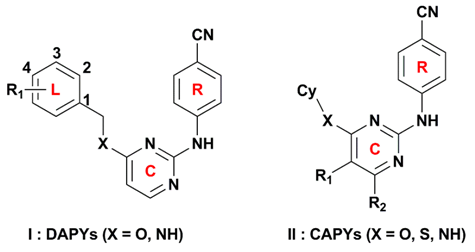
2.1. CoMFA and CoMSIA Statistical Results
The classical parameters of CoMFA and CoMSIA models, including
,
ONC,
,
,
SEE, and
F-values, are listed in
Table 1. The other important validation parameters, such as
RMSE,
MAE,
,
,
,
k,
k’,
,
, Δ
, and
are listed in
Table 2. The results from the CoMFA model indicated that
,
,
,
MAE,
RMSE, Δ
, and
were 0.679, 0.983, 0.884, 0.124, 0.160, 0.1215 (or 0.0026), and 0.7829 (or 0.9690), respectively. These data proved that the constructed CoMFA model was reliable, and its predictive accuracy was acceptable (
> 0.5). The steric and electrostatic fields contributions were 46.30% and 53.70%, respectively, indicating that the electrostatic fields gave an important contribution.
For the CoMSIA analysis, different combinations of descriptor fields were used to construct different CoMSIA models. All possible combinations of fields were performed to determine the optimal predictive model [
12]. According to the experimental data in
Table 1, it could be found that a model consisting of steric, electrostatic, hydrophobic, hydrogen-bond donor and hydrogen-bond acceptor fields led to relatively higher
,
,
and relatively lower
SEE. Therefore, the model (S + E + H + D + A,
Table 1) was considered to be the best possible combination, which assigned satisfactory values to the parameters
,
,
,
MAE,
RMSE, Δ
, and
, i.e., 0.734, 0.985, 0.891, 0.108, 0.155, 0.1387 (or 0.0171), and 0.7389 (or 0.9763), respectively. The corresponding contributions of steric, electrostatic, hydrophobic, hydrogen-bond acceptor, and hydrogen-bond donor fields were 12.30%, 41.40%, 27.60%, 7.5%, and 11.20%, respectively. Compared to the CoMFA model, the CoMSIA model seemed to show a somewhat better predictivity. It was also found that the electrostatic fields were significant contributors in the optimal CoMSIA model. These results indicate that the constructed models are powerful to predict the activity of DAPYs and CAPYs. The field contributions revealed that the electrostatic fields play important roles in the CoMFA and CoMSIA models.
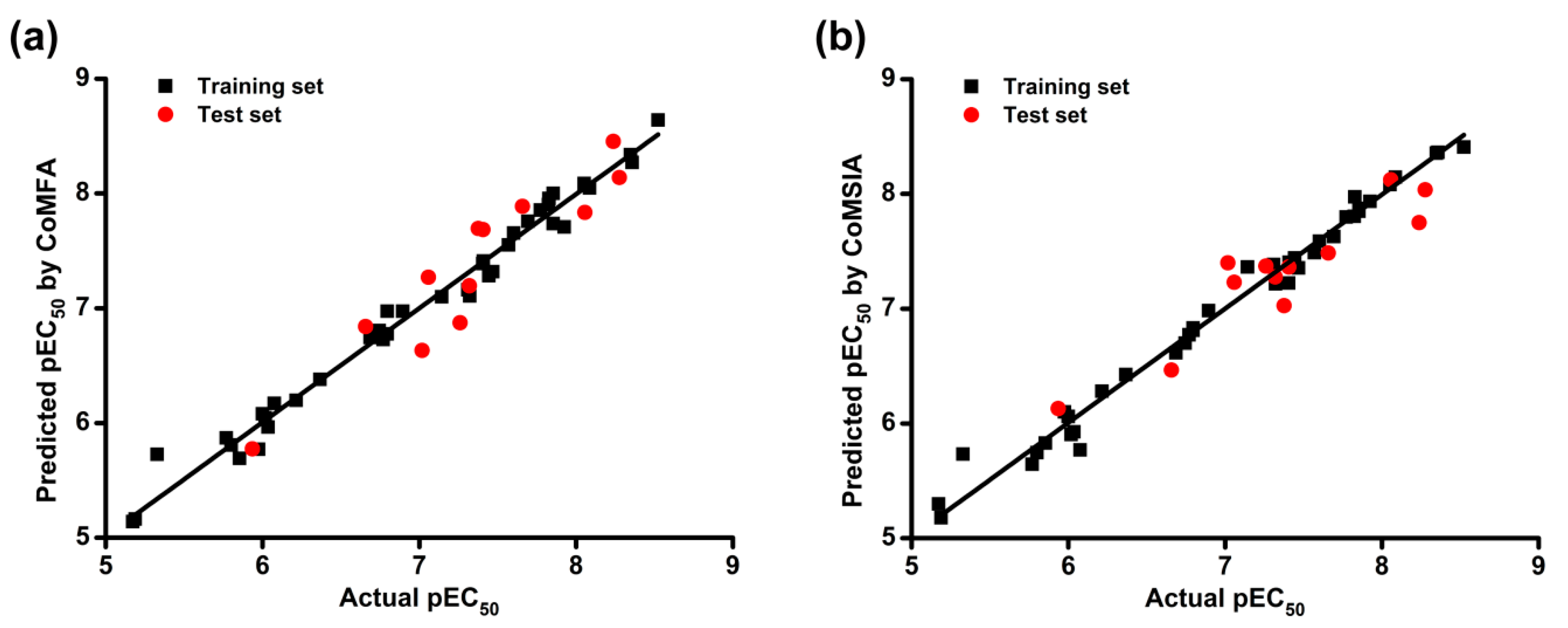
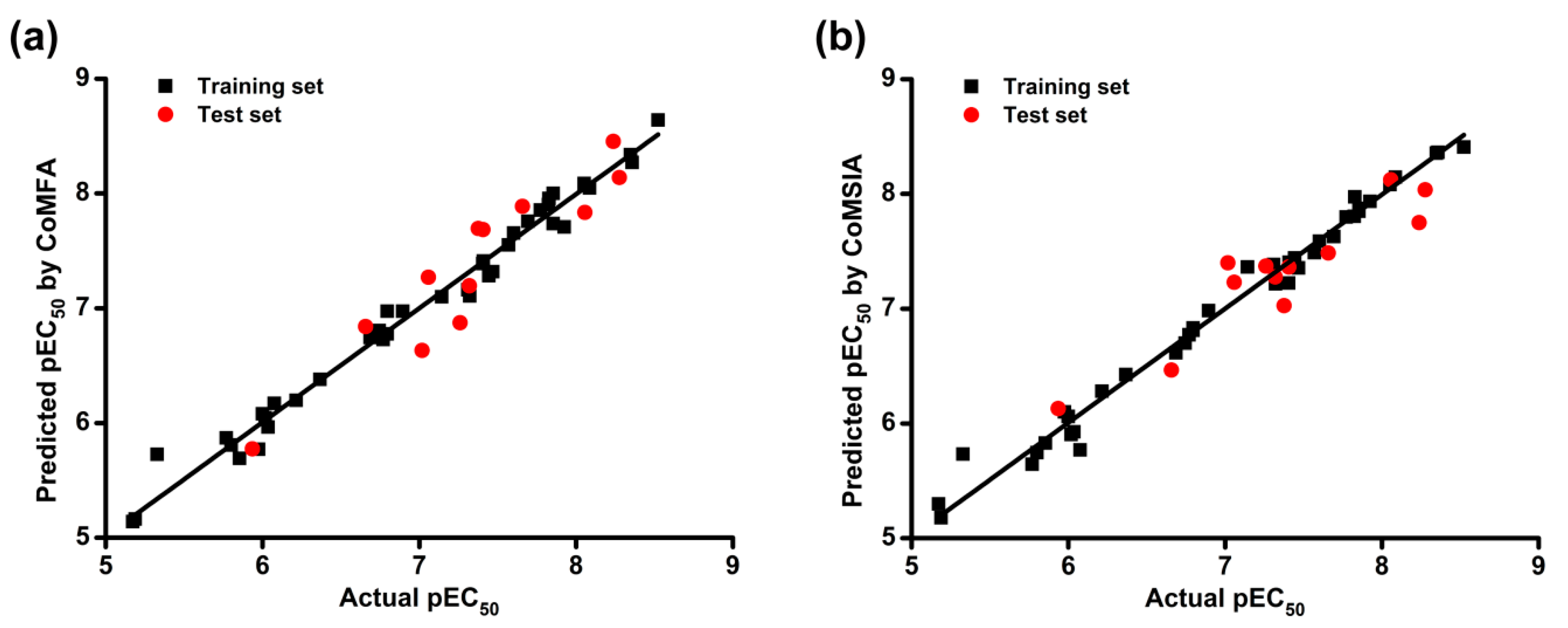
Then, we used the models to predict the activity of the training and test set compounds. The actual and predicted pEC
50 (−logEC
50) values of DAPYs and CAPYs are listed in
Table 3. The correlations between actual and predicted pEC
50 values are shown in
Figure 2. The predicted pEC
50 values were close to the experimental data, and most of the points were located on or near the trend line, which indicated the predictivity and reliability of both models. The above results indicate that constructed CoMFA and CoMSIA models are reasonable and have the ability to predict the anti-HIV-1 activity of the training and test compounds of DAPYs and CAPYs.
2.2. CoMFA and CoMSIA Contour Maps
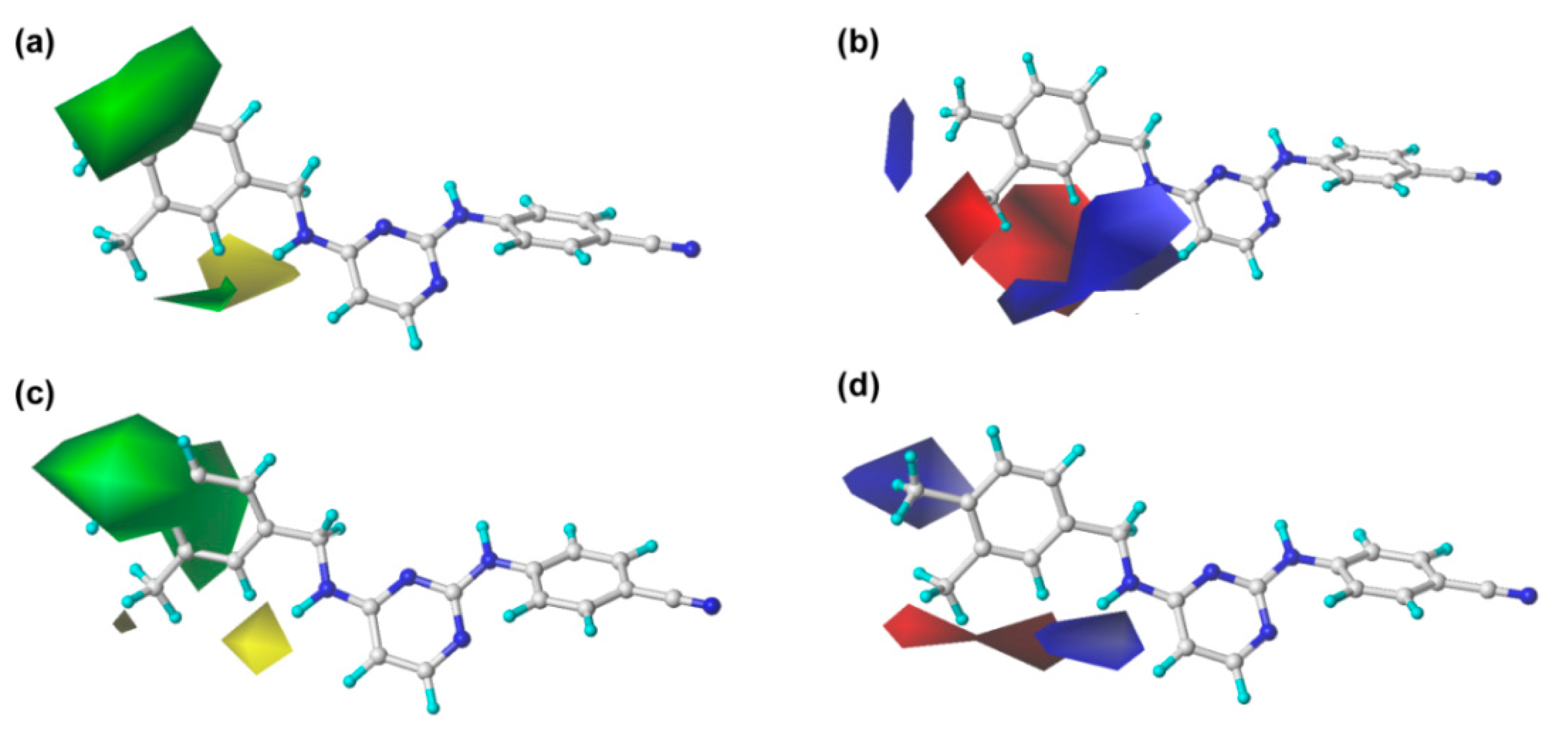
To visualize the different field effects in three-dimensional spaces where the modifications could increase the activity of the target compounds, contour maps were generated subsequently in the CoMFA and CoMSIA models. Compound 43 with the highest activity was used as a reference structure to illustrate all the contour maps.
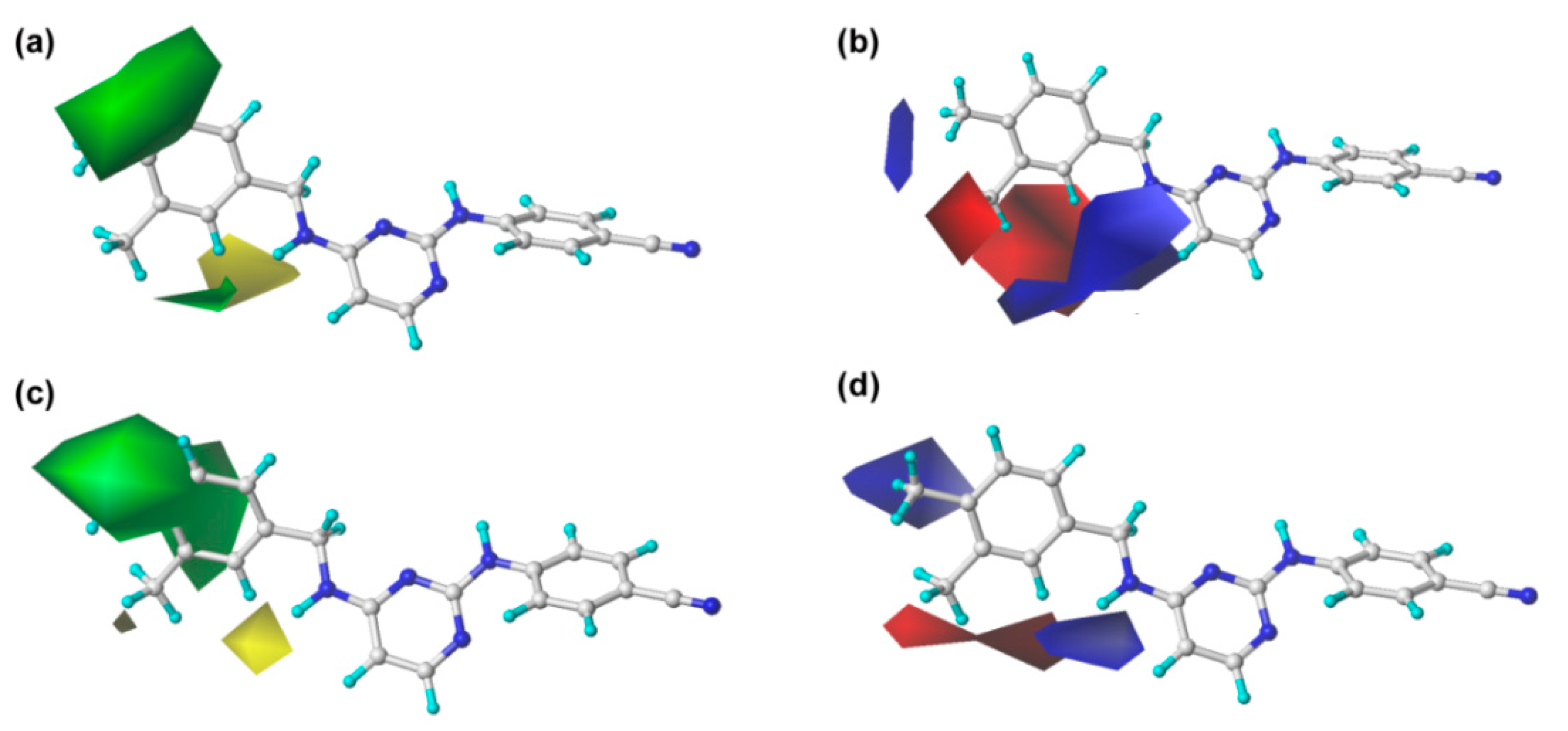
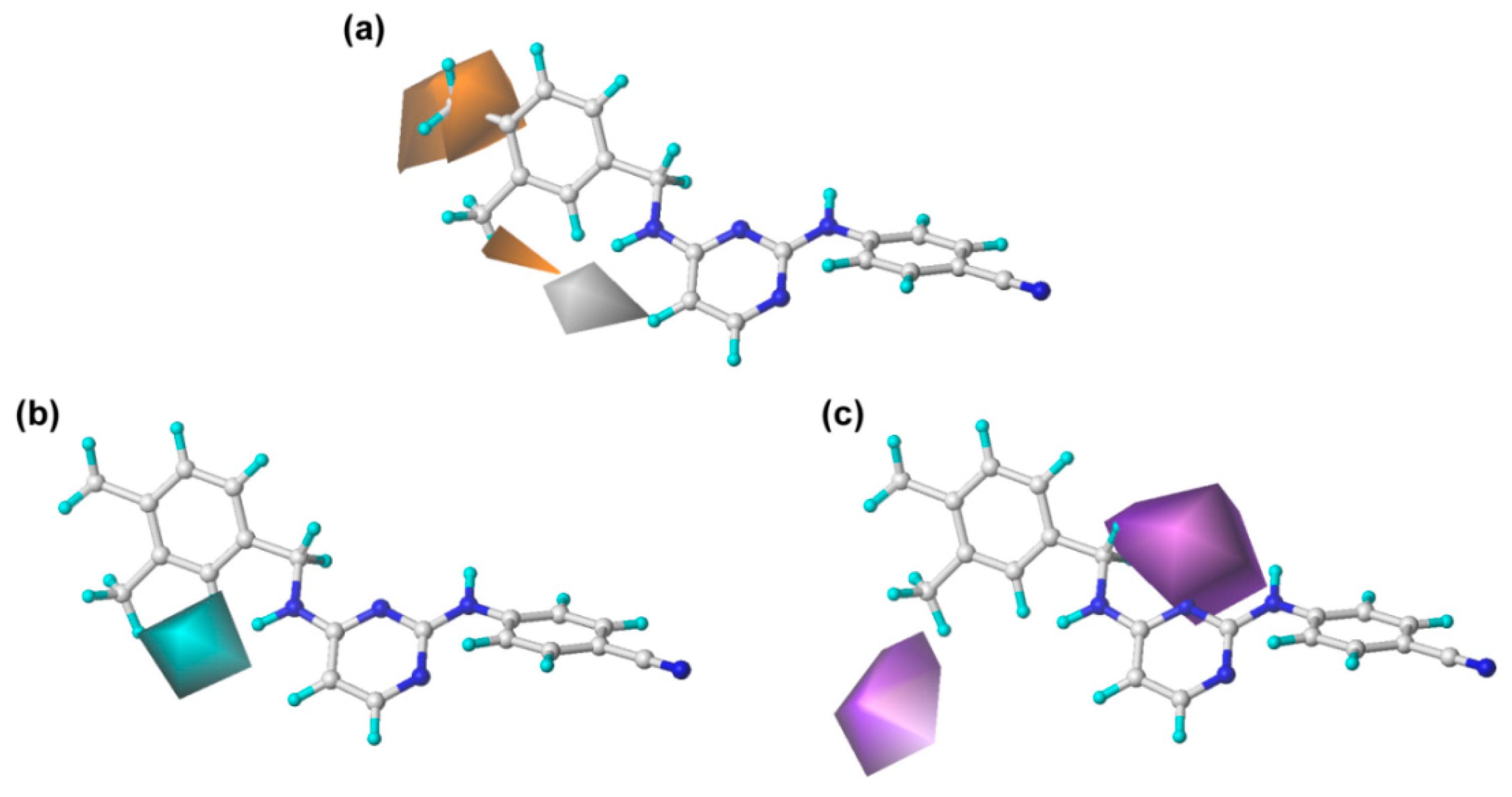
The steric and electrostatic contour maps of CoMFA and CoMSIA are shown in
Figure 3. In the steric fields, the green contours indicate sterically favorable bulky substituents, whereas the yellow contours indicate where the substituents are sterically unfavorable [
13]. It should be noted that the steric contour maps of CoMSIA are similar to those of the CoMFA model, which proves the consistency of the results. As shown in
Figure 3, a large green contour surrounding the C
3 (or C
5) and C
4 positions of the left phenyl ring indicates that bulky groups here are beneficial for enhancing the activity. This finding might explain why the activity of compound
43 is significantly higher than that of other compounds. However, the structure–activity relationships of several compounds with single substitution at the C
3 or C
4 position were not coincident with this finding, as seen, for example, for compounds
6 (4-Br) <
9 (4-F). This might have been caused by other properties of the substitutions or the effects of other fields and remains to be studied. On the other hand, there was a small yellow contour surrounding the C
2-position of the left phenyl ring, which suggested that bulky substituents at this site might be unfavorable for the activity, as observed for the following compounds in this order:
4 (2-Br) <
1 (2-Cl) <
7 (2-F) and
16 (2-CF
3) <
10 (2-CH
3) <
24 (2-H).
For the electrostatic maps, the blue contours denote regions where positively charged substituents will improve the inhibitory activity, whereas the red regions show negatively charged substituents are helpful for enhancing the activity. As shown in
Figure 3, a large blue contour around the C
4-position of the left phenyl ring reveals that the positively charged substituents at this position are favorable for increasing the inhibitory activity. This finding can be supported by examples as follows: compound
36 with a methyl substituent at the C
4-position showed higher inhibition activity compared to compounds
30 (4-Cl),
33 (4-Br), and
35 (4-F); the activity order was
36 (4-CH
3) >
33 (4-Br) >
30 (4-Cl) >
35 (4-F). Moreover, a big blue irregular contour near the linker between the left wing and the central pyrimidine indicates that the presence of a positively charged group in this region is beneficial to the bioactivity, which is consistent with the experimental data, showing, for example,
27 (linker = –NH) >
24 (linker = –O). Two small red contours surrounding the C
2 and C
3 positions of the left phenyl ring, respectively, indicating that the presence of negative charges in these regions will be favorable for bioactivity. For example, compounds
1 (2-Cl),
4 (2-Br), and
7 (2-F) showed higher activity than compound
10 (2-CH
3), and the order of their inhibitory activity was
7 (2-F) >
1 (2-Cl) >
4 (2-Br) >
10 (2-CH
3). This result is also reflected in the fact that the activity of compound
34, with a fluorine (negative charge) at the C
3-position, was increased significantly compared with that of compound
41 bearing a trifluoromethyl (positive charge).
As shown in
Table 1 and
Figure 4a, the hydrophobic field also plays an important role in the optimal CoMSIA model. Two large orange contours are located near the C
3 and C
4 positions of the left phenyl ring, indicating that a hydrophobic substituent in these two positions will be favorable to the inhibitory activity. For example, compound
43 with methyl groups in these two zones showed higher activity than compound
24 with hydrogens. Besides, there is a white contour close to the C
2-position of the left phenyl ring, which suggests that the presence of a hydrophilic group in this position might increase the inhibitory activity. For instance, the activity of compound
7 with a fluorine group at the C
2-position of the left phenyl ring was higher than that of compound
24 with a hydrogen at this position.
Furthermore, the hydrogen-bond donor and acceptor fields also play relatively important roles in the bioactivity of the compounds. As shown in
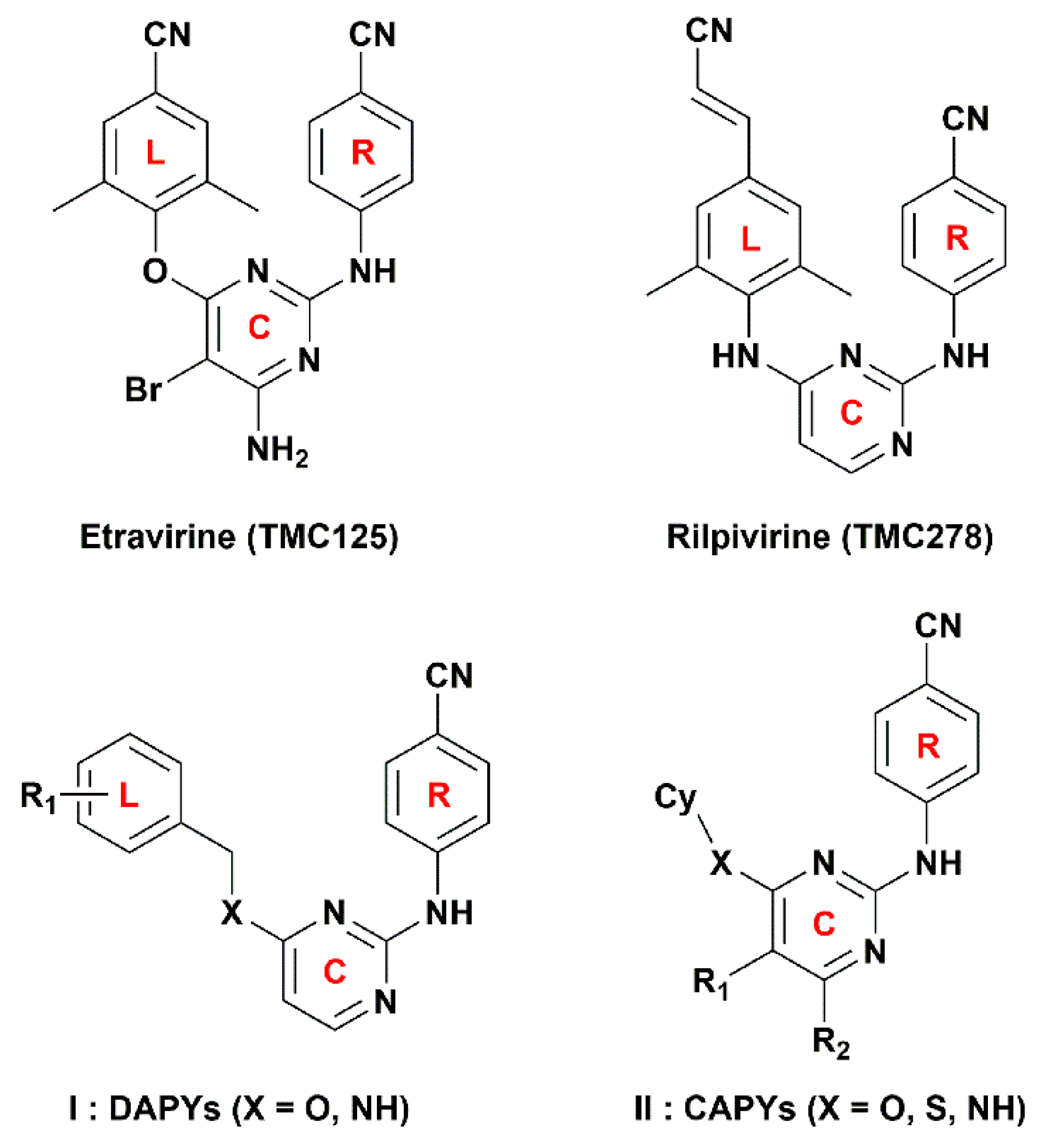
Figure 4b, c, a cyan contour surrounds the linker between the central pyrimidine and the left wing, which signifies that hydrogen-bond donor groups at this position will be beneficial to the bioactivity. This can be certified by the fact that the inhibitory activity of compound
27 (linker = –NH) was significantly higher than that of compound
24 (linker = –O). Moreover, it also can be observed that there are two violet contours around the C
3-position of the left phenyl ring and the X substituent of the linker (
Figure 1), respectively, indicating that hydrogen-bond acceptor groups in these positions are not beneficial for the biological activity. This result is supported by the biological activity of the compounds that contain an oxygen atom as X substituent, such as compounds
45,
46, and
47.
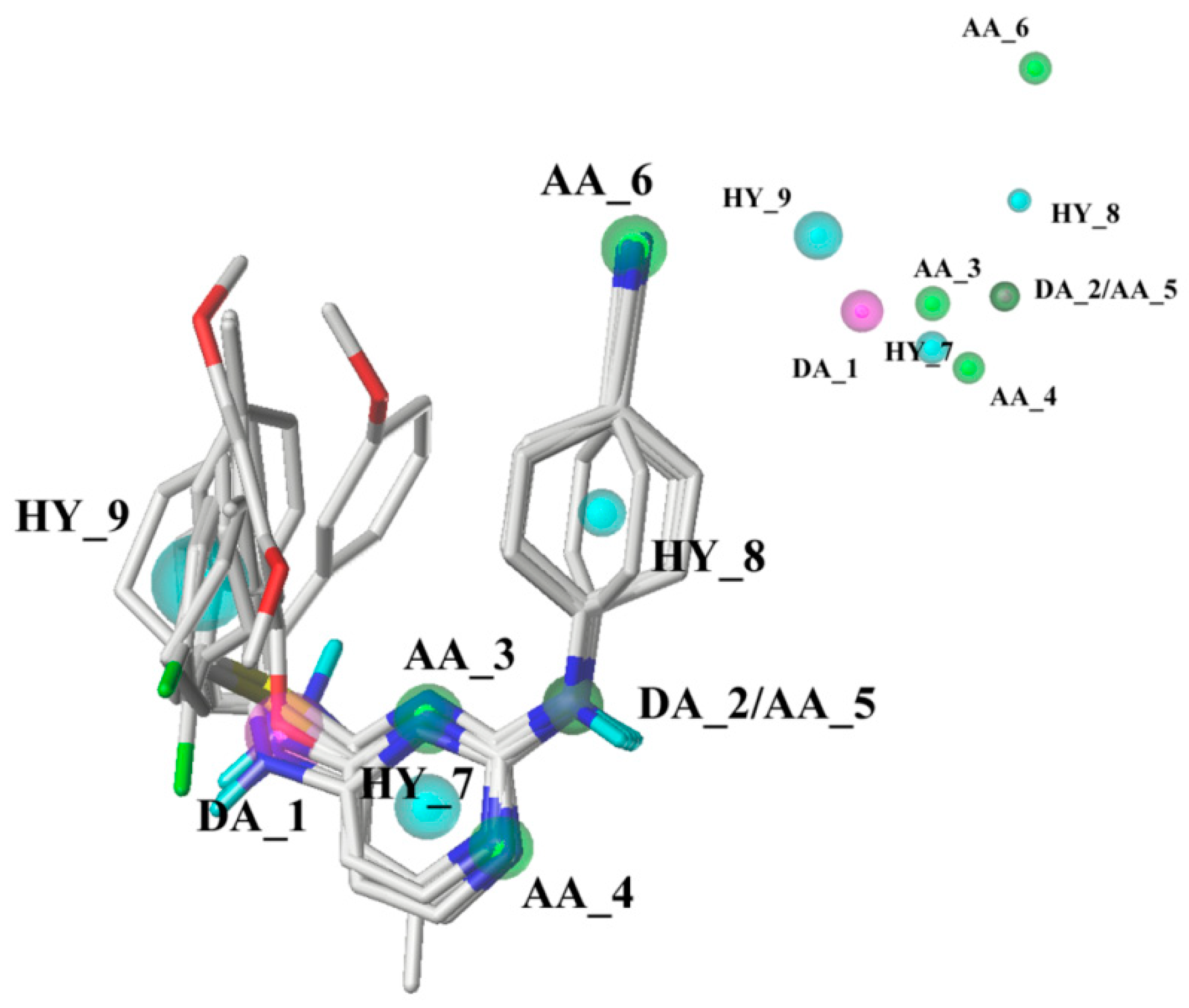
2.3. Pharmacophore Model
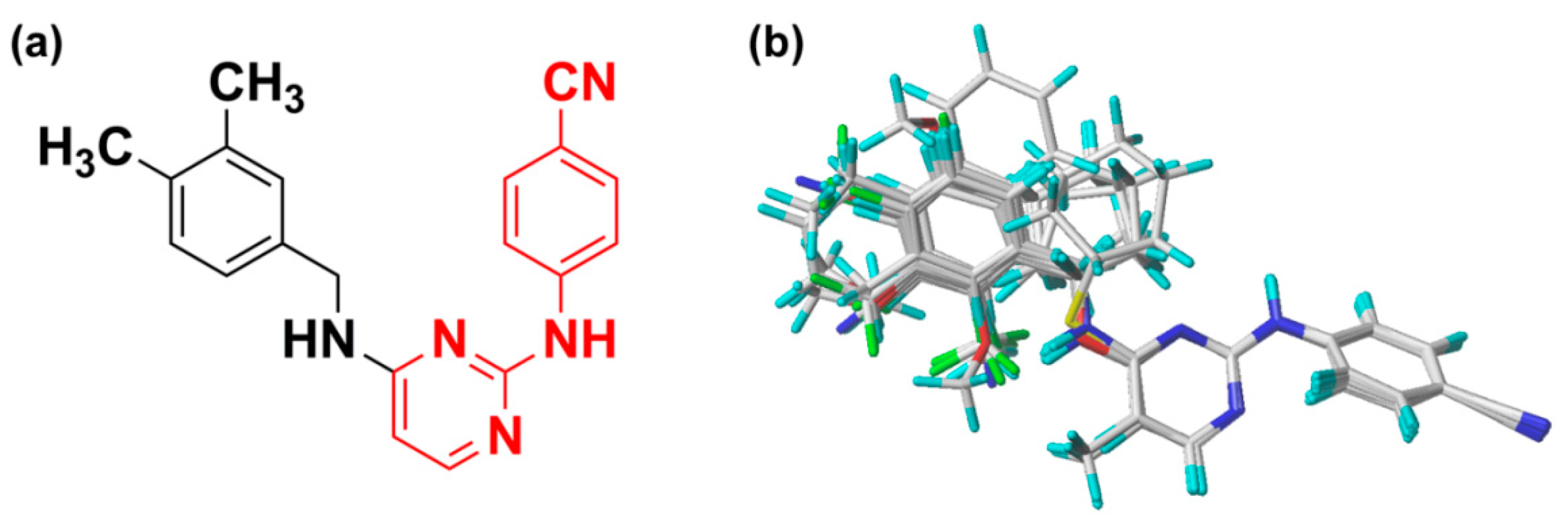
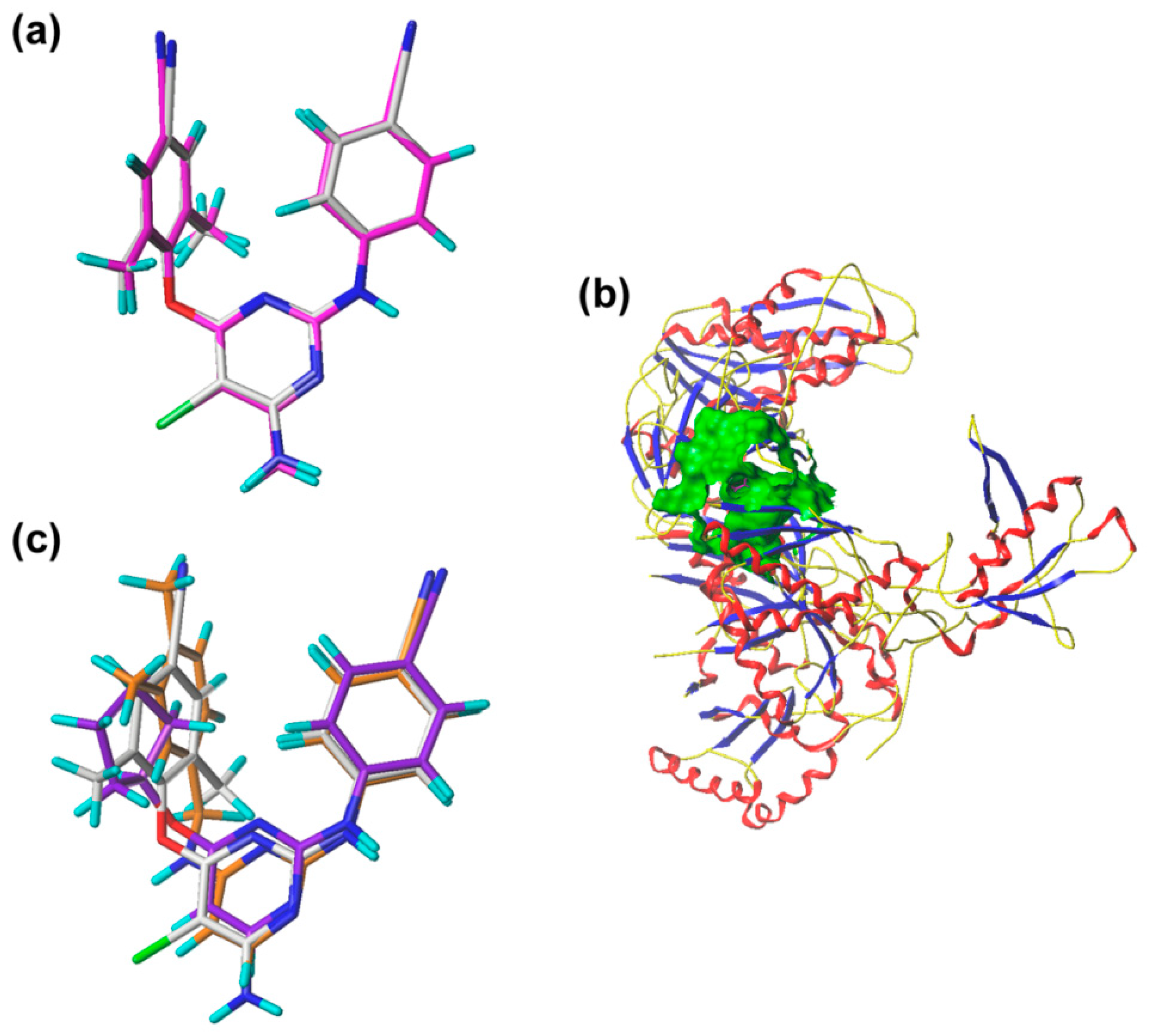
The pharmacophore model was constructed using nine compounds with diverse structures and relatively high activities as a training set. Twenty pharmacophore models were generated after Genetic Algorithm with Linear Assignment of Hypermolecular Alignment of Datasets (GALAHAD) run, each of which represented a different trade-off among the competing criteria. The lower the strain energy (SE) values, and the higher the steric overlap (SO) and pharmacophoric similarity (PhS) values, the better the model. According to the experimental results, it was found that the parameters of the best generated model were: SE = 2.982, SO = 255.80, and PhS = 123.30. The pharmacophore model with the alignment of nine compounds is shown in
Figure 5, indicating a satisfactory superimposition. As depicted in
Figure 5, the magenta, green, and cyan spheres represent hydrogen-bond donor atoms (DAs), hydrogen-bond acceptor atoms (AAs), and hydrophobes (HYs), respectively. The best model is formed by nine pharmacophore features: two hydrogen-bond DAs, four hydrogen-bond AAs, and three HY centers. One of the hydrogen-bond DAs is the nitrogen atom of an imine group, and the four hydrogen-bond AAs correspond to the nitrogen atoms of a pyrimidine ring, imine group, and nitrile group, respectively. These features reflect the importance of the DAPY/CAPY common scaffold for the inhibitory activity. Another hydrogen-bond DA is located at the linker atom, indicating that a hydrogen-bond donor groups such as –NH at this position may increase the inhibitory activity, which is in accordance with the hydrogen-bond donor fields results in the CoMSIA contour maps. The three hydrophobic centers are located at the center of the left phenyl ring, the center of the pyrimidine ring, and the center of the right phenyl ring, respectively, which suggests that a large hydrophobic structure on the left wing is favorable for the activity. These results are in agreement with the actual activities and the steric fields of the 3D-QSAR contour maps.
2.4. Molecular Docking Analysis
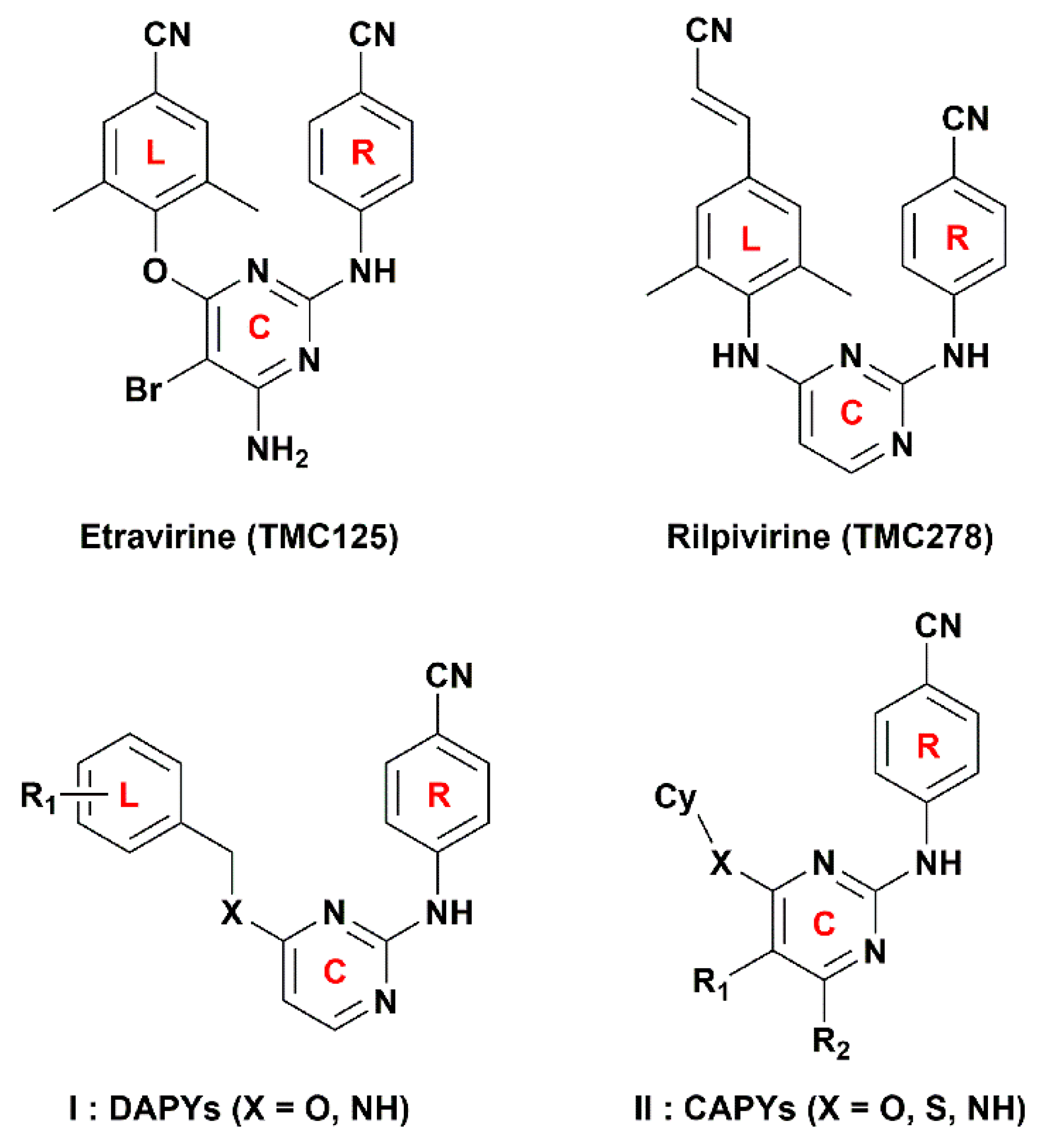
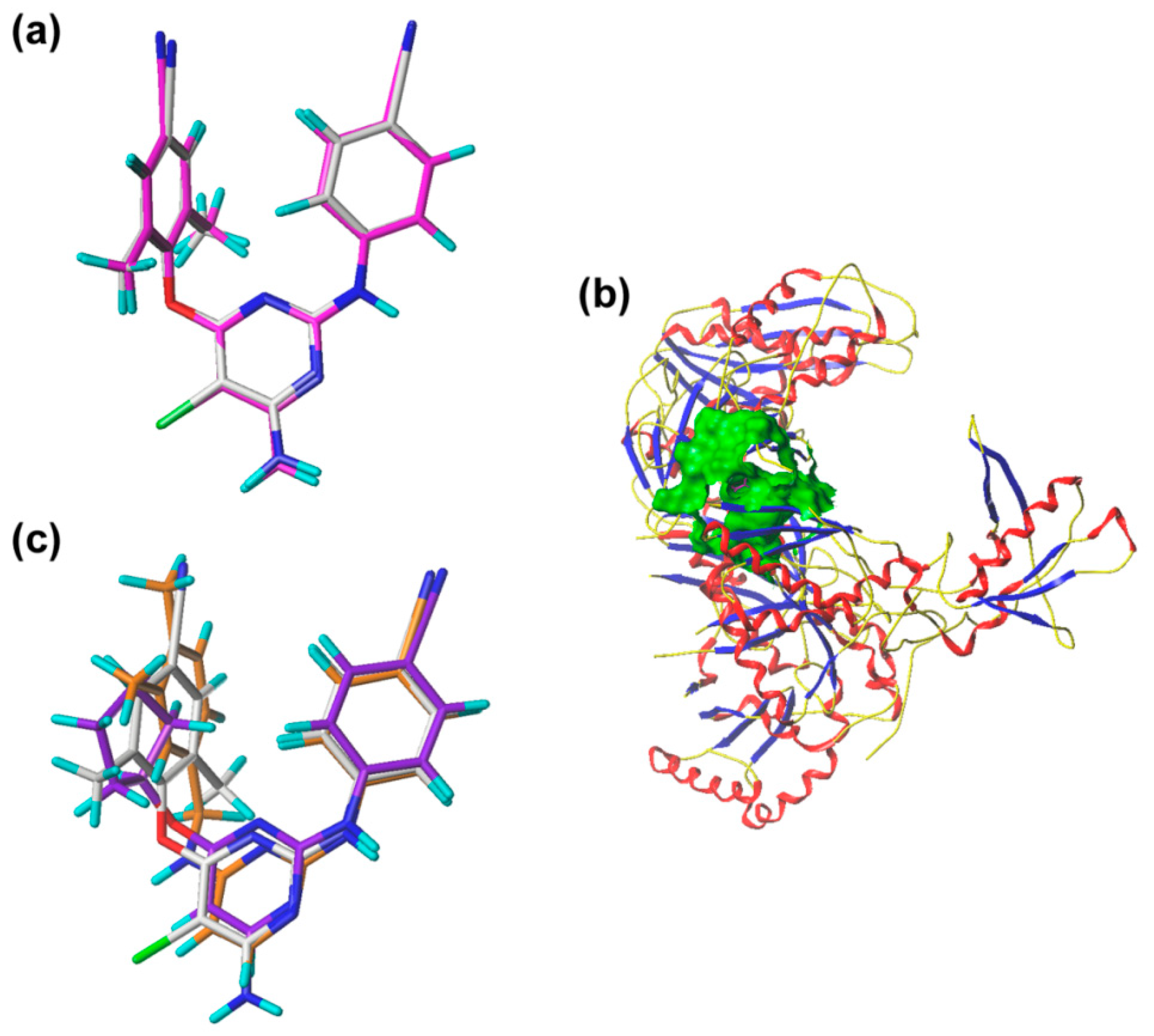
To validate the docking reliability, a cognate ligand, i.e., etravirine, which was extracted from the crystal structure of the WT HIV-1 RT (PDB ID: 3MEC), was first re-docked into the binding site using surflex-docking. The redocked conformation was compared with the original crystallographic conformation of the ligand [
14]. As shown in
Figure 6a, the redocked etravirine and the crystal etravirine in the complex are almost completely superimposable, and the root-mean-square deviation (RMSD) of the two conformations for all atoms is 0.25 Å. These results suggest that the surflex docking method and the used parameters are reasonable and reliable [
15]. The DAPYs and CAPYs were then docked into the binding site in the same way. The generated binding pocket is shown in
Figure 6b.
After validating the docking reliability, all the DAPYs and CAPYs were docked into the binding pocket. The superimposition of the most active compound
43 and the least active compound
46 with the redocked etravirine is shown in
Figure 6c. It should be noted that compound
43 superimposes with etravirine better than compound
46, although their docking conformations are in a similar orientation. The docking score of compound
43 (total-score = 9.5247) was higher than that of compound
46 (total-score = 8.2434), which is in agreement with their activities.
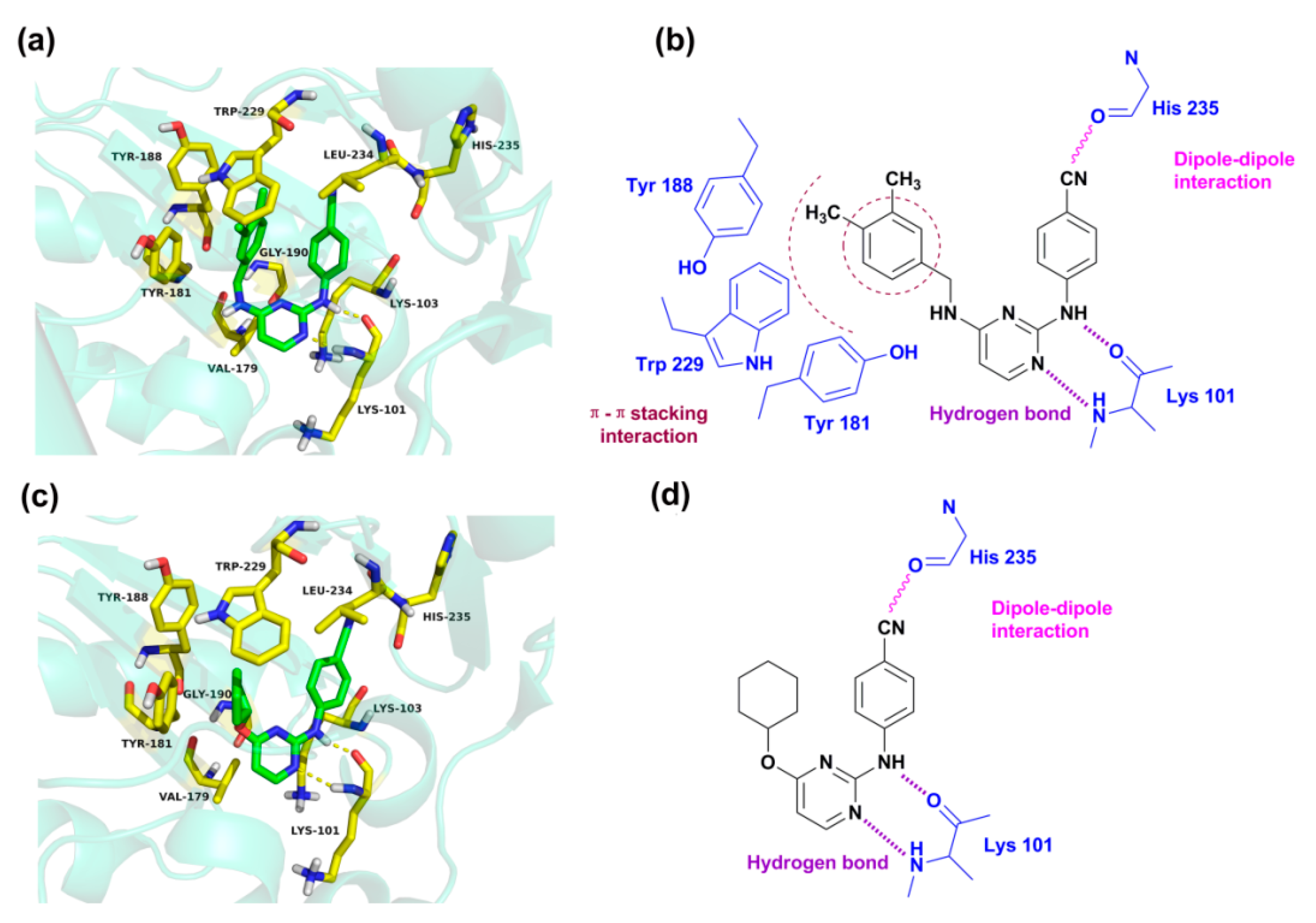
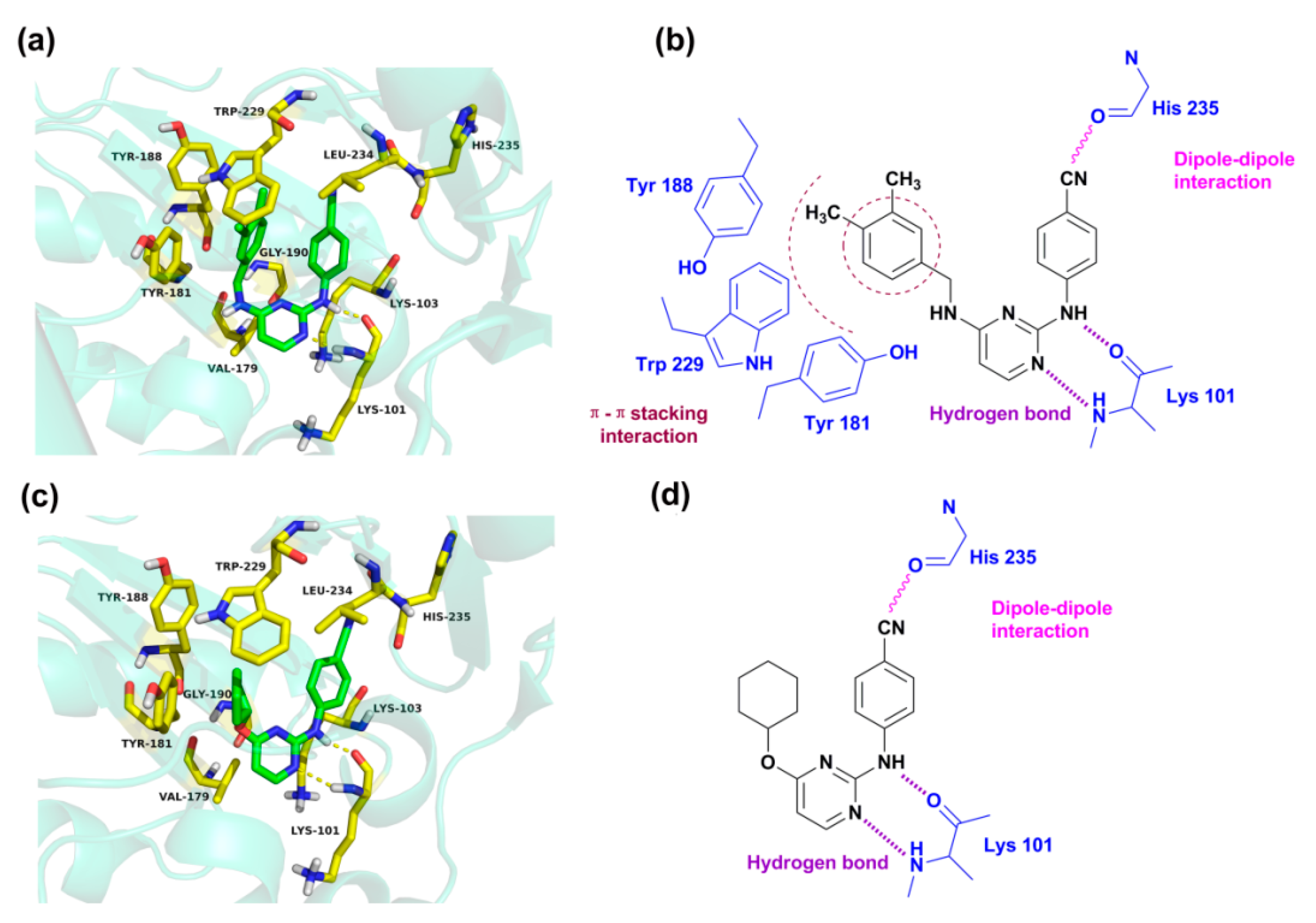
Figure 7 presents the detailed interacting modes of compounds
43 and
46 in the binding site of the HIV-1 RT (3MEC). As seen from
Figure 7a,c, the two compounds have the same orientation and adopt a horseshoe or a “U”-shaped conformation in the pocket, as previously reported [
16,
17]. As shown in
Figure 7a,b, the backbone of Lys 101 forms two hydrogen bonds with the nitrogen atoms of the pyrimidine and -NH linker of compound
43, respectively. This result is in agreement with our previous report that the residue Lys101 might interact with DAPYs and CAPYs through hydrogen bonds [
8]. The same interactions were also observed in the binding mode of compound
46. The hydrogen bond distances and angles are shown in
Table 4.
It was also found that some amino acid residues in the binding pocket, including Tyr318, Tyr232, Phe 227, Trp239, Trp229, Pro225, Pro226, Met230, Ile94, and Val189, formed hydrophobic interactions with compounds
43 and
46 [
18]. According to the pharmacophore model, it could also be concluded that bulky lipophilic substituents, such as an aromatic ring on the left wing of DAPYs, might make hydrophobic contacts with these amino acid residues. Moreover, van der Waals interactions could be established between the docked compounds and amino acid residues such as Leu100, Lys103, Val179, Gly190, and Leu234. The cyano group in the right aryl wing could establish a dipole–dipole interaction with the carbonyl of His235. These interactions might allow the inhibitors to maintain a horseshoe or a “U”-shaped conformation.
Additionally, π–π stacking interactions were found between the left phenyl ring of compound
43 and aromatic amino acid residues such as Tyr188, Tyr181, and Trp229 [
11,
19]. As shown in
Figure 7a, the left phenyl group is parallel to Tyr181 or Tyr188, and the 4-CH
3 on the phenyl ring points towards the highly conserved Trp229. However, π–π stacking interactions are not found in the docking results of compound
46 because of its lack of an aromatic ring on the left wing. The results indicate that the cyclohexyl or cyclopentyl substituents on the left wing of CAPYs are unfavorable for inhibitory activity, which might be due to the loss of the π–π stacking interactions.
2.5. Newly Designed DAPYs
Based on the combination analysis of the 3D-QSAR, pharmacophore, and molecular docking results, structure–activity relationships of DAPYs were obtained and subsequently utilized to design new DAPYs as potential HIV-1 NNRTIs. Ten novel DAPYs were designed, and their anti-HIV-1 activities were predicted by the CoMFA and the best CoMSIA models, as seen in
Table 5. Several principles were considered in the design of these novel DAPYs. First, the left phenyl ring was retained as a core moiety in the designed compounds because it is able to participate in π–π stacking interactions with the aromatic amino acid residues in the binding pocket. Second, the contour maps of the hydrogen-bond donor fields and pharmacophore features indicate that hydrogen-bond donors located at the left linker are preferred to enhance the activity, thus an imino group was retained as a linker instead of an oxygen atom. Third, different substitutions were introduced into the left phenyl group according to the contour maps analysis as follows: (a) a bulky, positively charged, and/or hydrophobic substituent, such as –CH
2CH
3, –CH(CH
3)
2, –C(CH
3)
3, and –NH
2, at the C
4-position; (b) a negatively charged and/or hydrophobic group, such as –CN, –NO
2, and –OOCCH
3, at the C
3-position; (c) a small, negatively charged, and/or hydrophilic substituent, such as –OH and –F, at the C
2-position.
As shown in
Table 5, the predicted activities of the newly designed molecules are all remarkable. Three compounds (
54,
60, and
62) demonstrated a higher activity in the CoMFA and the optimal CoMSIA models than the most active compound
43. These results indicate that the molecular simulation study was able to provide a reference for optimizing the structure and evaluating new potent DAPYs. However, future studies on synthesis methods, activity assays, and pharmacokinetic tests of these newly designed DAPYs are necessary.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}