Mir-34a-5p Mediates Cross-Talk between M2 Muscarinic Receptors and Notch-1/EGFR Pathways in U87MG Glioblastoma Cells: Implication in Cell Proliferation


 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
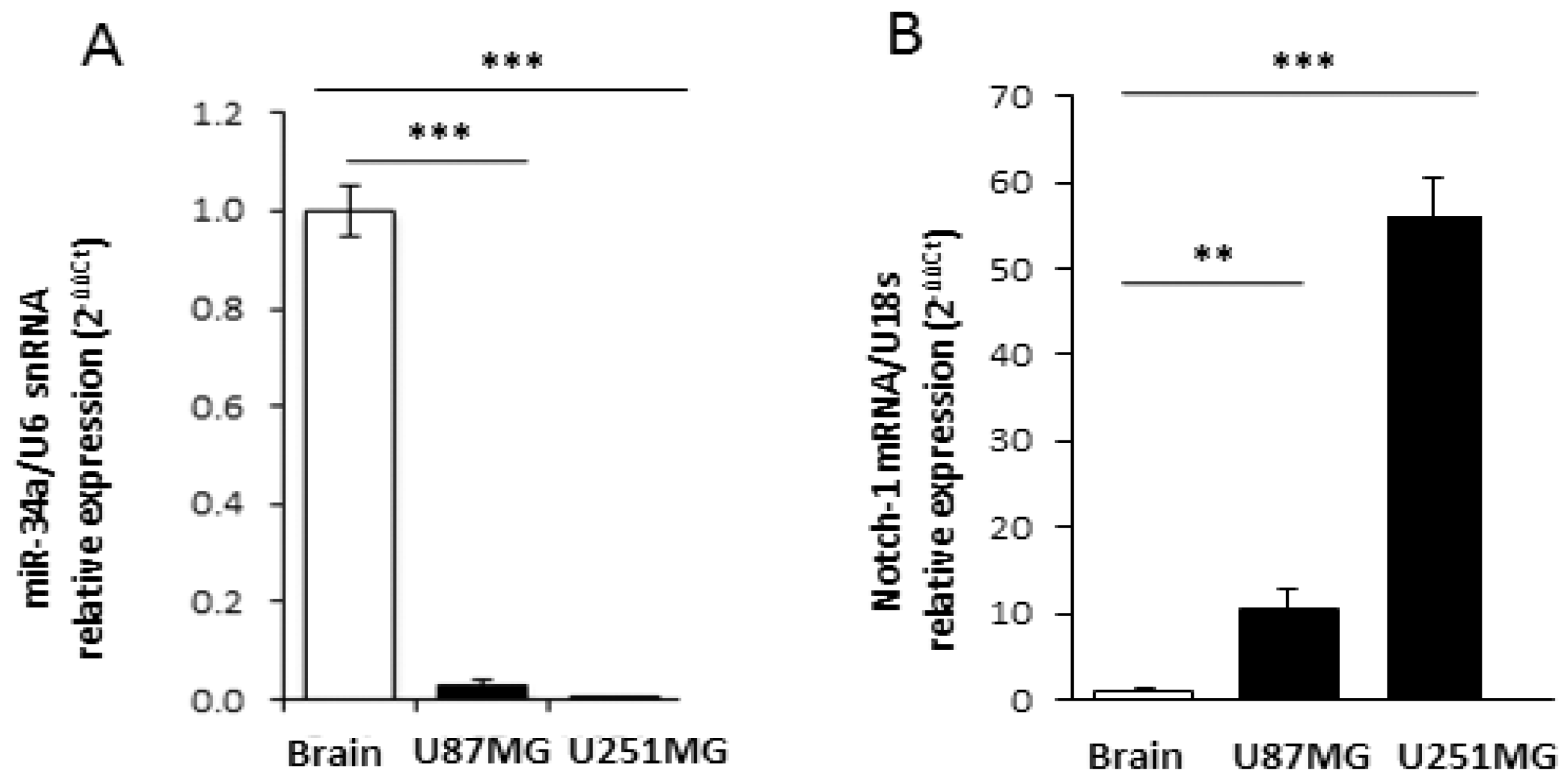
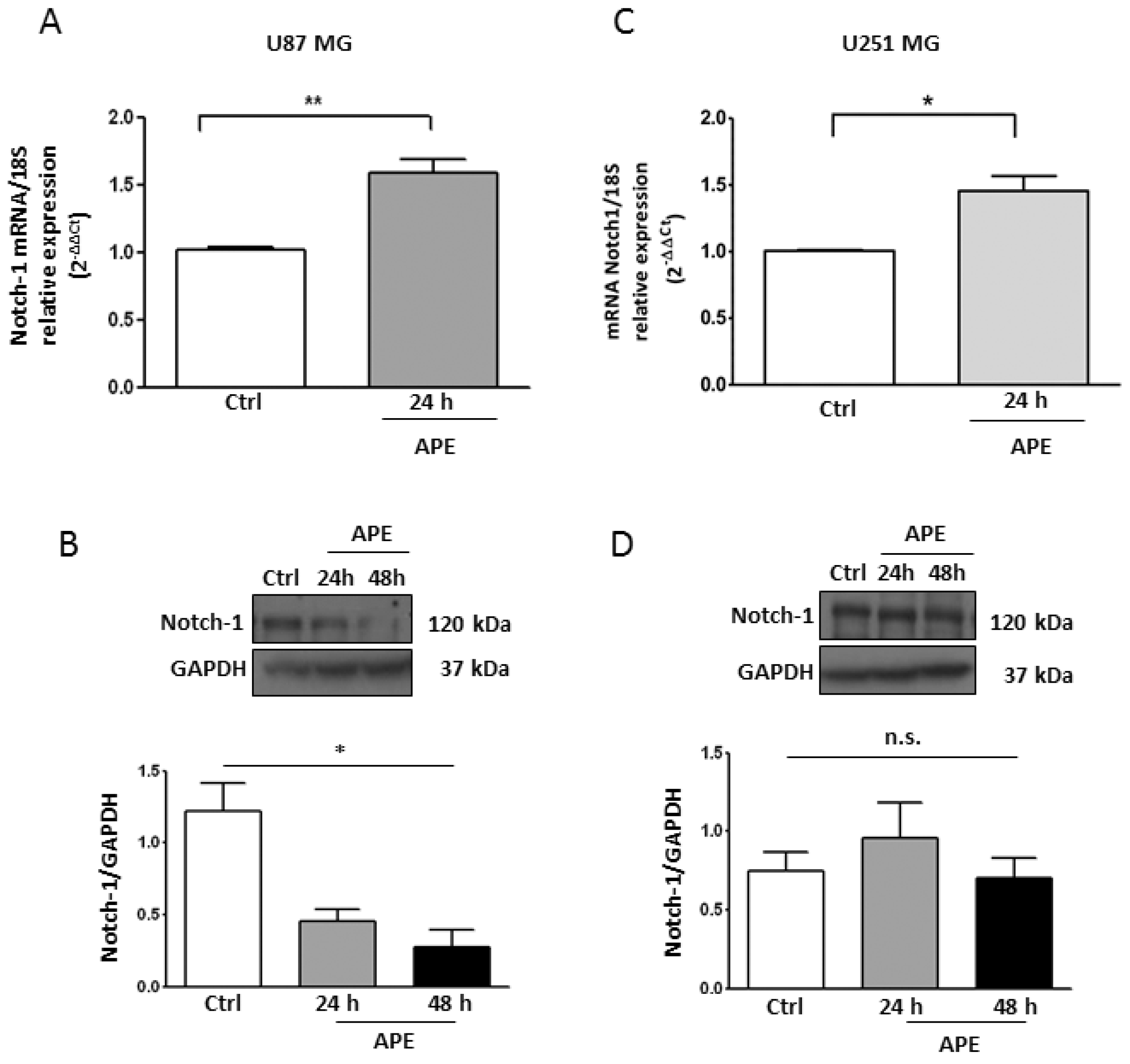
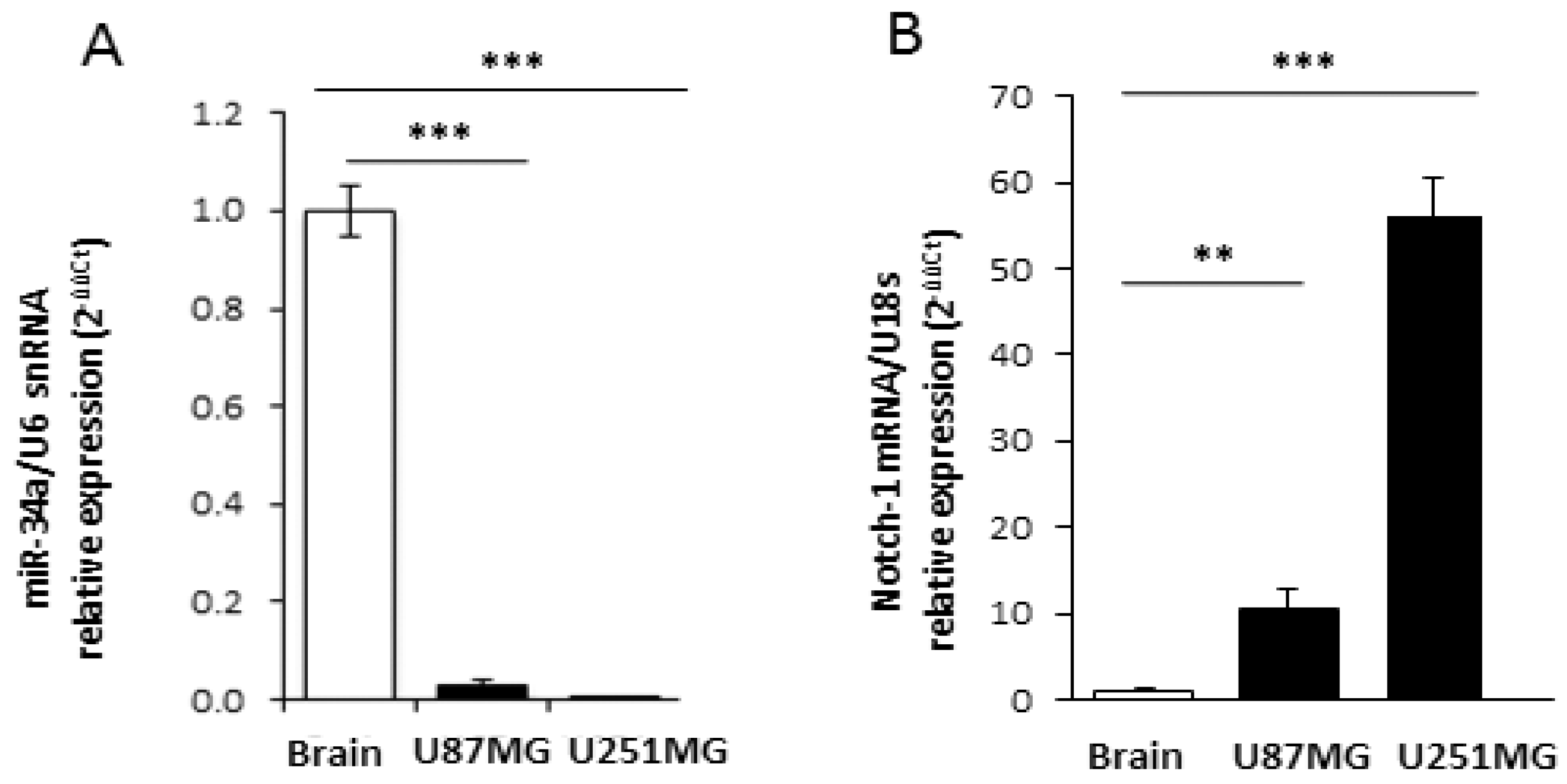
2.1. M2 Receptor Activation Modulates Notch-1 Expression
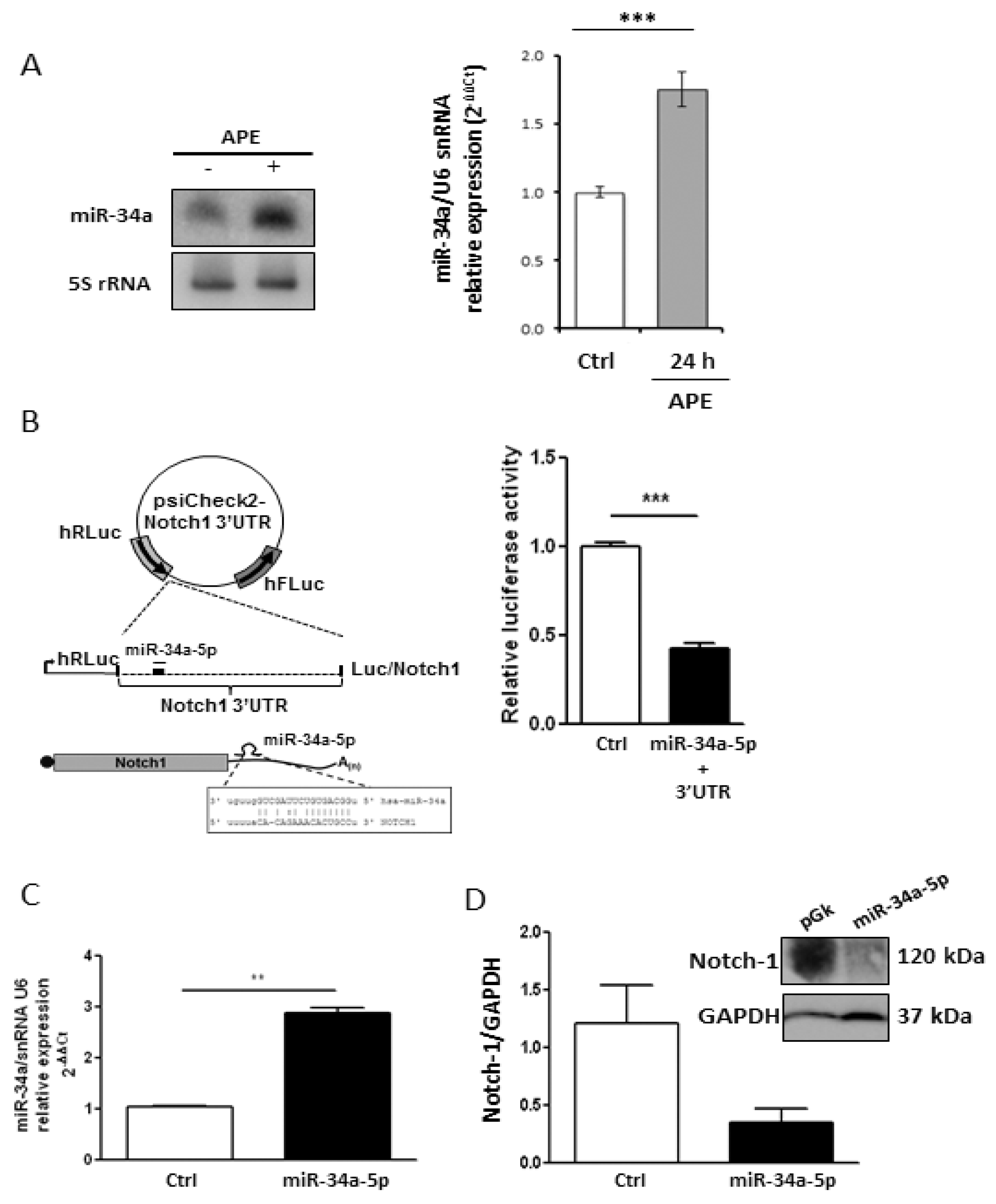
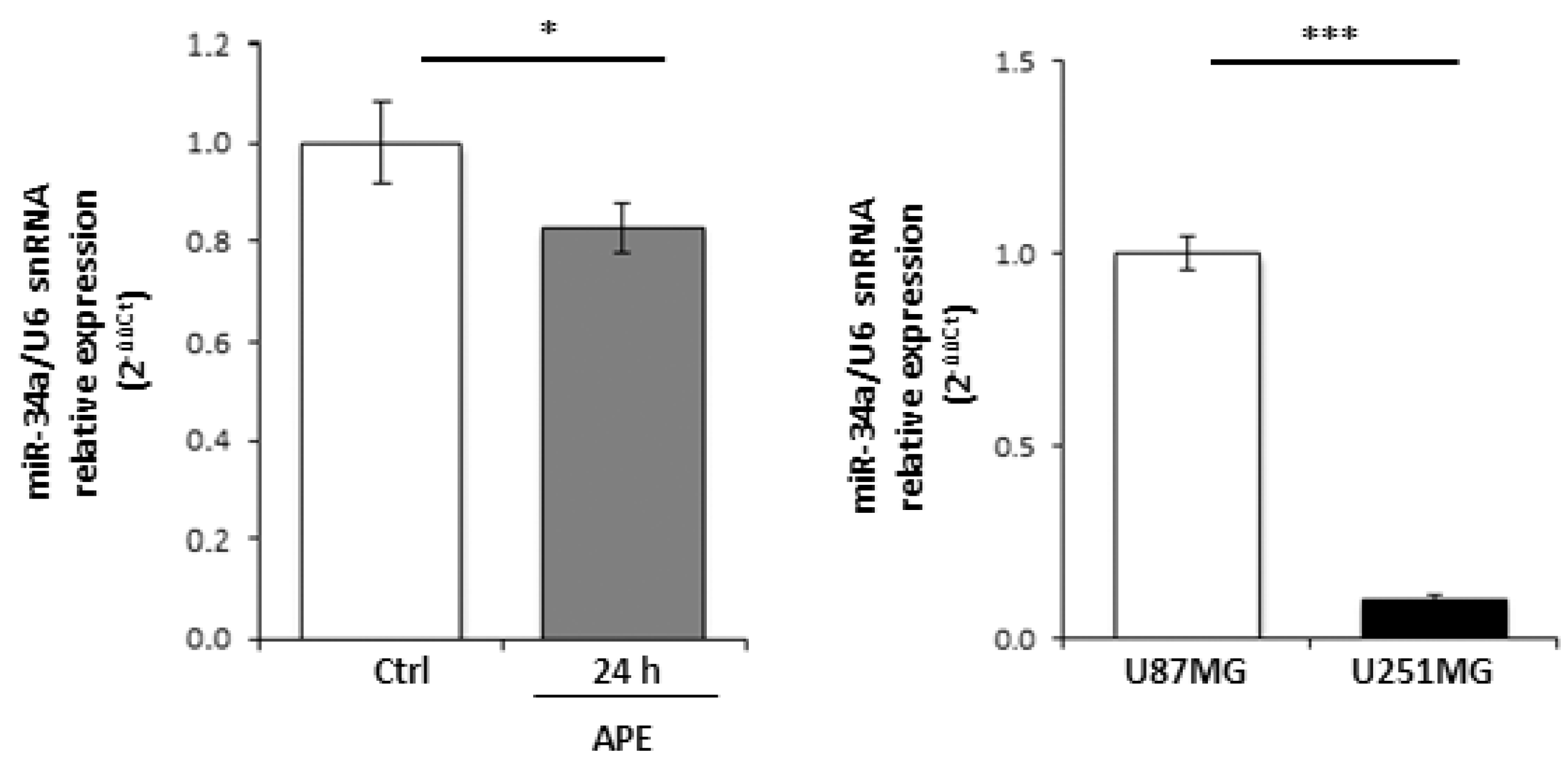
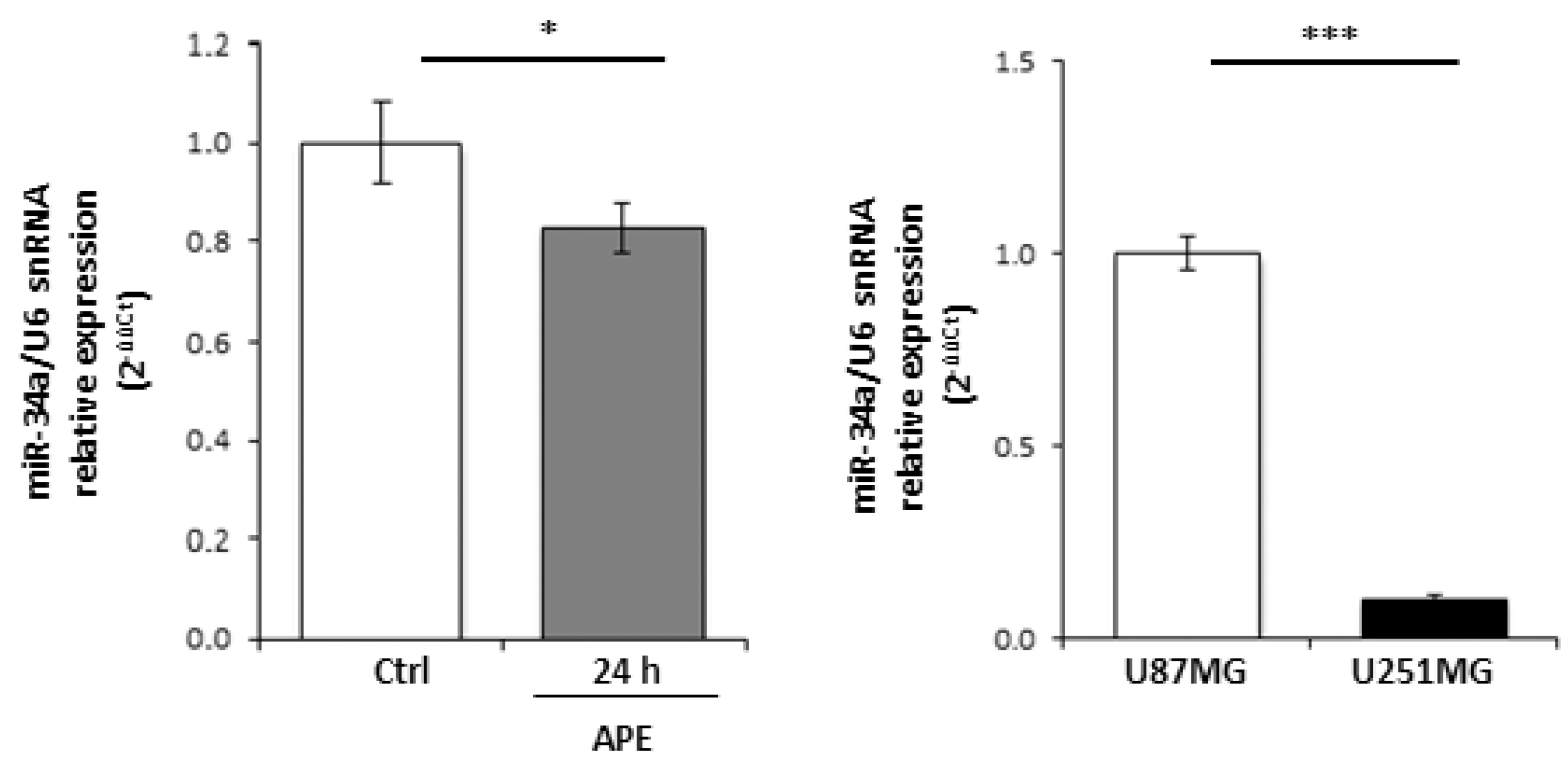
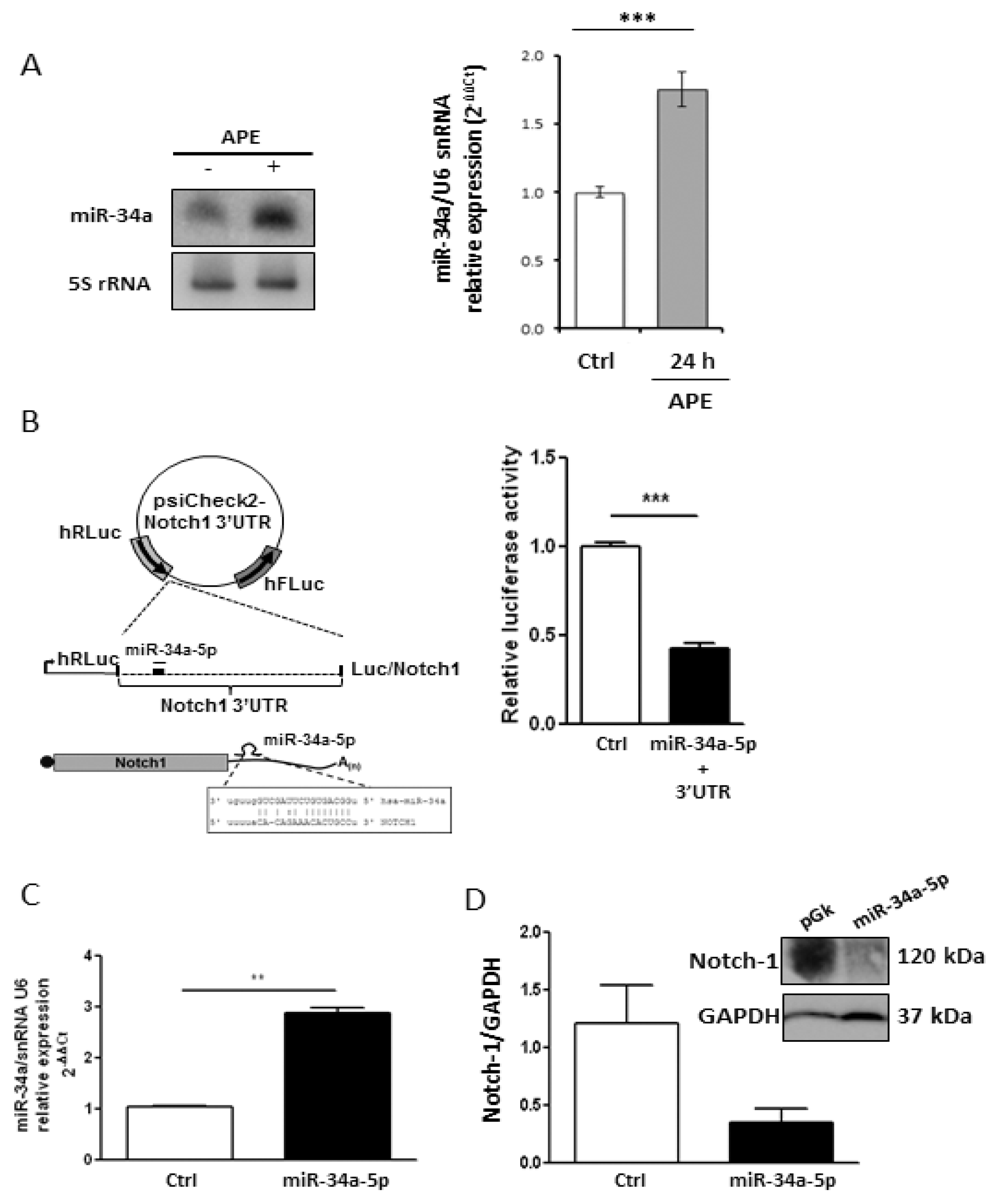
2.2. M2 Receptor Activation Induces Mir-34a-5p Expression in U87MG Cells
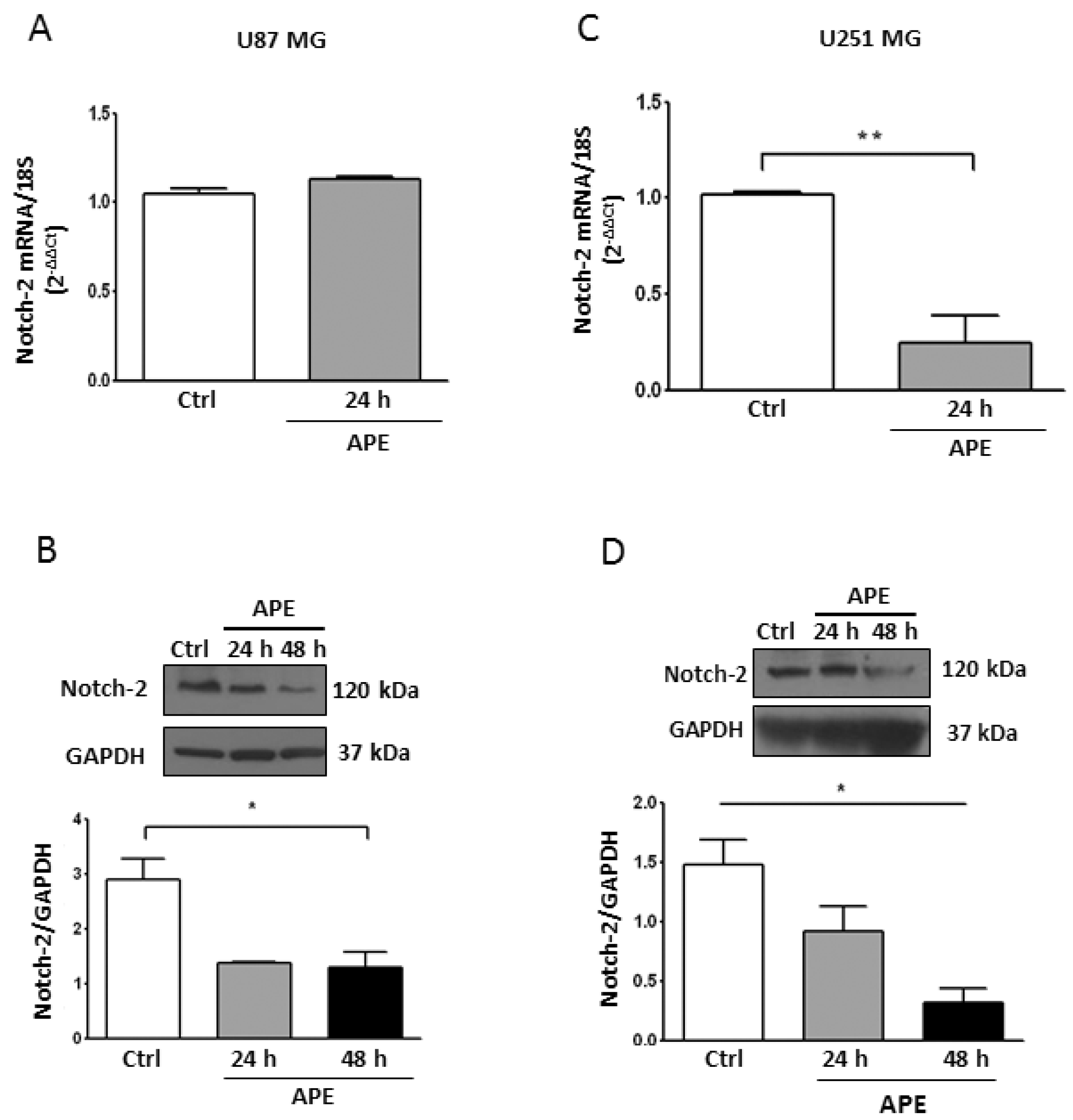
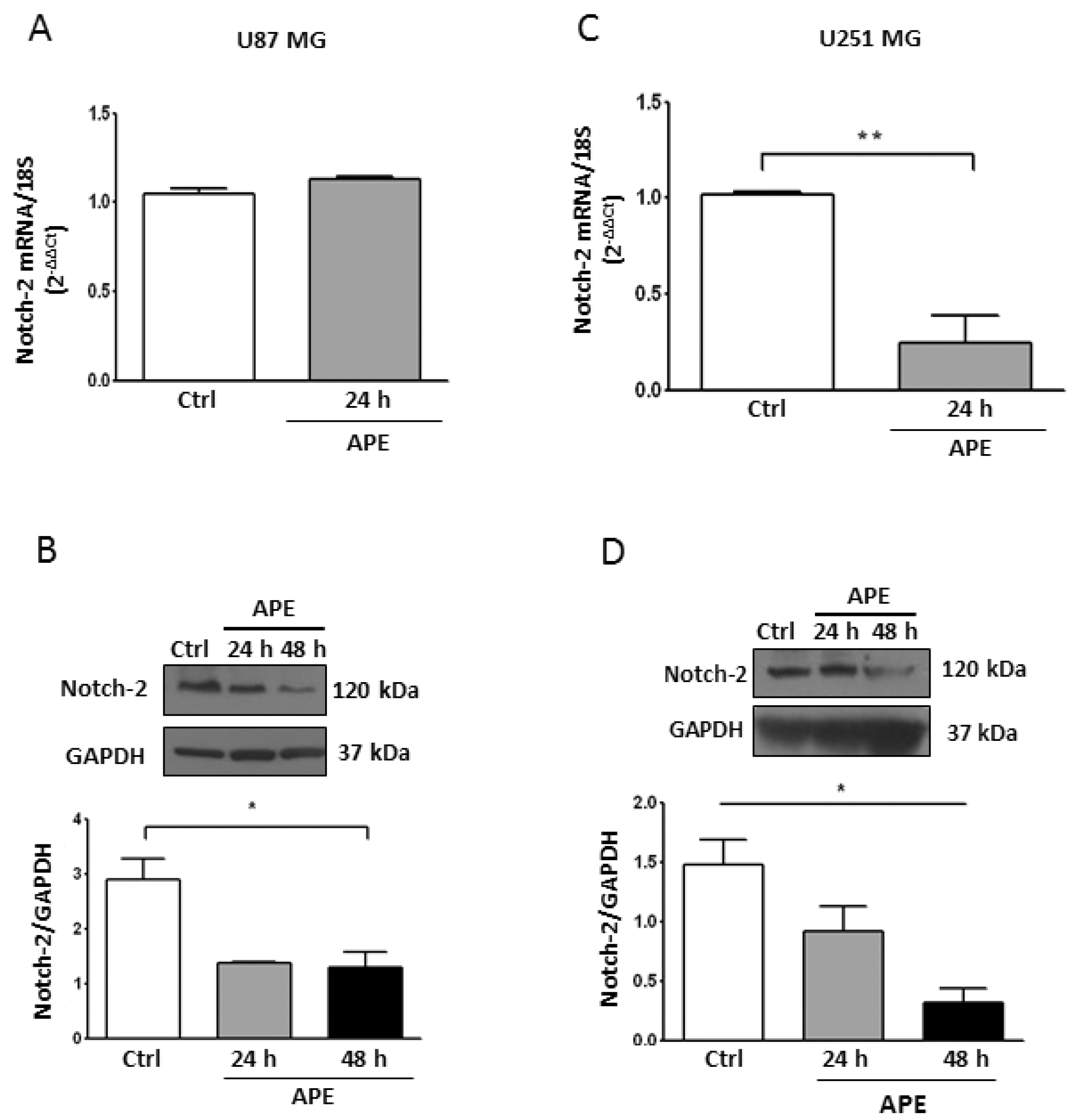
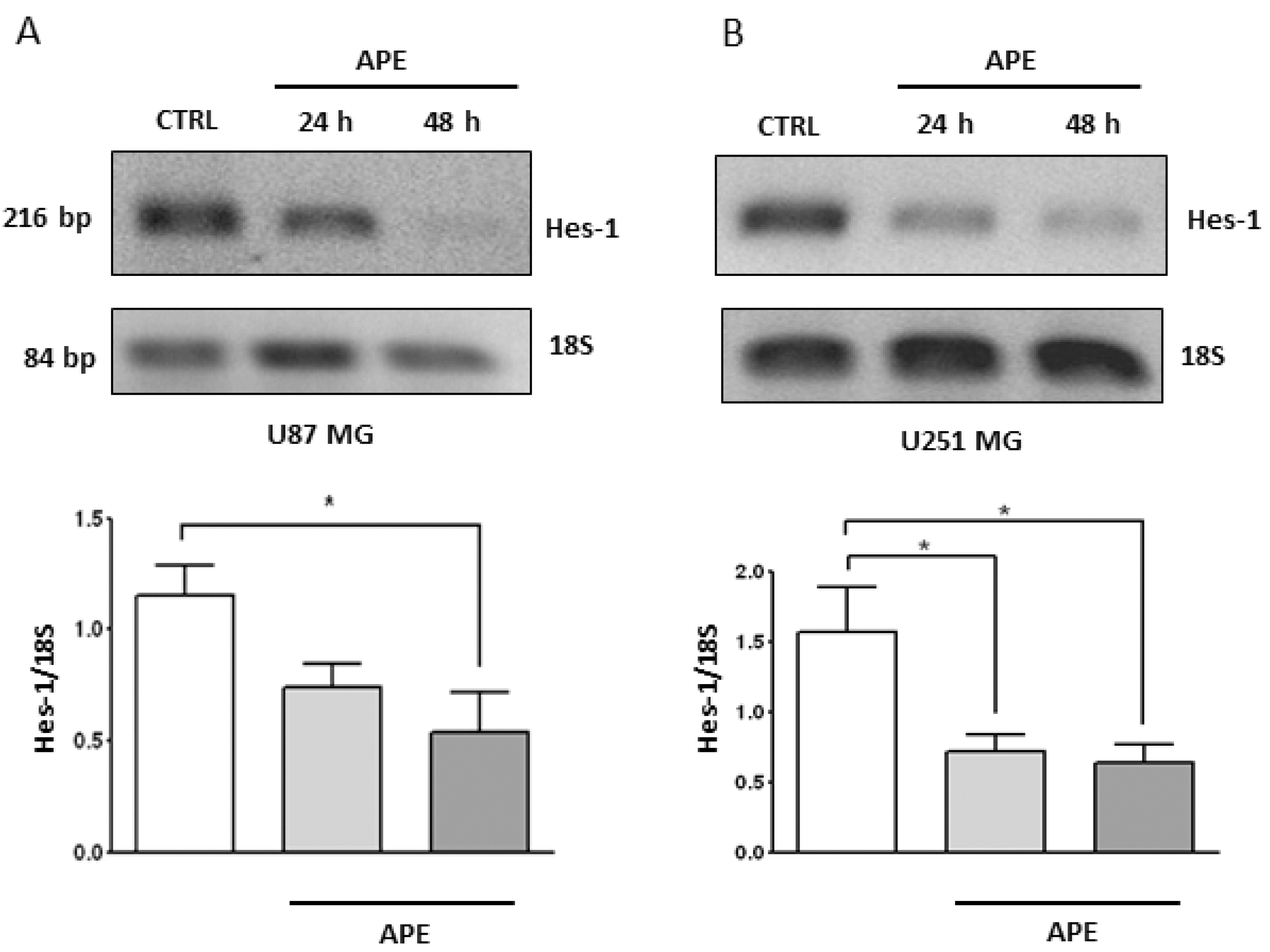
2.3. M2 Receptor Activation Modulates Notch-2 and Hes-1 Expression
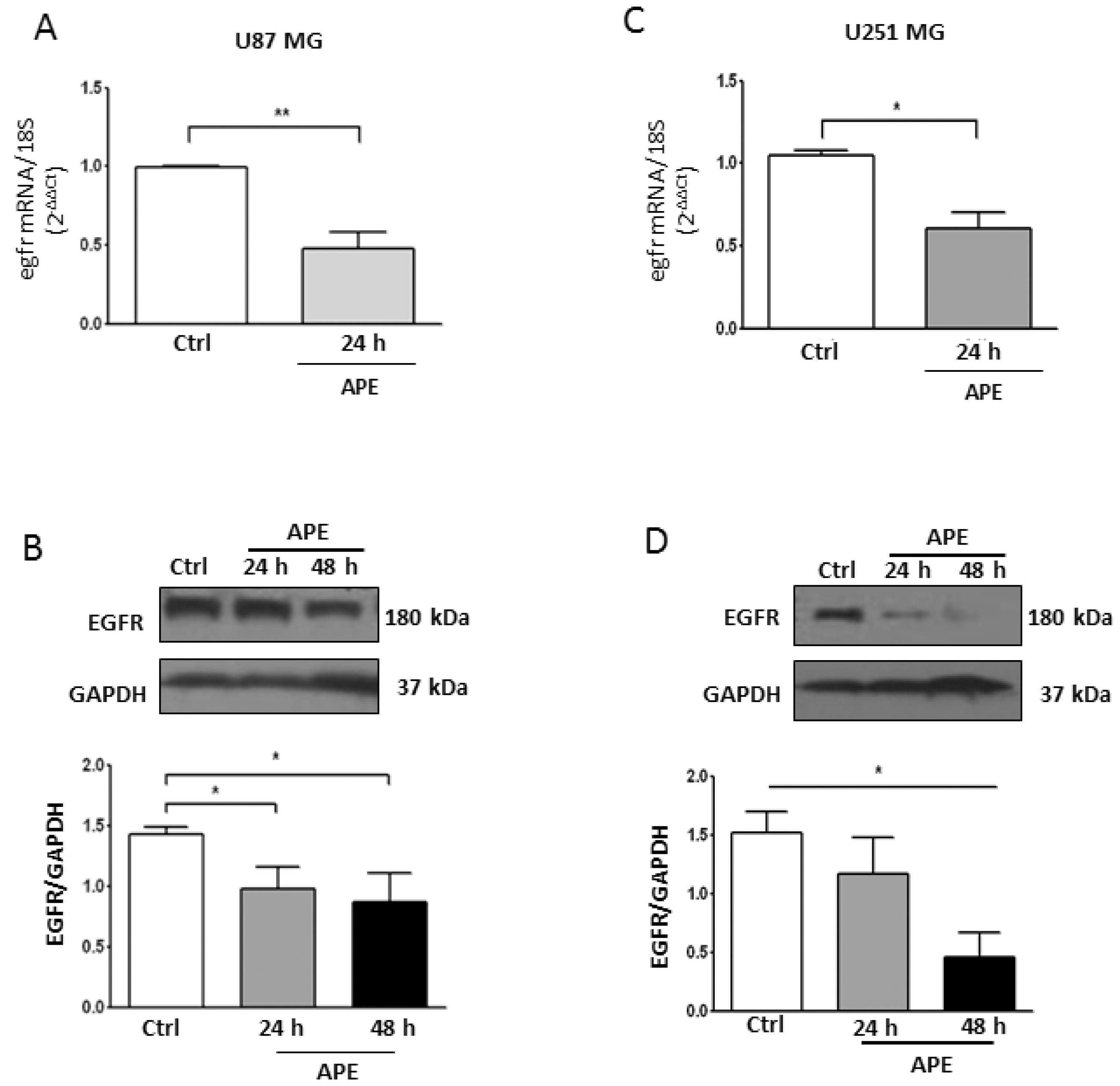
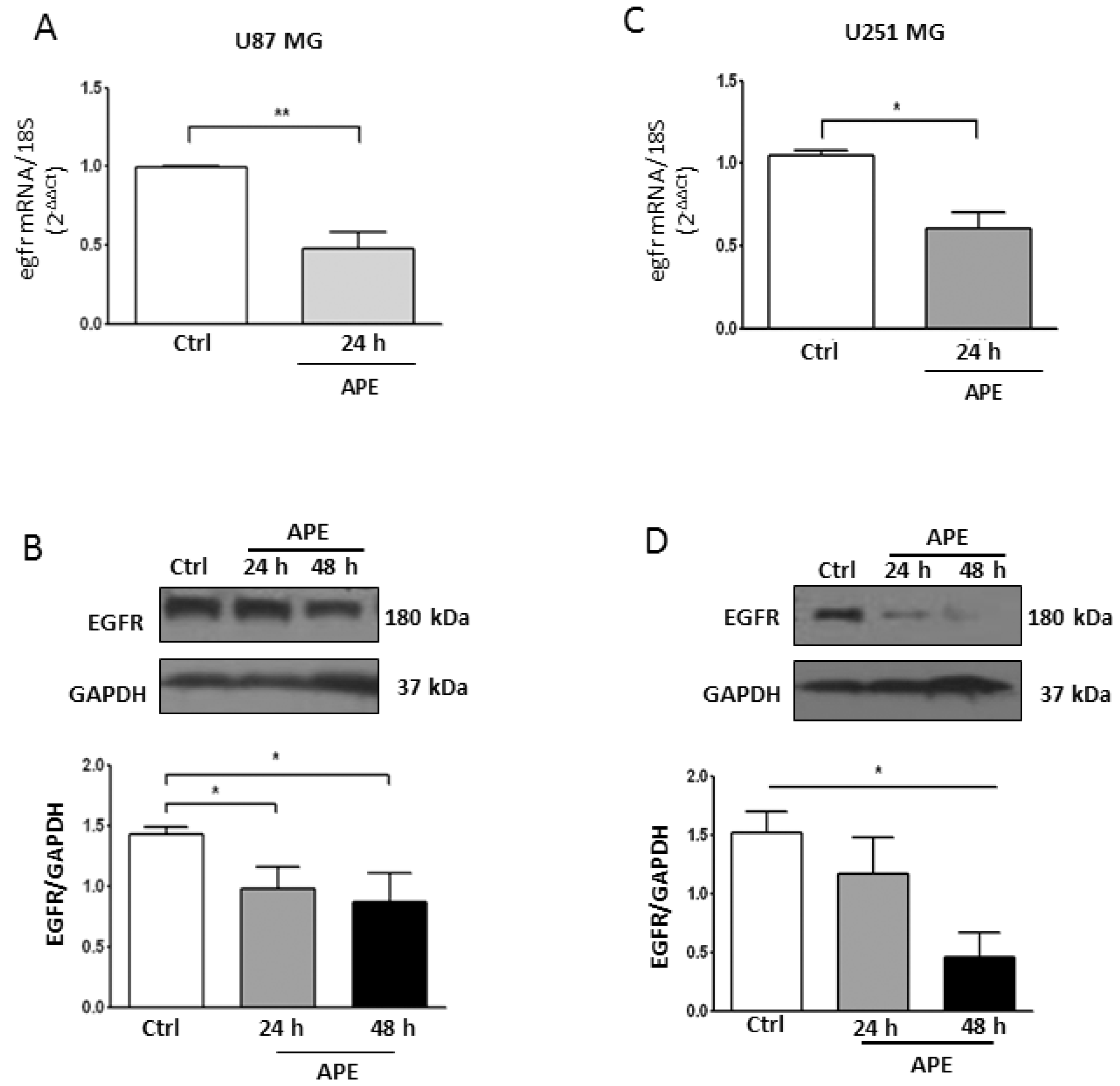
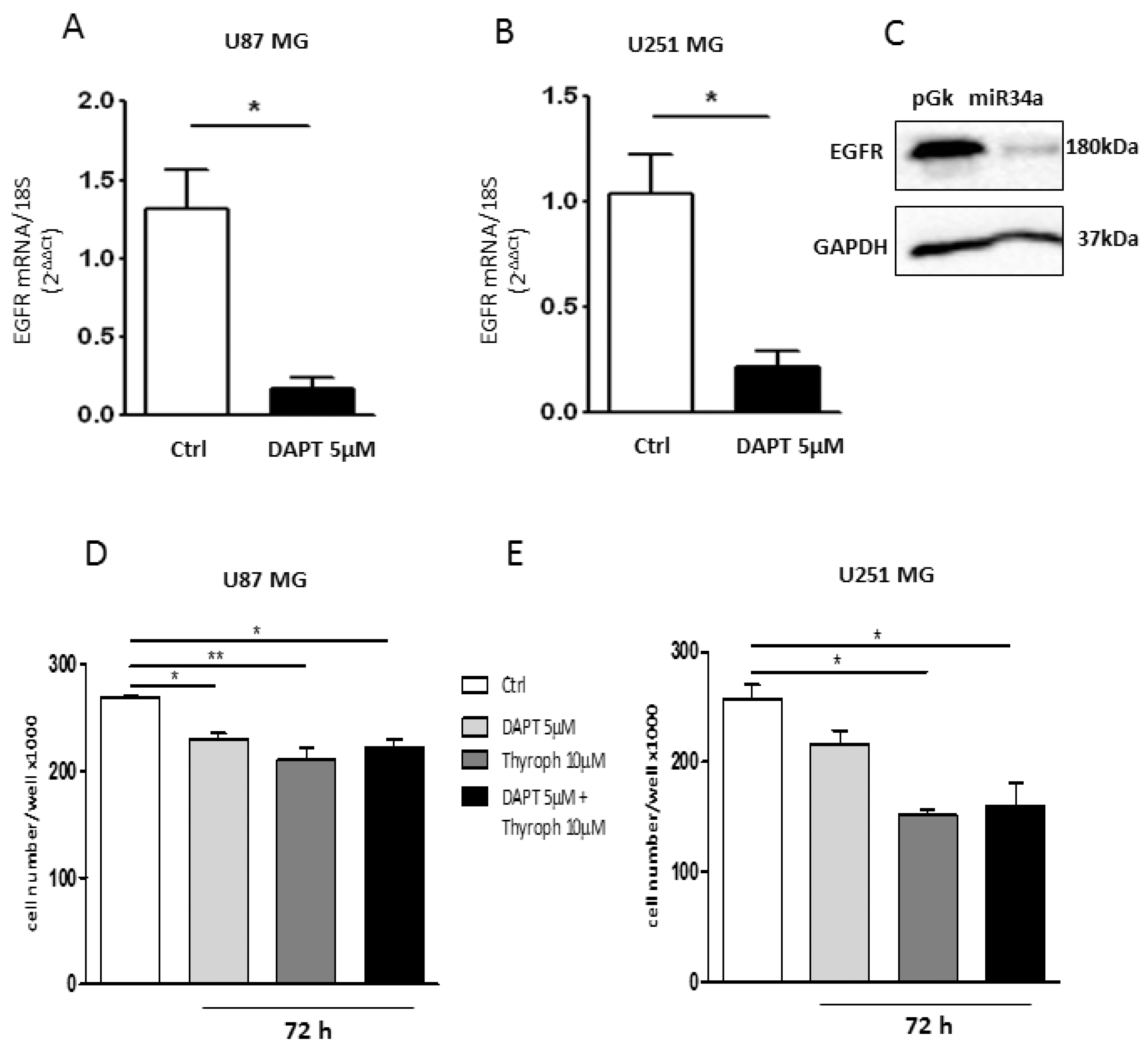
2.4. M2 Agonist Treatment Negatively Modulates EGFR Expression
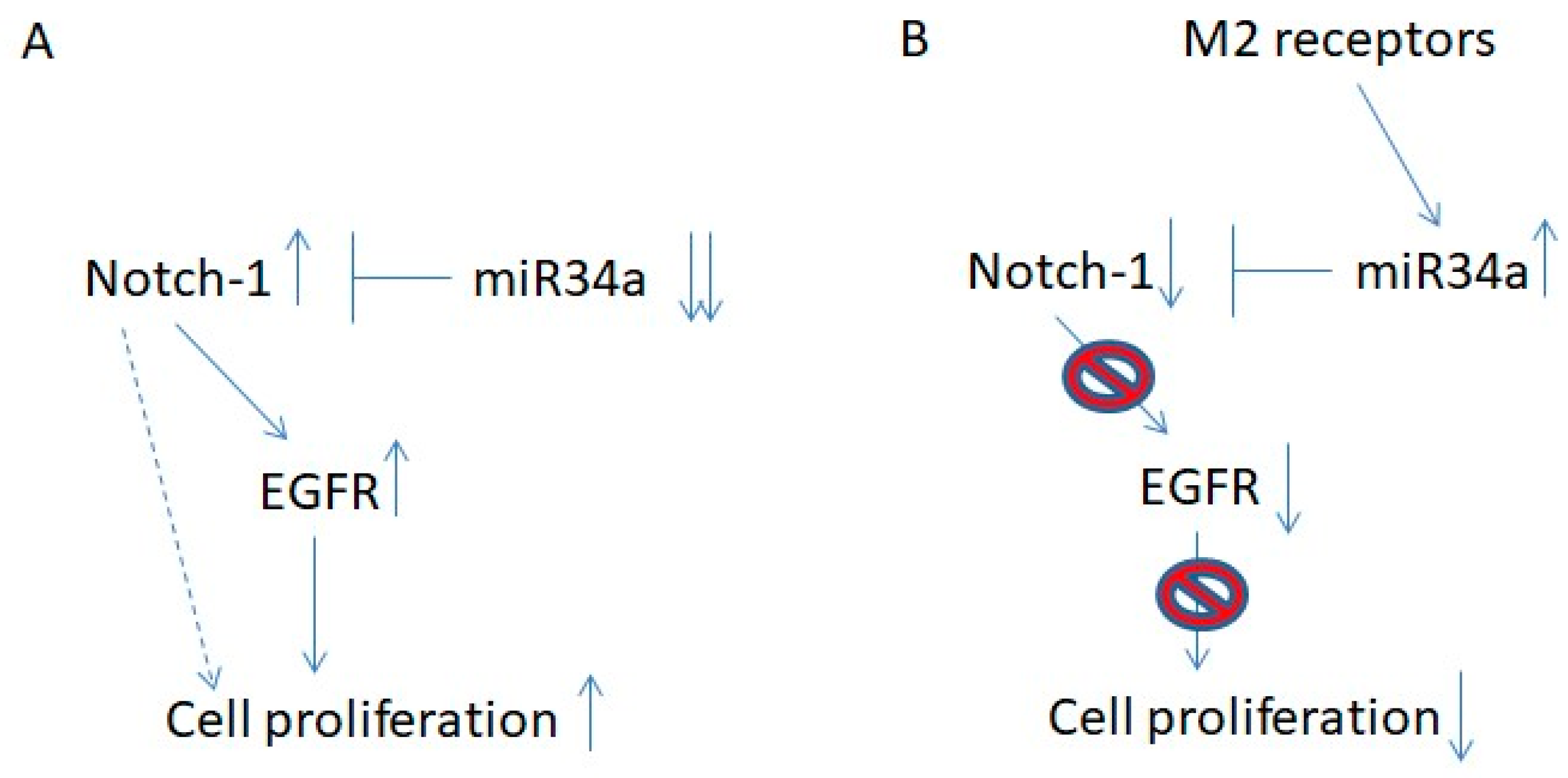
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Pharmacological Treatments
4.3. Cell Viability
4.4. Western Immunoblot
4.5. RNA Extraction and Semi-Quantitative RT-PCR Analysis
4.6. Real Time PCR Analysis
4.7. Northern Blot Analysis
4.8. Expression Vectors and Transfections
4.9. Statistical Analysis
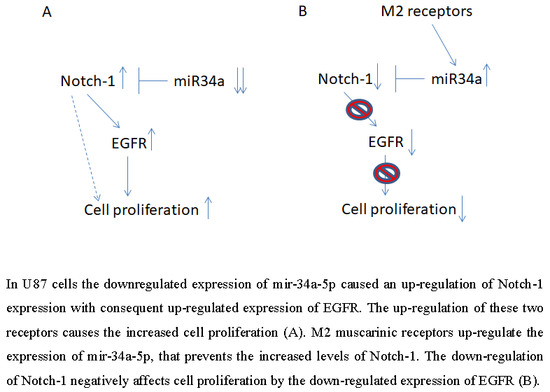
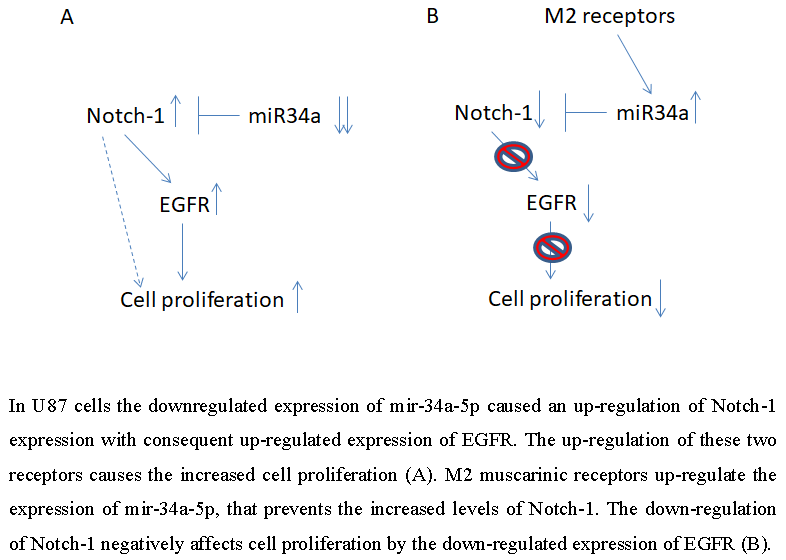
5. Conclusions
Author Contribution
Acknowledgments
Conflicts of Interest
Appendix A
References
- Kleihues, P.; Louis, D.N.; Scheithauer, B.W.; Rorke, L.B.; Reifenberger, G.; Burger, P.C.; Cavenee, W.K. The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Lebrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wu, H.; Xu, H.; Xiong, H.; Chu, Q.; Yu, S.; Wu, G.S.; Wu, K. Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 2015, 369, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Jarriault, S.; Brou, C.; Logeat, F.; Schroeter, E.H.; Kopan, R.; Israel, A. Signalling downstream of activated mammalian Notch. Nature 1995, 377, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Yahyanejad, S.; King, H.; Iglesias, V.S.; Granton, P.V.; Barbeau, L.M.O.; van Hoof, S.J.; Groot, A.J.; Habets, R.; Prickaerts, J.; Chalmers, A.J.; et al. NOTCH blockade combined with radiation therapy and temozolomide prolongs survival of orthotopic glioblastoma. Oncotarget 2016, 7, 41251–41264. [Google Scholar] [CrossRef] [PubMed]
- Purow, B.W.; Haque, R.M.; Noel, M.W.; Su, Q.; Burdick, M.J.; Lee, J.; Sundaresan, T.; Pastorino, S.; Park, J.K.; Mikolaenko, I.; et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005, 65, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Cao, Y.; Yue, Z.; Liu, J. Gamma-secretase inhibitor DAPT suppresses glioblastoma growth via uncoupling of tumor vessel density from vessel function. Clin. Exp. Med. 2013, 13, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.J.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Nishikawa, R.; Ji, X.D.; Harmon, R.C.; Lazar, C.S.; Gill, G.N.; Cavenee, W.K.; Huang, H.J. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA 1994, 91, 7727–7731. [Google Scholar] [CrossRef] [PubMed]
- Purow, B.W.; Sundaresan, T.K.; Burdick, M.J.; Kefas, B.A.; Comeau, L.D.; Hawkinson, M.P.; Su, Q.; Kotliarov, Y.; Lee, J.; Zhang, W.; et al. Notch-1 regulates transcription of the epidermal growth factor receptor through p53. Carcinogenesis 2008, 29, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, V.K.; Viale, A.; Socci, N.D.; Wiedmann, M.; Hu, X.; Holland, E.C. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol. Cell 2003, 12, 889–901. [Google Scholar] [CrossRef]
- Wess, J. Molecular biology of muscarinic acetylcholine receptors. Crit. Rev. Neurobiol. 1996, 10, 69–99. [Google Scholar] [CrossRef] [PubMed]
- Haga, T. Molecular properties of muscarinic acetylcholine receptors. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 226–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhi, X.; Zhang, Q.; Wei, S.; Li, Z.; Zhou, J.; Jiang, J.; Zhu, Y.; Yang, L.; Xu, H.; et al. Muscarinic receptor M3 mediates cell proliferation induced by acetylcholine and contributes to apoptosis in gastric cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 2105–2117. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Shang, C.; Zhao, J.; Han, Y.; Liu, J.; Chen, K.; Shi, W. Activation of M3 muscarinic receptor by acetylcholine promotes non-small cell lung cancer cell proliferation and invasion via EGFR/PI3K/AKT pathway. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 4091–4100. [Google Scholar] [CrossRef] [PubMed]
- Patanè, S. M3 muscarinic acetylcholine receptor in cardiology and oncology. Int. J. Cardiol. 2014, 177, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.; Fabbiano, C.; Di Bari, M.; Ponti, D.; Calogero, A.; Tata, A.M. M2 muscarinic receptors inhibit cell proliferation in human glioblastoma cell lines. Life Sci. 2012, 91, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.; Fabbiano, C.; Di Bari, M.; Conte, C.; Castigli, E.; Sciaccaluga, M.; Ponti, D.; Ruggieri, P.; Raco, A.; Ricordy, R.; et al. M2 receptor activation inhibits cell cycle progression and survival in human glioblastoma cells. J. Cell. Mol. Med. 2013, 17, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Cristofaro, I.; Di Bari, M.; Zasso, J.; Conti, L.; Tata, A.M. The activation of M2 muscarinic receptor inhibits cell growth and survival in human glioblastoma cancer stem cells. Int. Immunopharmacol. 2015, 29, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.; Tombolillo, V.; Conte, C.; Castigli, E.; Sciaccaluga, M.; Iorio, E.; Carpinelli, G.; Ricordy, R.; Fiore, M.; Degrassi, F.; et al. Cytotoxic and genotoxic effects mediated by M2 muscarinic receptor activation in human glioblastoma cells. Neurochem. Int. 2015, 90, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2007, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guessous, F.; Zhang, Y.; Dipierro, C.; Kefas, B.; Johnson, E.; Marcinkiewicz, L.; Jiang, J.; Yang, Y.; Schmittgen, T.D.; et al. MicroRNA- 34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009, 69, 7569–7576. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Thomas, M.P.; Altschuler, G.; Navarro, F.; O’Day, E.; Li, X.L.; Concepcion, C.; Han, Y.-C.; Thiery, J.; Rajani, D.K.; et al. Capture of microRNA-bound mRNAs identifies the tumor suppressor miR-34a as a regulator of growth factor signaling. PLoS Genet. 2011, 7, e1002363. [Google Scholar] [CrossRef] [PubMed]
- Pang, R.T.K.; Leung, C.O.N.; Ye, T.-M.; Liu, W.; Chiu, P.C.N.; Lam, K.K.W.; Lee, K.-F.; Yeung, W.S.B. MicroRNA-34a suppresses invasion through downregulation of Notch1 and Jagged1 in cervical carcinoma and choriocarcinoma cells. Carcinogenesis 2010, 31, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ji, M.; Zhong, S.; Zha, Q.; Xu, J.; Zhao, J.; Tang, J. MicroRNA-34a modulates chemosensitivity of breast cancer cells to adriamycin by targeting Notch1. Arch. Med. Res. 2012, 43, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, W.; Kong, F.; Xiao, X.; Kuang, H.; Zhu, Y. microRNA-34a overexpression inhibits cell migration and invasion via regulating SIRT1 in hepatocellular carcinoma. Oncol. Lett. 2017, 14, 6950–6954. [Google Scholar] [CrossRef] [PubMed]
- Guessous, F.; Zhang, Y.; Kofman, A.; Catania, A.; Li, Y.; Schiff, D.; Purow, B.; Abounader, R. microRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle Georget. Tex. 2010, 9, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Boulay, J.-L.; Miserez, A.R.; Zweifel, C.; Sivasankaran, B.; Kana, V.; Ghaffari, A.; Luyken, C.; Sabel, M.; Zerrouqi, A.; Wasner, M.; et al. Loss of NOTCH2 positively predicts survival in subgroups of human glial brain tumors. PLoS ONE 2007, 2, e576. [Google Scholar] [CrossRef] [PubMed]
- Nandagopal, N.; Santat, L.A.; LeBon, L.; Sprinzak, D.; Bronner, M.E.; Elowitz, M.B. Dynamic Ligand Discrimination in the Notch Signaling Pathway. Cell 2018, 172, 869.e19–880.e19. [Google Scholar] [CrossRef] [PubMed]
- Binesh, A.; Devaraj, S.N.; Devaraj, H. Inhibition of nuclear translocation of notch intracellular domain (NICD) by diosgenin prevented atherosclerotic. Biochimie 2018. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Anastasiadou, E.; Esteller, M.; He, L.; Slack, F.J. The Inescapable Influence of Noncoding RNAs in Cancer. Cancer Res. 2015, 75, 5206–5210. [Google Scholar] [CrossRef] [PubMed]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 1985, 313, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Cristofaro, I.; Spinello, Z.; Matera, C.; Fiore, M.; Conti, L.; De Amici, M.; Dallanoce, C.; Tata, A.M. Activation of M2 muscarinic acetylcholine receptors by a hybrid agonist enhances cytotoxic effects in GB7 glioblastoma cancer stem cells. Neurochem. Int. 2018, 118, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Camp, S.; De Jaco, A.; Zhang, L.; Marquez, M.; De la Torre, B.; Taylor, P. Acetylcholinesterase expression in muscle is specifically controlled by a promoter-selective enhancesome in the first intron. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 2459–2470. [Google Scholar] [CrossRef] [PubMed]
- Pacini, L.; De Falco, E.; Di Bari, M.; Coccia, A.; Siciliano, C.; Ponti, D.; Pastore, A.L.; Petrozza, V.; Carbone, A.; Tata, A.M.; et al. M2muscarinic receptors inhibit cell proliferation and migration in urothelial bladder cancer cells. Cancer Biol. Ther. 2014, 15, 1489–1498. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Bari, M.; Bevilacqua, V.; De Jaco, A.; Laneve, P.; Piovesana, R.; Trobiani, L.; Talora, C.; Caffarelli, E.; Tata, A.M. Mir-34a-5p Mediates Cross-Talk between M2 Muscarinic Receptors and Notch-1/EGFR Pathways in U87MG Glioblastoma Cells: Implication in Cell Proliferation. Int. J. Mol. Sci. 2018, 19, 1631. https://doi.org/10.3390/ijms19061631
Di Bari M, Bevilacqua V, De Jaco A, Laneve P, Piovesana R, Trobiani L, Talora C, Caffarelli E, Tata AM. Mir-34a-5p Mediates Cross-Talk between M2 Muscarinic Receptors and Notch-1/EGFR Pathways in U87MG Glioblastoma Cells: Implication in Cell Proliferation. International Journal of Molecular Sciences. 2018; 19(6):1631. https://doi.org/10.3390/ijms19061631
Chicago/Turabian StyleDi Bari, Maria, Valeria Bevilacqua, Antonella De Jaco, Pietro Laneve, Roberta Piovesana, Laura Trobiani, Claudio Talora, Elisa Caffarelli, and Ada Maria Tata. 2018. "Mir-34a-5p Mediates Cross-Talk between M2 Muscarinic Receptors and Notch-1/EGFR Pathways in U87MG Glioblastoma Cells: Implication in Cell Proliferation" International Journal of Molecular Sciences 19, no. 6: 1631. https://doi.org/10.3390/ijms19061631
APA StyleDi Bari, M., Bevilacqua, V., De Jaco, A., Laneve, P., Piovesana, R., Trobiani, L., Talora, C., Caffarelli, E., & Tata, A. M. (2018). Mir-34a-5p Mediates Cross-Talk between M2 Muscarinic Receptors and Notch-1/EGFR Pathways in U87MG Glioblastoma Cells: Implication in Cell Proliferation. International Journal of Molecular Sciences, 19(6), 1631. https://doi.org/10.3390/ijms19061631