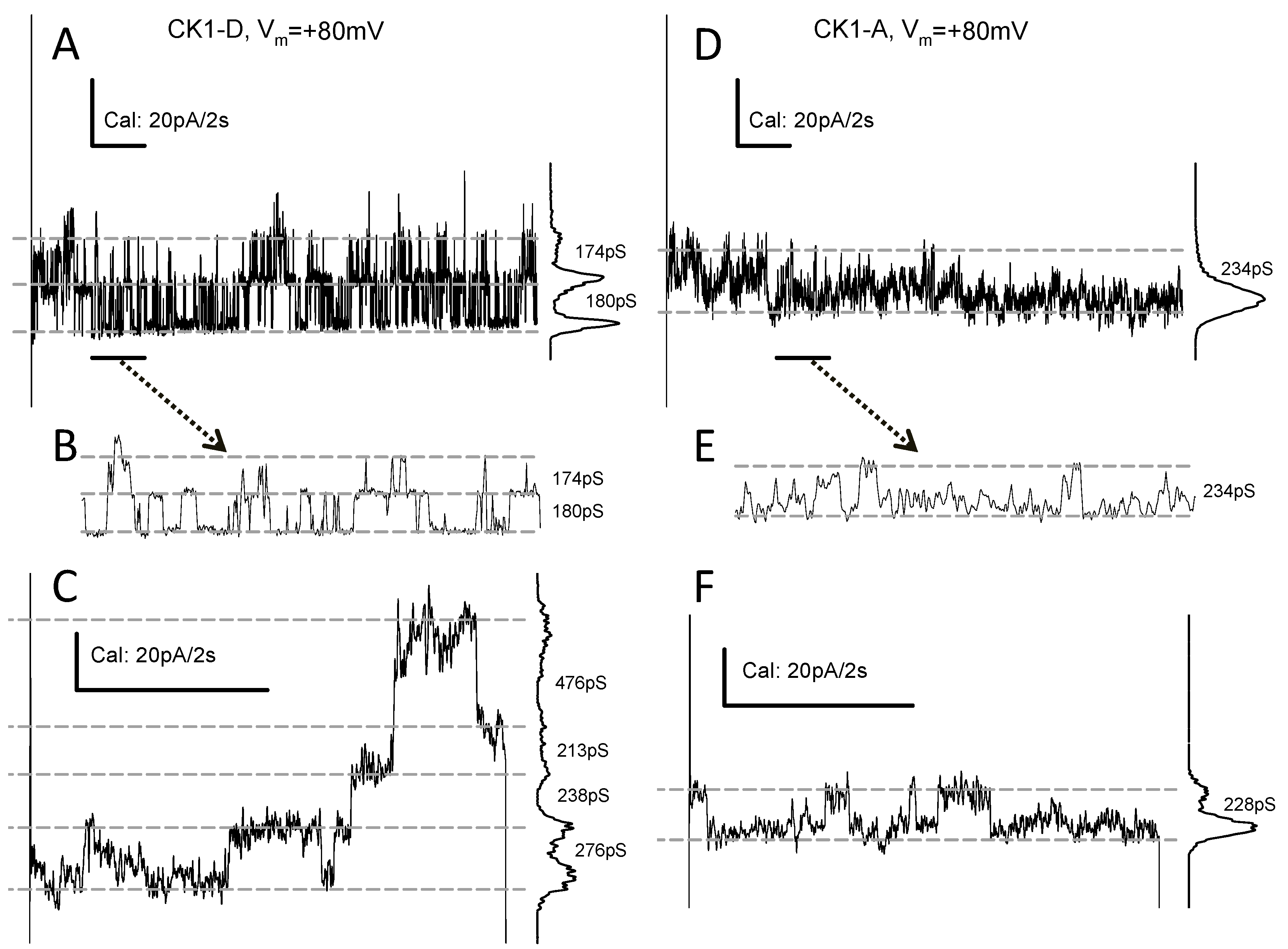
2.2. Cx43-CK1-D and Cx43-CK1-A Display Highly Conductive, Vj-Sensitive, Channel Transition Amplitudes
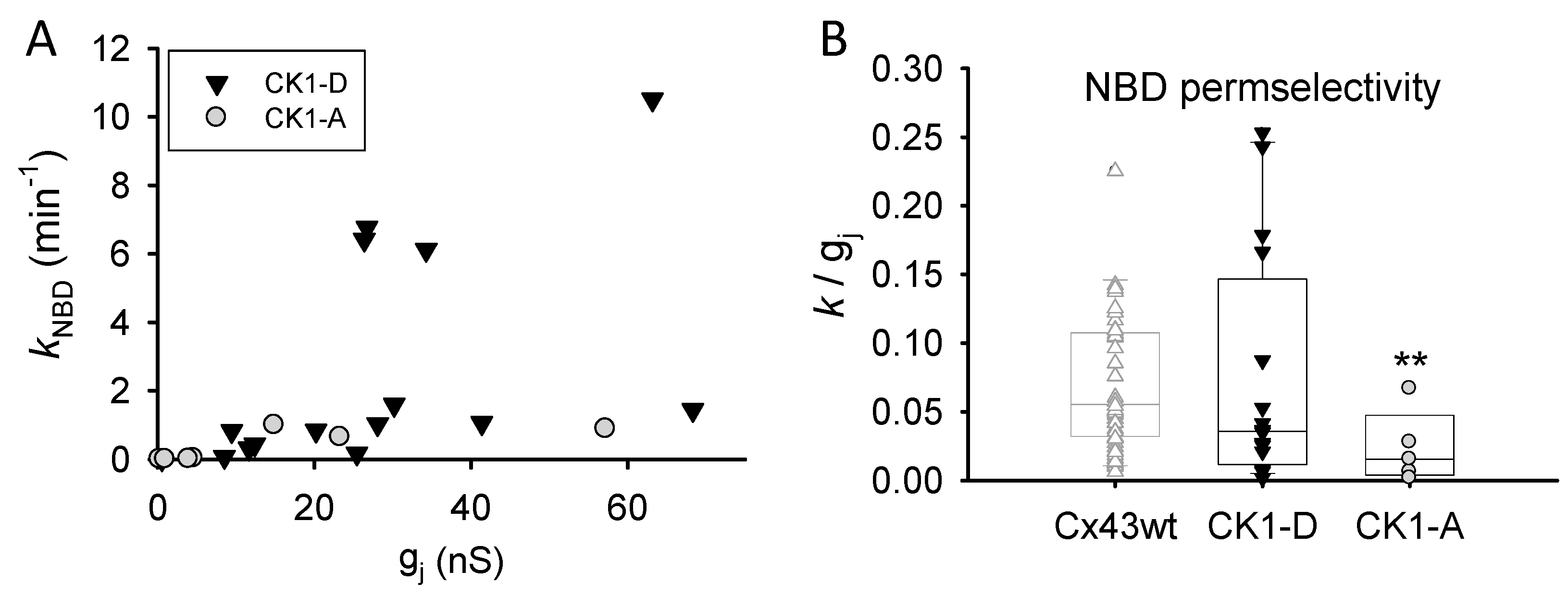
2.3. Cx43-CK1-A Displayed Lower Permselectivity than Cx43-CK1-D
2.4. Cx43-CK1-D and Cx43-CK1-A GJs are Resistant to Intracellular Acidification
2.5. Cx43-CK1-D and -CK1-A Hemichannels Open Frequently
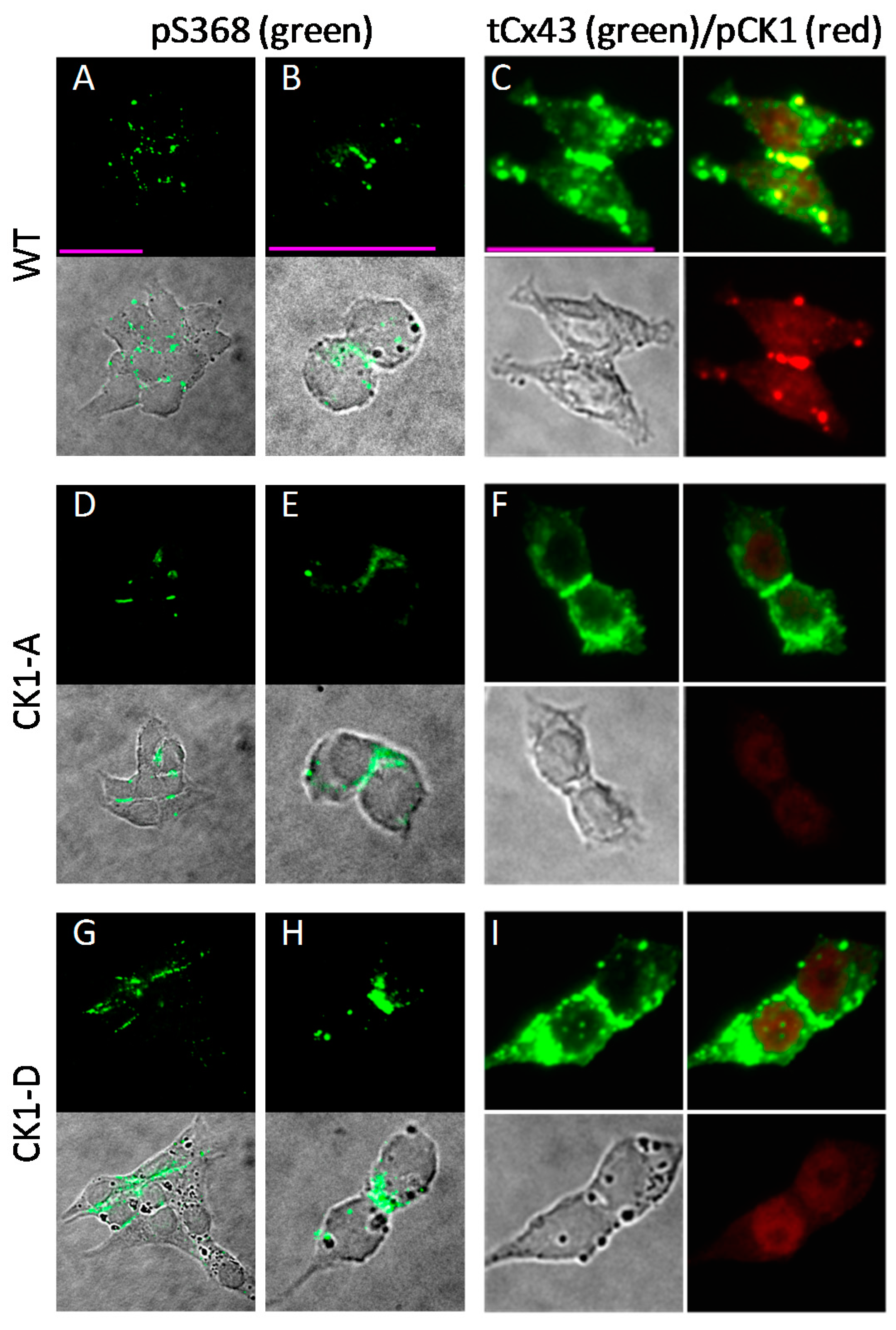
2.6. Cx43-CK1-D and -CK1-A are Phosphorylated at S368
3. Discussion
3.1. On the Role Cx43 of Phosphorylation
3.2. What the Data Suggest
3.2.1. Voltage Gating and Channel States
3.2.2. pH-Gating
3.2.3. Channel Selectivity
3.2.4. Hemichannels
3.2.5. Further Cx43 Phosphorylation
3.2.6. Implications of CK1-Phospho-Mimicking Mutants for a Channel Gating Model
4. Materials and Methods
4.1. Plasmid Construction
4.2. Cell Culture, Transfections and Protein Expression
4.3. Electrophysiology
4.4. Permselectivity
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CK1 | Casein Kinase 1 |
| Cx | Connexin |
| Cx43 | Connexin 43 |
| CT | Carboxyl Terminus |
| CL | Cytoplasmic Loop |
| S3A | Transgenic mouse line expressing mutant Cx43-S325,328,330A |
| S3E | Transgenic mouse line expressing mutant Cx43-S325,328,330E |
| TAC | Transverse Aortic Constriction |
| GJR | Gap Junction Remodelling |
| ID(s) | Intercalated disc(s) |
| GJCh(s) | Gap Junction Channel(s) |
| HCh(s) | Hemichannel(s) |
| Rin | Rat insulinoma |
| WCVC | Whole Cell Voltage Clamp |
| pS | PicoSiemens |
| nS | nanoSiemens |
| pHi | internal pH |
| pHo | external pH |
| DIC | Differential Interference Contrast |
| CT | Carboxyl Terminus |
| CL | Cytoplasmic Loop |
| ZO-1 | Zonula Occludens 1 |
References
- Vozzi, C.; Dupont, E.; Coppen, S.R.; Yeh, H.I.; Severs, N.J. Chamber-related differences in connexin expression in the human heart. J. Mol. Cell. Cardiol. 1999, 31, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, T.; Finet, J.E.; Takeuchi, A.; Fujino, Y.; Strom, M.; Greener, I.D.; Rosenbaum, D.S.; Donahue, J.K. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation 2012, 125, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Plum, A.; Hallas, G.; Magin, T.; Dombrowski, F.; Hagendorff, A.; Schumacher, B.; Wolpert, C.; Kim, J.; Lamers, W.H.; Evert, M.; et al. Unique and shared functions of different connexins in mice. Curr. Biol. 2000, 10, 1083–1091. [Google Scholar] [CrossRef]
- Betsuyaku, T.; Nnebe, N.S.; Sundset, R.; Patibandla, S.; Krueger, C.M.; Yamada, K.A. Overexpression of cardiac connexin45 increases susceptibility to ventricular tachyarrhythmias in vivo. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H163–H171. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; Lau, A.F. The effects of connexin phosphorylation on gap junctional communication. Int. J. Biochem. Cell Biol. 2004, 36, 1171–1186. [Google Scholar] [CrossRef] [Green Version]
- Pogoda, K.; Kameritsch, P.; Retamal, M.A.; Vega, J.L. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: A revision. BMC Cell Biol. 2016, 17 (Suppl. 1), 137–150. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.D.; Lampe, P.D. Casein kinase 1 regulates connexin-43 gap junction assembly. J. Biol. Chem. 2002, 277, 44962–44968. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; Cooper, C.D.; King, T.J.; Burt, J.M. Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J. Cell Sci. 2006, 119, 3435–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remo, B.F.; Qu, J.; Volpicelli, F.M.; Giovannone, S.; Shin, D.; Lader, J.; Liu, F.Y.; Zhang, J.; Lent, D.S.; Morley, G.E.; et al. Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ. Res. 2011, 108, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Volpicelli, F.M.; Garcia, L.I.; Sandeep, N.; Zhang, J.; Marquez-Rosado, L.; Lampe, P.D.; Fishman, G.I. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ. Res. 2009, 104, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Z.; Li, J.; Lemanski, L.F.; Veenstra, R.D. Gating of mammalian cardiac gap junction channels by transjunctional voltage. Biophys. J. 1992, 63, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, M.; Costa, M.; Gao, Y.; Fort, A.; Fishman, G.I.; Spray, D.C. Voltage dependence of macroscopic and unitary currents of gap junction channels formed by mouse connexin50 expressed in rat neuroblastoma cells. J. Physiol. 1999, 517, 673–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ek Vitorin, J.F.; Pontifex, T.K.; Burt, J.M. Determinants of Cx43 Channel Gating and Permeation: The Amino Terminus. Biophys. J. 2016, 110, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.P.; Fishman, G.I.; Spray, D.C. Phosphorylation shifts unitary conductance and modifies voltage dependent kinetics of human connexin43 gap junction channels. Biophys. J. 1992, 62, 51–53. [Google Scholar] [CrossRef] [Green Version]
- Moreno, A.P.; Rook, M.B.; Fishman, G.I.; Spray, D.C. Gap junction channels: Distinct voltage-sensitivie and -insensitive conductance states. Biophys. J. 1994, 67, 113–119. [Google Scholar] [CrossRef]
- Gonzalez, D.; Gomez-Hernandez, J.M.; Barrio, L.C. Molecular basis of voltage dependence of connexin channels: An integrative appraisal. Prog. Biophys. Mol. Biol. 2007, 94, 66–106. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.P.; Saez, J.C.; Fishman, G.I.; Spray, D.C. Human Connexin43 gap junction channels: Regulation of unitary conductances by phosphorylation. Circ. Res. 1994, 74, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.P.; Chanson, M.; Elenes, S.; Anumonwo, J.; Scerri, I.; Gu, H.; Taffet, S.M.; Delmar, M. Role of the carboxyl terminal of connexin43 in transjunctional fast voltage gating. Circ. Res. 2002, 90, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.K.; Beyer, E.C.; Burt, J.M. Characterization of gap junction channels in A7r5 vascular smooth muscle cells. Am. J. Physiol. 1991, 260, C975–C981. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; Tenbroek, E.M.; Burt, J.M.; Kurata, W.E.; Johnson, R.G.; Lau, A.F. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 2000, 149, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, G.T.; Lin, R.; Warn-Cramer, B.J.; Lau, A.F.; Burt, J.M. Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am. J. Physiol. Cell Physiol. 2003, 284, C511–C520. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.K.; Sorgen, P.L.; Burt, J.M. Carboxy terminus and pore-forming domain properties specific to Cx37 are necessary for Cx37-mediated suppression of insulinoma cell proliferation. Am. J. Physiol. Cell Physiol. 2013, 305, C1246–C1256. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, N.L.; Pontifex, T.K.; Li, H.; Solan, J.L.; Lampe, P.D.; Sorgen, P.L.; Burt, J.M. Regulation of Cx37 channel and growth-suppressive properties by phosphorylation. J. Cell Sci. 2017, 130, 3308–3321. [Google Scholar] [CrossRef] [PubMed]
- Gemel, J.; Nelson, T.K.; Burt, J.M.; Beyer, E.C. Inducible coexpression of connexin37 or connexin40 with connexin43 selectively affects intercellular molecular transfer. J. Membr. Biol. 2012, 245, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Ek-Vitorin, J.F.; Taffet, S.M.; Delmar, M. UltraRapid communication: Coexpression of connexins 40 and 43 enhances the pH sensitivityof gap junctions: A model for synergistic interactions among connexins. Circ. Res. 2000, 86, e98–e103. [Google Scholar] [CrossRef] [PubMed]
- Ek-Vitorin, J.F.; Burt, J.M. Quantification of Gap Junction Selectivity. Am. J. Physiol. Cell Physiol. 2005, 289, C1535–C1546. [Google Scholar] [CrossRef] [PubMed]
- Ek-Vitorin, J.F.; King, T.J.; Heyman, N.S.; Lampe, P.D.; Burt, J.M. Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ. Res. 2006, 98, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Gadian, D.G.; Hoult, D.I.; Radda, G.K.; Seeley, P.J.; Chance, B.; Barlow, C. Phosphorus nuclear magnetic resonance studies on normoxic and ischemic cardiac tissue. Proc. Natl. Acad. Sci. USA 1976, 73, 4446–4448. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wetsel, W.; Steenbergen, C.; Murphy, E. Effect of ischemic preconditioning and PKC activation on acidification during ischemia in rat heart. J. Mol. Cell. Cardiol. 1996, 28, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.; Gross, W.; Gebhard, M.M. Hearts during ischemia with or without HTK-protection analysed by dielectric spectroscopy. Physiol. Meas. 2018, 39, 025002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Meana, M.; Garcia-Dorado, D.; Lane, S.; Pina, P.; Inserte, J.; Mirabet, M.; Soler-Soler, J. Persistence of gap junction communication during myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2563–H2571. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.R. Ischaemia-induced cellular electrical uncoupling and ventricular fibrillation. Neth. Heart J. 2002, 10, 360–365. [Google Scholar] [PubMed]
- Jain, S.K.; Schuessler, R.B.; Saffitz, J.E. Mechanisms of delayed electrical uncoupling induced by ischemic preconditioning. Circ. Res. 2003, 92, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Cascio, W.E.; Yang, H.; Muller-Borer, B.J.; Johnson, T.A. Ischemia-induced arrhythmia: The role of connexins, gap junctions, and attendant changes in impulse propagation. J. Electrocardiol. 2005, 38, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, T.-F.; Lazrak, A.; Peracchia, C.; Goldberg, G.S.; Lampe, P.D.; Johnson, C.M. Properties and regulation of gap junctional hemichannels in the plasma membranes of cultured cells. J. Cell Biol. 1996, 134, 1019–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, S.A.; Kondo, R.; Wang, S.Y.; Goldhaber, J.I.; Weiss, J.N. Connexin-43 hemichannels opened by metabolic inhibition. J. Biol. Chem. 1999, 274, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Shahidullah, M.; Delamere, N.A. Connexins form functional hemichannels in porcine ciliary epithelium. Exp. Eye Res. 2014, 118, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, J.E.; Saez, J.C.; Bukauskas, F.F.; Bennett, M.V. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc. Natl. Acad. Sci. USA 2003, 100, 11388–11393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampe, P.D.; Lau, A.F. Regulation of gap junctions by phosphorylation of connexins. Arch. Biochem. Biophys. 2000, 384, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Beardslee, M.A.; Lerner, D.L.; Tadros, P.N.; Laing, J.G.; Beyer, E.C.; Yamada, K.A.; Kleber, A.G.; Schuessler, R.B.; Saffitz, J.E. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ. Res. 2000, 87, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim. Biophys. Acta 2005, 1711, 154–163. [Google Scholar] [CrossRef] [PubMed]
- King, T.J.; Lampe, P.D. Temporal regulation of connexin phosphorylation in embryonic and adult tissues. Biochim. Biophys. Acta 2005, 1719, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Marquez-Rosado, L.; Solan, J.L.; Dunn, C.A.; Norris, R.P.; Lampe, P.D. Connexin43 phosphorylation in brain, cardiac, endothelial and epithelial tissues. Biochim. Biophys. Acta 2012, 1818, 1985–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solan, J.L.; Lampe, P.D. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014, 588, 1423–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solan, J.L.; Lampe, P.D. Kinase programs spatiotemporally regulate gap junction assembly and disassembly: Effects on wound repair. Semin. Cell Dev. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Reynhout, J.K.; Lampe, P.D.; Johnson, R.G. An activator of protein kinase C inhibits gap junction communication between cultured bovine lens cells. Exp. Cell Res. 1992, 198, 337–342. [Google Scholar] [CrossRef]
- Stein, L.S.; Boonstra, J.; Burghardt, R.C. Reduced cell-cell communication between mitotic and nonmitotic coupled cells. Exp. Cell Res. 1992, 198, 1–7. [Google Scholar] [CrossRef]
- Solan, J.L.; Fry, M.D.; Tenbroek, E.M.; Lampe, P.D. Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J. Cell Sci. 2003, 116, 2203–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burt, J.M.; Nelson, T.K.; Simon, A.M.; Fang, J.S. Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am. J. Physiol. Cell Physiol. 2008, 295, C1103–C1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, T.S.; Dunn, C.A.; Carter, W.G.; Usui, M.L.; Olerud, J.E.; Lampe, P.D. Protein kinase C spatially and temporally regulates gap junctional communication during human wound repair via phosphorylation of connexin43 on serine368. J. Cell Biol. 2004, 167, 555–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.S.; Angelov, S.N.; Simon, A.M.; Burt, J.M. Cx37 deletion enhances vascular growth and facilitates ischemic limb recovery. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1872–H1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.S.; Angelov, S.N.; Simon, A.M.; Burt, J.M. Cx40 is required for, and Cx37 limits, postischemic hindlimb perfusion, survival and recovery. J. Vasc. Res. 2012, 49, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.Y.; Cooper, E.S.; Waldo, K.; Kirby, M.L.; Gilula, N.B.; Lo, C.W. Gap junction-mediated cell-cell communication modulates mouse neural crest migration. J. Cell Biol. 1998, 143, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Van Rijen, H.V.; Van Kempen, M.J.; Postma, S.; Jongsma, H.J. Tumour necrosis factor alpha alters the expression of connexin43, connexin40, and connexin37 in human umbilical vein endothelial cells. Cytokine 1998, 10, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.R.; Pepper, M.S.; Gros, D.B.; Meda, P. Inhibition of endothelial wound repair by dominant negative connexin inhibitors. Mol. Biol. Cell 2001, 12, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akita, N.; Kawamoto, E.; Hayashi, T.; Suzuki, K.; Shimaoka, M. Endothelial connexin32 enhances angiogenesis by positively regulating tube formation and cell migration. Exp. Cell Res. 2014, 321, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Sawey, M.J.; Goldschmidt, M.H.; Risek, B.; Gilula, N.B.; Lo, C.W. Perturbation in connexin 43 and connexin 26 gap-junction expression in mouse skin hyperplasia and neoplasia. Mol. Carcinog. 1996, 17, 49–61. [Google Scholar] [CrossRef]
- Hirschi, K.K.; Burt, J.M.; Hirschi, K.D.; Dai, C. Gap junction communication mediates transforming growth factor-β activation and endothelial-induced mural cell differentiation. Circ. Res. 2003, 93, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.S.; Dai, C.; Kurjiaka, D.T.; Burt, J.M.; Hirschi, K.K. Connexin45 regulates endothelial-induced mesenchymal cell differentiation toward a mural cell phenotype. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Lerner, D.L.; Yamada, K.A.; Schuessler, R.B.; Saffitz, J.E. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation 2000, 101, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Procida, K.; Jorgensen, L.; Schmitt, N.; Delmar, M.; Taffet, S.M.; Holstein-Rathlou, N.H.; Nielsen, M.S.; Braunstein, T.H. Phosphorylation of connexin43 on serine 306 regulates electrical coupling. Heart Rhythm 2009, 6, 1632–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morley, G.E.; Ek-Vitorin, J.F.; Taffet, S.M.; Delmar, M. Structure of connexin43 and its regulation by pHi. J. Cardiovasc. Electrophysiol. 1997, 8, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Duffy, H.S.; Sorgen, P.L.; Girvin, M.E.; O’Donnell, P.; Coombs, W.; Taffet, S.M.; Delmar, M.; Spray, D.C. pH-dependent intramolecular binding and structure involving Cx43 cytoplasmic domains. J. Biol. Chem. 2002, 277, 36706–36714. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R. Gap-junctional single channel permeability for fluorescent tracers in mammalian cell cultures. Biophys. J. 2006, 91, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.G.; Peracchia, C. Connexin 32/38 chimeras suggest a role for the second half of inner loop in gap junction gating by low pH. Am. J. Physiol. 1996, 271, C1743–C1749. [Google Scholar] [CrossRef] [PubMed]
- Bukauskas, F.F.; Peracchia, C. Two distinct gating mechanisms in gap junction channels: CO2-sensitive and voltage-sensitive. Biophys. J. 1997, 72, 2137–2142. [Google Scholar] [CrossRef]
- Stergiopoulos, K.; Alvarado, J.L.; Mastroianni, M.; Ek-Vitorin, J.F.; Taffet, S.M.; Delmar, M. Hetero-domain interactions as a mechanism for the regulation of connexin channels. Circ. Res. 1999, 84, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Francis, D.; Stergiopoulos, K.; Ek-Vitorin, J.F.; Cao, F.L.; Taffet, S.M.; Delmar, M. Connexin diversity and gap junction regulation by pHi. Dev. Genet. 1999, 24, 123–136. [Google Scholar] [CrossRef]
- Liu, S.; Taffet, S.; Stoner, L.; Delmar, M.; Vallano, M.L.; Jalife, J. A structural basis for the unequal sensitivity of the major cardiac and liver gap junctions to intracellular acidification: The carboxyl tail length. Biophys. J. 1993, 64, 1422–1433. [Google Scholar] [CrossRef]
- Morley, G.E.; Taffet, S.M.; Delmar, M. Intramolecular interactions mediate pH regulation of connexin43 channels. Biophys. J. 1996, 70, 1294–1302. [Google Scholar] [CrossRef] [Green Version]
- Inserte, J.; Barba, I.; Hernando, V.; Abellan, A.; Ruiz-Meana, M.; Rodriguez-Sinovas, A.; Garcia-Dorado, D. Effect of acidic reperfusion on prolongation of intracellular acidosis and myocardial salvage. Cardiovasc. Res. 2008, 77, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Heyman, N.S.; Burt, J.M. Hindered diffusion through an aqueous pore describes invariant dye selectivity of Cx43 junctions. Biophys. J. 2008, 94, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Kondo, R.P.; Wang, S.Y.; John, S.A.; Weiss, J.N.; Goldhaber, J.I. Metabolic inhibition activates a non-selective current through connexin hemichannels in isolated ventricular myocytes. J. Mol. Cell Cardiol. 2000, 32, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Shintani-Ishida, K.; Uemura, K.; Yoshida, K. Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1714–H1720. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.C.; Williams, O.J.; Martin, P.E.; Evans, W.H. ATP release by cardiac myocytes in a simulated ischaemia model: Inhibition by a connexin mimetic and enhancement by an antiarrhythmic peptide. Eur. J. Pharmacol. 2009, 605, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Hawat, G.; Benderdour, M.; Rousseau, G.; Baroudi, G. Connexin 43 mimetic peptide Gap26 confers protection to intact heart against myocardial ischemia injury. Pflugers Arch. 2010, 460, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavan, R.; Lin, J.; Hoeker, G.S.; Keener, J.P.; Gourdie, R.G.; Poelzing, S. Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: An experimental and modeling study. Pflugers Arch. 2015. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sugishita, K.; Su, Z.; Ueda, I.; Barry, W.H. Activation of connexin-43 hemichannels can elevate [Ca2+]i and [Na+]i in rabbit ventricular myocytes during metabolic inhibition. J. Mol. Cell Cardiol. 2001, 33, 2145–2155. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Marquez-Rosado, L.; Sorgen, P.L.; Thornton, P.J.; Gafken, P.R.; Lampe, P.D. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J. Cell Biol. 2007, 179, 1301–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, J.W.; Zhang, S.S.; Sanchez, J.M.; Lamouille, S.; Vogan, J.M.; Hesketh, G.G.; Hong, T.; Tomaselli, G.F.; Shaw, R.M. A 14-3-3 mode-1 binding motif initiates gap junction internalization during acute cardiac ischemia. Traffic 2014, 15, 684–699. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cole, P.A. Synthetic approaches to protein phosphorylation. Curr. Opin. Chem. Biol. 2015, 28, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dissmeyer, N.; Schnittger, A. Use of phospho-site substitutions to analyze the biological relevance of phosphorylation events in regulatory networks. Methods Mol. Biol. 2011, 779, 93–138. [Google Scholar] [CrossRef] [PubMed]
- Thevenin, A.F.; Margraf, R.A.; Fisher, C.G.; Kells-Andrews, R.M.; Falk, M.M. Phosphorylation regulates connexin43/ZO-1 binding and release, an important step in gap junction turnover. Mol. Biol. Cell 2017, 28, 3595–3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doble, B.W.; Ping, P.; Kardami, E. The epsilon subtype of protein kinase C is required for cardiomyocyte connexin-43 phosphorylation. Circ. Res. 2000, 86, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Cross, H.R.; Murphy, E.; Bolli, R.; Ping, P.; Steenbergen, C. Expression of activated PKC epsilon (PKC epsilon) protects the ischemic heart, without attenuating ischemic H+ production. J. Mol. Cell Cardiol. 2002, 34, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Doble, B.W.; Dang, X.; Ping, P.; Fandrich, R.R.; Nickel, B.E.; Jin, Y.; Cattini, P.A.; Kardami, E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J. Cell Sci. 2004, 117, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Srisakuldee, W.; Jeyaraman, M.M.; Nickel, B.E.; Tanguy, S.; Jiang, Z.S.; Kardami, E. Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc. Res. 2009, 83, 672–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patton, C. 2018. 2018. Available online: https://web.stanford.edu/~cpatton/CaEGTA-TS.htm (accessed on 17 April 2018).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | Gj max | Gj min | V0 (mV) | A |
|---|---|---|---|---|
| Cx43WT | 0.98/0.99 | 0.32/0.34 | −64/+70 | 4.8/4.1 |
| Cx43-CK1-D | 1.1/1.0 | 0.21/0.34 | −49/+46 | 12.1/5.4 |
| Cx43-CK1-A | 1.0/0.98 | 0.35/0.40 | −71/+70 | 8.4/4.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ek-Vitorín, J.F.; Pontifex, T.K.; Burt, J.M. Cx43 Channel Gating and Permeation: Multiple Phosphorylation-Dependent Roles of the Carboxyl Terminus. Int. J. Mol. Sci. 2018, 19, 1659. https://doi.org/10.3390/ijms19061659
Ek-Vitorín JF, Pontifex TK, Burt JM. Cx43 Channel Gating and Permeation: Multiple Phosphorylation-Dependent Roles of the Carboxyl Terminus. International Journal of Molecular Sciences. 2018; 19(6):1659. https://doi.org/10.3390/ijms19061659
Chicago/Turabian StyleEk-Vitorín, José F., Tasha K. Pontifex, and Janis M. Burt. 2018. "Cx43 Channel Gating and Permeation: Multiple Phosphorylation-Dependent Roles of the Carboxyl Terminus" International Journal of Molecular Sciences 19, no. 6: 1659. https://doi.org/10.3390/ijms19061659
APA StyleEk-Vitorín, J. F., Pontifex, T. K., & Burt, J. M. (2018). Cx43 Channel Gating and Permeation: Multiple Phosphorylation-Dependent Roles of the Carboxyl Terminus. International Journal of Molecular Sciences, 19(6), 1659. https://doi.org/10.3390/ijms19061659