Autophagic Regulation of p62 is Critical for Cancer Therapy


Abstract
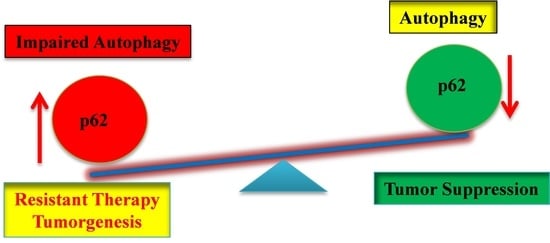
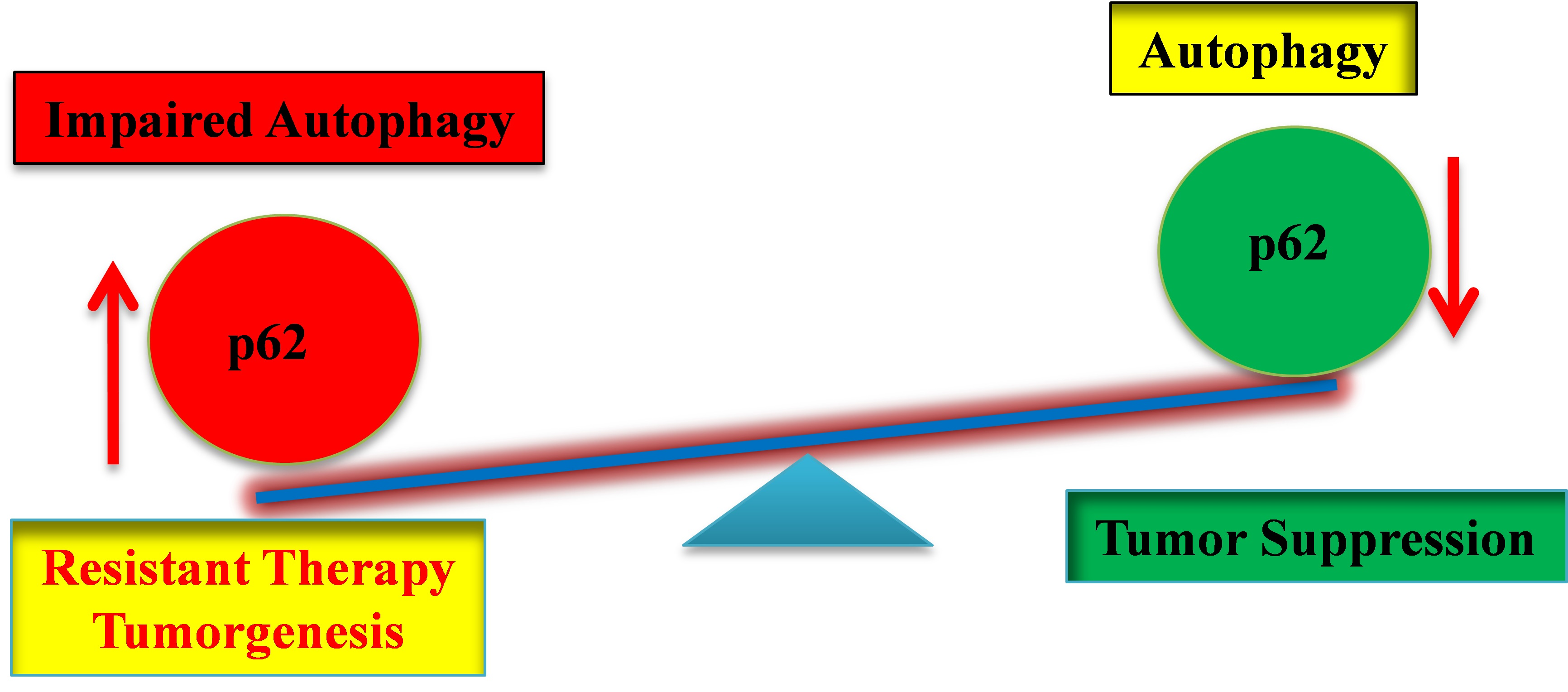
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
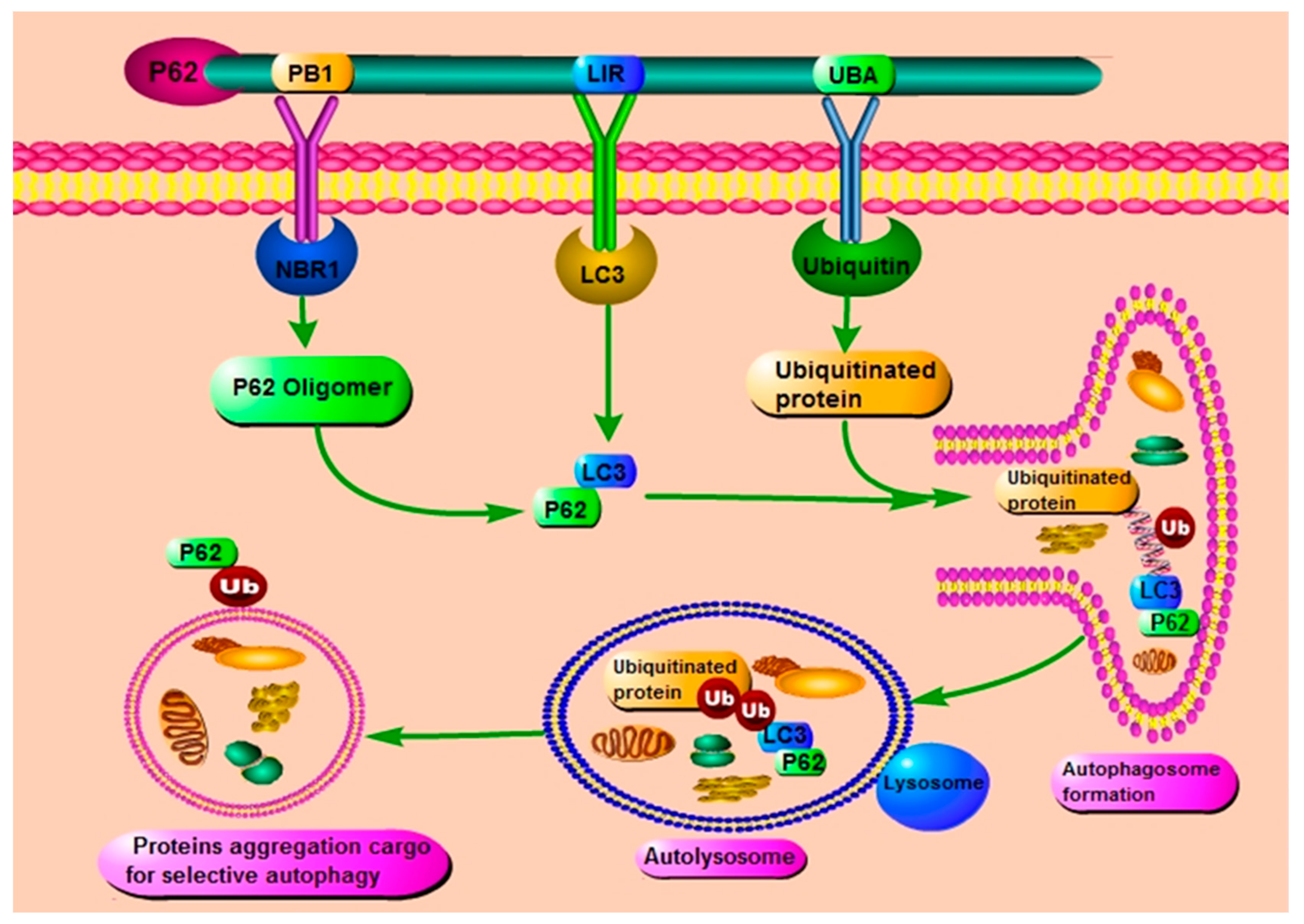
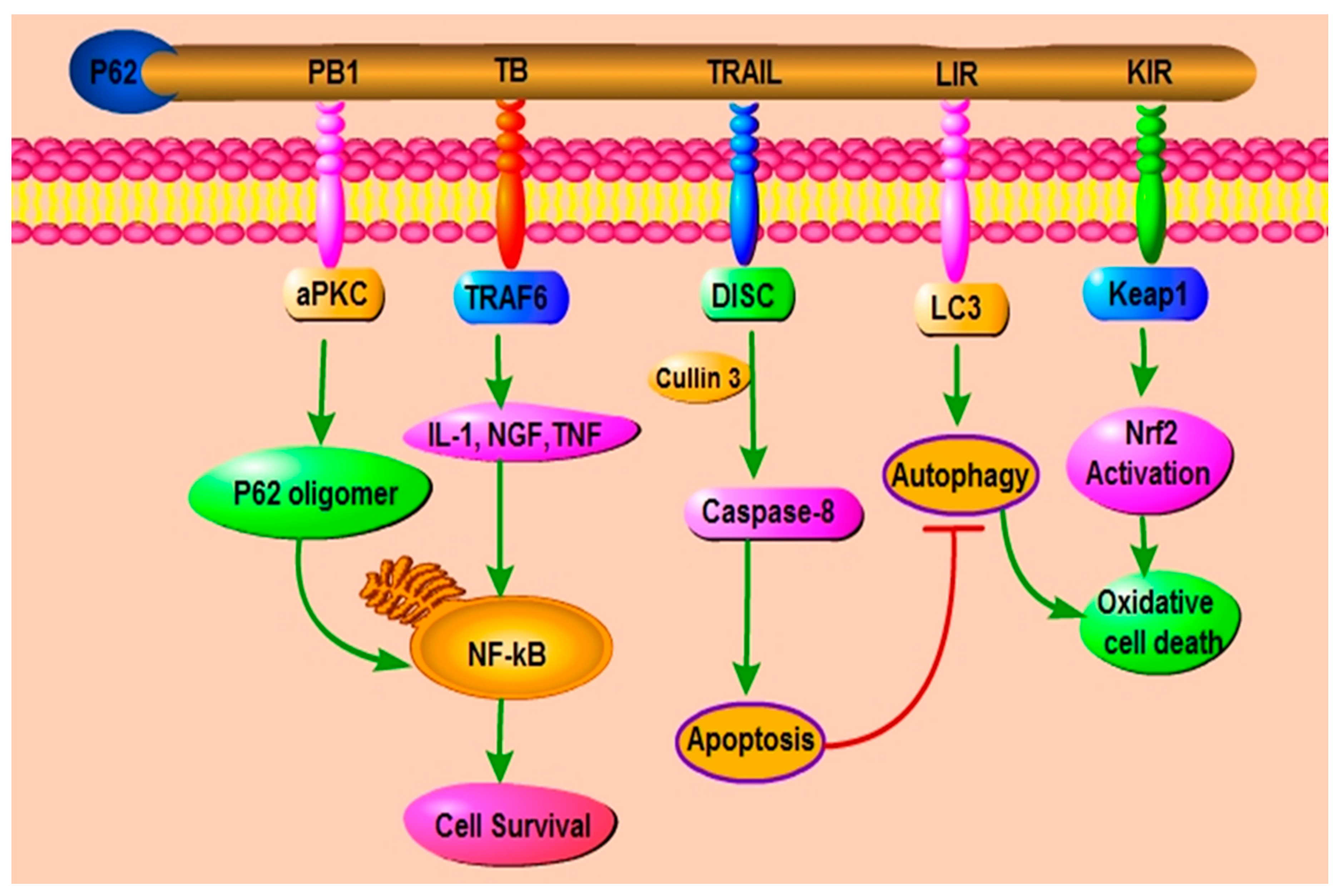
2. The Role and Function of p62/ SQSTM1 through Its Different Domains
2.1. The Role of PB1 Domain of p62
2.2. The Role of ZZ Domain of p62
2.3. The Role of LIR Domain of p62
2.4. The Role of KIR Domain of p62
2.5. The Role of UBA Domain of p62
3. P62 and Autophagy
4. P62 and Cancer
5. P62 and Apoptosis
6. Concluding Remarks and Future Direction
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Christian, F.; Krause, E.; Houslay, M.D.; Baillie, G.S. PKA phosphorylation of p62/SQSTM1 regulates PB1 domain interaction partner binding. Biochim. Biophys. Acta 2014, 1843, 2765–2774. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Perander, M.; Outzen, H.; Kristiansen, K.; Øvervatn, A.; Michaelsen, E.; Bjørkøy, G.; Johansen, T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J. Biol. Chem. 2003, 278, 34568–34581. [Google Scholar] [CrossRef] [PubMed]
- Wooten, M.W.; Geetha, T.; Seibenhener, M.L.; Babu, J.R.; Diaz-Meco, M.T.; Moscat, J. The p62 scaffold regulates nerve growth factor-induced NF-κB activation by influencing TRAF6 polyubiquitination. J. Biol. Chem. 2005, 280, 35625–35629. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, S.; Zhao, Y.; Ma, X.; Zhang, K.; He, X.; Wang, Z. Interaction domains of p62: a bridge between p62 and selective autophagy. DNA Cell Biol. 2013, 32, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Chamoux, E.; McManus, S.; Laberge, G.; Bisson, M.; Roux, S. Involvement of kinase PKC-zeta in the p62/p62 P392L-driven activation of NF-κB in human osteoclasts. Biochim. Biophys. Acta 2013, 1832, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Schläfli, A.M.; Adams, O.; Galván, J.A.; Gugger, M.; Savic, S.; Bubendorf, L.; Schmid, R.A.; Becker, K.-F.; Tschan, M.P.; Langer, R. Prognostic value of the autophagy markers LC3 and p62/SQSTM1 in early-stage non-small cell lung cancer. Oncotarget 2016, 7, 39544. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Williams, J.A.; Ding, W.-X. Role of p62/SQSTM1 in liver physiology and pathogenesis. Exp. Biol. Med. 2013, 238, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Galvez, A.S.; Duran, A.; Linares, J.F.; Pathrose, P.; Castilla, E.A.; Abu-Baker, S.; Leitges, M.; Diaz-Meco, M.T.; Moscat, J. Protein kinase Cζ represses the interleukin-6 promoter and impairs tumorigenesis in vivo. Mol. Cell. Biol. 2009, 29, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Okamoto, K.; Yu, C.; Sinicrope, F.A. p62/sequestosome-1 up-regulation promotes ABT-263-induced caspase-8 aggregation/activation on the autophagosome. J. Biol. Chem. 2013, 288, 33654–33666. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kominami, E.; Tanaka, K.; Komatsu, M. Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 2008, 4, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, B.; Zaffagnini, G.; Fracchiolla, D.; Turco, E.; Abert, C.; Romanov, J.; Martens, S. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. eLife 2015, 4, e08941. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M. Potential role of p62 in tumor development. Autophagy 2011, 7, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [PubMed]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.-D.; Zhang, J. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.; Diaz-Meco, M.T.; Nakano, H.; Moscat, J. The atypical PKC-interacting protein p62 channels NF-κB activation by the IL-1–TRAF6 pathway. EMBO J. 2000, 19, 1576–1586. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-Y.; Nigro, P.; Fujiwara, K.; Abe, J.-I.; Berk, B.C. p62 binding to protein kinase C ζ regulates tumor necrosis factor α–induced apoptotic pathway in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2974–2980. [Google Scholar] [CrossRef]
- Nakamura, K.; Kimple, A.J.; Siderovski, D.P.; Johnson, G.L. PB1 domain interaction of p62/sequestosome 1 and MEKK3 regulates NF-κB activation. J. Biol. Chem. 2010, 285, 2077–2089. [Google Scholar] [CrossRef] [PubMed]
- Linares, J.F.; Duran, A.; Reina-Campos, M.; Aza-Blanc, P.; Campos, A.; Moscat, J.; Diaz-Meco, M.T. Amino acid activation of mTORC1 by a PB1-domain-driven kinase complex cascade. Cell Rep. 2015, 12, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Dai, Q.; Meng, H.; Sun, A.; Wei, J.; Peng, K.; Childress, C.; Chen, M.; Shao, G.; Yang, W. The HECT E3 ubiquitin ligase NEDD4 interacts with and ubiquitylates SQSTM1 for inclusion body autophagy. J. Cell Sci. 2017, 130, 3839–3850. [Google Scholar] [CrossRef] [PubMed]
- Regan, C.P.; Li, W.; Boucher, D.M.; Spatz, S.; Su, M.S.; Kuida, K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. USA 2002, 99, 9248–9253. [Google Scholar] [CrossRef] [PubMed]
- Sohn, S.J.; Sarvis, B.K.; Cado, D.; Winoto, A. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J. Biol. Chem. 2002, 277, 43344–43351. [Google Scholar] [CrossRef] [PubMed]
- Watson, F.L.; Heerssen, H.M.; Bhattacharyya, A.; Klesse, L.; Lin, M.Z.; Segal, R.A. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat. Neurosci. 2001, 4, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, K.; Lobel-Rice, K.; Sun, W.; Lapadat, R.; Webb, S.; Johnson, G.L.; Garrington, T.P. MEKK2 regulates the coordinate activation of ERK5 and JNK in response to FGF-2 in fibroblasts. J. Cell. Physiol. 2004, 199, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. 2015, 6, e1980. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, M.-N.; Ryu, K.-Y. Effect of p62/SQSTM1 polyubiquitination on its autophagic adaptor function and cellular survival under oxidative stress induced by arsenite. Biochem. Biophys. Res. Commun. 2017, 486, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. p62-and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.; Sanchez, P.; Lallena, M.J.; Diaz-Meco, M.T.; Moscat, J. The interaction of p62 with RIP links the atypical PKCs to NF-κB activation. EMBO J. 1999, 18, 3044–3053. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.B.; Kielczewska, A.; Rozek, A.; Takenaka, S.; Li, Y.; Thorson, L.; Hancock, R.E.; Guarna, M.M.; North, J.R.; Foster, L.J. Sequestosome-1/p62 is the key intracellular target of innate defense regulator peptide. J. Biol. Chem. 2009, 284, 36007–36011. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.Y.; Zhang, Y.; Zhang, J.J.; Zhang, L.C.; Liu, Y.N.; Wu, Y.; Xue, Y.N.; Lu, S.Y.; Su, J.; Sun, L.K. p62/SQSTM1 as an oncotarget mediates cisplatin resistance through activating RIP1-NF-κB pathway in human ovarian Cancer Cells. Cancer Sci. 2017, 108, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Teramachi, J.; Silbermann, R.; Yang, P.; Zhao, W.; Mohammad, K.S.; Guo, J.; Anderson, J.L.; Zhou, D.; Feng, R.; Myint, K.-Z. Blocking the ZZ domain of sequestosome1/p62 suppresses myeloma growth and osteoclast formation in vitro and induces dramatic bone formation in myeloma-bearing bones in vivo. Leukemia 2016, 30, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Teramachi, J.; Myint, K.Z.Y.; Feng, R.; Xie, X.; Windle, J.J.; Roodman, D.; Kurihara, N. Blocking the ZZ Domain of Sequestosome 1/p62 suppress the enhancement of myeloma cell growth and osteoclast formation by marrow stromal cells. Blood. 2011, 118, 888. [Google Scholar]
- Jiang, J.; Parameshwaran, K.; Seibenhener, M.L.; Kang, M.-G.; Suppiramaniam, V.; Huganir, R.L.; Diaz-Meco, M.T.; Wooten, M.W. AMPA receptor trafficking and synaptic plasticity require SQSTM1/p62. Hippocampus 2009, 19, 392. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.J.; Kim, H.M.; Koo, J.S. Expression of Autophagy-Related Proteins in Hürthle Cell Neoplasm Is Different from That in Follicular Neoplasm. Dis. Mark. 2017, 2017, 1372387. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Birgisdottir, Å.; Huber, J.; Kniss, A.; Dötsch, V.; Kirkin, V.; Rogov, V. Chapter Nine-Methods for Studying Interactions Between Atg8/LC3/GABARAP and LIR-Containing Proteins. Methods Enzymol. 2017, 587, 143–169. [Google Scholar] [PubMed]
- Maruyama, Y.; Sou, Y.-S.; Kageyama, S.; Takahashi, T.; Ueno, T.; Tanaka, K.; Komatsu, M.; Ichimura, Y. LC3B is indispensable for selective autophagy of p62 but not basal autophagy. Biochem. Biophys. Res. Commun. 2014, 446, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Lamark, T.; Sou, Y.-S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.-A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.K.; Jun, Y.W.; Choi, H.E.; Huh, Y.H.; Kaang, B.K.; Jang, D.J.; Lee, J.A. Development of LC3/GABARAP sensors containing a LIR and a hydrophobic domain to monitor autophagy. EMBO J. 2017, 36, 1100–1116. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Liu, M.; Lin, G.; Mao, B.; Cheng, W.; Liu, H.; Gal, J.; Zhu, H.; Yuan, Z.; Deng, W. Tumor suppressor Spred2 interaction with LC3 promotes autophagosome maturation and induces autophagy-dependent cell death. Oncotarget 2016, 7, 25652. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Kim, D.-S.; Kim, K.-H.; Yoo, B.C.; Kim, S.-H.; Gong, Y.-D.; Kim, S.-Y. Transglutaminase 2 inhibition found to induce p53 mediated apoptosis in renal cell carcinoma. FASEB J. 2013, 27, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kumanomidou, T.; Sou, Y.-S.; Mizushima, T.; Ezaki, J.; Ueno, T.; Kominami, E.; Yamane, T.; Tanaka, K.; Komatsu, M. Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 2008, 283, 22847–22857. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Darvekar, S.R.; Elvenes, J.; Brenne, H.B.; Johansen, T.; Sjøttem, E. SPBP is a sulforaphane induced transcriptional coactivator of NRF2 regulating expression of the autophagy receptor p62/SQSTM1. PLoS ONE 2014, 9, e85262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Kang, D.H.; Bae, S.H. PF-4708671, a specific inhibitor of p70 ribosomal S6 kinase 1, activates Nrf2 by promoting p62-dependent autophagic degradation of Keap1. Biochem. Biophys. Res. Commun. 2015, 466, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Isogai, S.; Morimoto, D.; Arita, K.; Unzai, S.; Tenno, T.; Hasegawa, J.; Sou, Y.-S.; Komatsu, M.; Tanaka, K.; Shirakawa, M. Crystal structure of the ubiquitin-associated (UBA) domain of p62 and its interaction with ubiquitin. J. Biol. Chem. 2011, 286, 31864–31874. [Google Scholar] [CrossRef] [PubMed]
- Goode, A.; Layfield, R. Recent advances in understanding the molecular basis of Paget disease of bone. J. Clin. Pathol. 2010, 63, 199–203. [Google Scholar] [CrossRef] [PubMed]
- McManus, S.; Roux, S. The adaptor protein p62/SQSTM1 in osteoclast signaling pathways. J. Mol. Signal. 2012, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Laurin, N.; Brown, J.P.; Morissette, J.; Raymond, V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 2002, 70, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Hocking, L.J.; Lucas, G.J.; Daroszewska, A.; Mangion, J.; Olavesen, M.; Cundy, T.; Nicholson, G.C.; Ward, L.; Bennett, S.T.; Wuyts, W. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum. Mol. Genet. 2002, 11, 2735–2739. [Google Scholar] [CrossRef] [PubMed]
- Durán, A.; Serrano, M.; Leitges, M.; Flores, J.M.; Picard, S.; Brown, J.P.; Moscat, J.; Diaz-Meco, M.T. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev. Cell 2004, 6, 303–309. [Google Scholar] [CrossRef]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The signaling adaptor p62 is an important NF-κB mediator in tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zheng, Z.; Ling, L.; Yang, X.; Zhang, N.; Wang, X.; Hu, M.; Xia, Y.; Ma, Y.; Yang, H. w09, a novel autophagy enhancer, induces autophagy-dependent cell apoptosis via activation of the EGFR-mediated RAS-RAF1-MAP2K-MAPK1/3 pathway. Autophagy 2017, 13, 1093–1112. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.-Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Cha-Molstad, H.; Yu, J.E.; Feng, Z.; Lee, S.H.; Kim, J.G.; Yang, P.; Han, B.; Sung, K.W.; Yoo, Y.D.; Hwang, J. p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat. Commun. 2017, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.K.; Hailey, D.W.; Mullen, R.T.; Lippincott-Schwartz, J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. USA 2008, 105, 20567–20574. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Weihl, C.C. Regulation of SQSTM1/p62 via UBA domain ubiquitination and its role in disease. Autophagy 2017, 13, 1615–1616. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Chou, T.-F.; Pittman, S.K.; Keith, A.L.; Razani, B.; Weihl, C.C. Keap1/Cullin3 modulates p62/SQSTM1 activity via UBA domain ubiquitination. Cell Rep. 2017, 19, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.-S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.-I.; Ezaki, J.; Murata, S. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Mizushima, N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J. Cell Biol. 2011, 192, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ni, H.-M.; Guo, F.; Ding, Y.; Shi, Y.-H.; Lahiri, P.; Fröhlich, L.F.; Rülicke, T.; Smole, C.; Schmidt, V.C. Sequestosome 1/p62 protein is associated with autophagic removal of excess hepatic endoplasmic reticulum in mice. J. Biol. Chem. 2016, 291, 18663–18674. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Yoshizumi, T.; Yoshida, Y.; Bekki, Y.; Matsumoto, Y.; Yoshiya, S.; Toshima, T.; Ikegami, T.; Itoh, S.; Harimoto, N. p62 Promotes Amino Acid Sensitivity of mTOR Pathway and Hepatic Differentiation in Adult Liver Stem/Progenitor Cells. J. Cell. Physiol. 2017, 232, 2112–2124. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; Li, S.; Liang, Q.; Zhang, X.; Wang, H.; Herbert, T.P.; Jenkins, T.A.; Xu, A.; Ye, J.-M. Chronic activation of PPARα with fenofibrate reduces autophagic proteins in the liver of mice independent of FGF21. PLoS ONE 2017, 12, e0173676. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fukui, K.; Koike, T.; Zheng, X. Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur. J. Neurosci. 2007, 26, 2979–2988. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, X.; Le, W. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 2008, 4, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Padman, B.S.; Nguyen, T.N.; Lazarou, M. Autophagosome formation and cargo sequestration in the absence of LC3/GABARAPs. Autophagy 2017, 13, 772–774. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Cai-McRae, X.; Zhong, H.; Karantza, V. Sequestosome 1/p62 facilitates HER2-induced mammary tumorigenesis through multiple signaling pathways. Oncogene 2015, 34, 2968. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes. Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Yamachika, S.; He, F.; Karin, M. p62/SQSTM1—Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett. 2016, 590, 2375–2397. [Google Scholar] [CrossRef] [PubMed]
- Bokun, R.; Bakotin, J.; Milasinović, D. Semiquantitative cytochemical estimation of glucose-6-phosphate dehydrogenase activity in benign diseases and carcinoma of the breast. Acta Cytol. 1987, 31, 249–252. [Google Scholar] [PubMed]
- Wang, J.; Yuan, W.; Chen, Z.; Wu, S.; Chen, J.; Ge, J.; Hou, F.; Chen, Z. Overexpression of G6PD is associated with poor clinical outcome in gastric cancer. Tumor Biol. 2012, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Zampella, E.J.; Bradley, E.L.; Pretlow, T.G. Glucose-6-phosphate dehydrogenase: A possible clinical indicator for prostatic carcinoma. Cancer 1982, 49, 384–387. [Google Scholar] [CrossRef]
- Aishima, S.; Fujita, N.; Mano, Y.; Iguchi, T.; Taketomi, A.; Maehara, Y.; Oda, Y.; Tsuneyoshi, M. p62+ Hyaline inclusions in intrahepatic cholangiocarcinoma associated with viral hepatitis or alcoholic liver disease. Am. J. Clin. Pathol. 2010, 134, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Yu, H.; Gu, S.; Xu, Y.; Su, J.; Li, H.; Kang, J.; Cui, M. p62/SQSTM1 is involved in cisplatin resistance in human ovarian Cancer Cells via the Keap1-Nrf2-ARE system. Int. J. Oncol. 2014, 45, 2341–2348. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Su, J.; Xu, Y.; Kang, J.; Li, H.; Zhang, L.; Yi, H.; Xiang, X.; Liu, F.; Sun, L. p62/SQSTM1 involved in cisplatin resistance in human ovarian Cancer Cells by clearing ubiquitinated proteins. Eur. J. Cancer 2011, 47, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pan, B.-Q.; Shi, F.; Xie, Z.-H.; Jiang, Y.-Y.; Shang, L.; Zhang, Y.; Xu, X.; Cai, Y.; Hao, J.-J. Sequestosome 1 protects esophageal squamous carcinoma cells from apoptosis via stabilizing SKP2 under serum starvation condition. Oncogene 2018, 1. [Google Scholar] [CrossRef] [PubMed]
- Valencia, T.; Kim, J.Y.; Abu-Baker, S.; Moscat-Pardos, J.; Ahn, C.S.; Reina-Campos, M.; Duran, A.; Castilla, E.A.; Metallo, C.M.; Diaz-Meco, M.T. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 2014, 26, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, W.-G.; Zhao, Y. Autophagy substrate SQSTM1/p62 regulates chromatin ubiquitination during the DNA damage response. Autophagy 2017, 13, 212–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, N.; Zhang, L.; Li, R.; Fu, W.; Ma, K.; Li, X.; Wang, L.; Wang, J.; Zhang, H. Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol. Cell 2016, 63, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, S.; Zhou, W.; Kang, Z.; Zhang, Q.; Kamran, M.; Xu, J.; Liang, D.; Wang, C.; Hou, Z. p62/SQSTM1 enhances breast cancer stem-like properties by stabilizing MYC mRNA. Oncogene 2017, 36, 304. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Debnath, J. ‘Doubling down’on the autophagy pathway to suppress tumor growth. Genes. Dev. 2014, 28, 1137–1139. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.K.; Wen, J.; Chen, S.; Guan, J.-L. Autophagy differentially regulates distinct breast cancer stem-like cells in murine models via EGFR/Stat3 and Tgfβ/Smad signaling. Cancer Res. 2016, 76, 3397–3410. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Sheng, J.; Shen, L.; Su, J.; Xu, Y.; Xie, Q.; Wu, Y.; Zhang, X.; Sun, L. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS ONE 2017, 12, e0173712. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Yu, H.; Cui, N.; Kong, X.; Liu, X.; Chang, Y.; Wu, Y.; Sun, L.; Wang, G. ABT737 enhances cholangiocarcinoma sensitivity to cisplatin through regulation of mitochondrial dynamics. Exp. Cell Res. 2015, 335, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; He, W.; Shi, H.; Huang, X.; Ji, G. Natural compound oblongifolin C inhibits autophagic flux, and induces apoptosis and mitochondrial dysfunction in human cholangiocarcinoma QBC939 cells. Mol. Med. Rep. 2016, 14, 3179–3183. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Zhang, A.; Qi, L.; Na, C.; Jiang, R.; Fan, Z.; Chen, J. FOXO1, a Potential Therapeutic Target, Regulates Autophagic Flux, Oxidative Stress, Mitochondrial Dysfunction, and Apoptosis in Human Cholangiocarcinoma QBC939 Cells. Cell. Physiol. Biochem. 2018, 45, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T.; Albert, A.; Campuzano, S. Cell signaling and function organized by PB1 domain interactions. Mol. Cell 2006, 23, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Sooro, M.A.; Zhang, N.; Zhang, P. Targeting EGFR-mediated autophagy as a potential strategy for cancer therapy. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liu, Y.; Wang, D.; Wang, L.; Zhou, H.; Xu, G.; Xie, L.; Wu, M.; Lin, Z.; Yu, Y. Galectin-3 regulates metastatic capabilities and chemotherapy sensitivity in epithelial ovarian carcinoma via NF-κB pathway. Tumor Biol. 2016, 37, 11469–11477. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, S.M.; Thakur, B.; Sakpal, A.; Singh, R.K.; Ray, P. Differential activation of NF-κB signaling is associated with platinum and taxane resistance in MyD88 deficient epithelial ovarian Cancer Cells. Int. J. Biochem. Cell Biol. 2015, 61, 90–102. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.A.; Sooro, M.A.; Zhang, P. Autophagic Regulation of p62 is Critical for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 1405. https://doi.org/10.3390/ijms19051405
Islam MA, Sooro MA, Zhang P. Autophagic Regulation of p62 is Critical for Cancer Therapy. International Journal of Molecular Sciences. 2018; 19(5):1405. https://doi.org/10.3390/ijms19051405
Chicago/Turabian StyleIslam, Md. Ariful, Mopa Alina Sooro, and Pinghu Zhang. 2018. "Autophagic Regulation of p62 is Critical for Cancer Therapy" International Journal of Molecular Sciences 19, no. 5: 1405. https://doi.org/10.3390/ijms19051405
APA StyleIslam, M. A., Sooro, M. A., & Zhang, P. (2018). Autophagic Regulation of p62 is Critical for Cancer Therapy. International Journal of Molecular Sciences, 19(5), 1405. https://doi.org/10.3390/ijms19051405