Immune Ecosystem of Virus-Infected Host Tissues


{kind=link}
Abstract
:1. Introduction
2. Immune Sensing of Viral Infection
3. Intercellular Immune Ecosystem of Virus-Infected Tissues
4. Intracellular Immune Ecosystem of Virus-Infected Cells
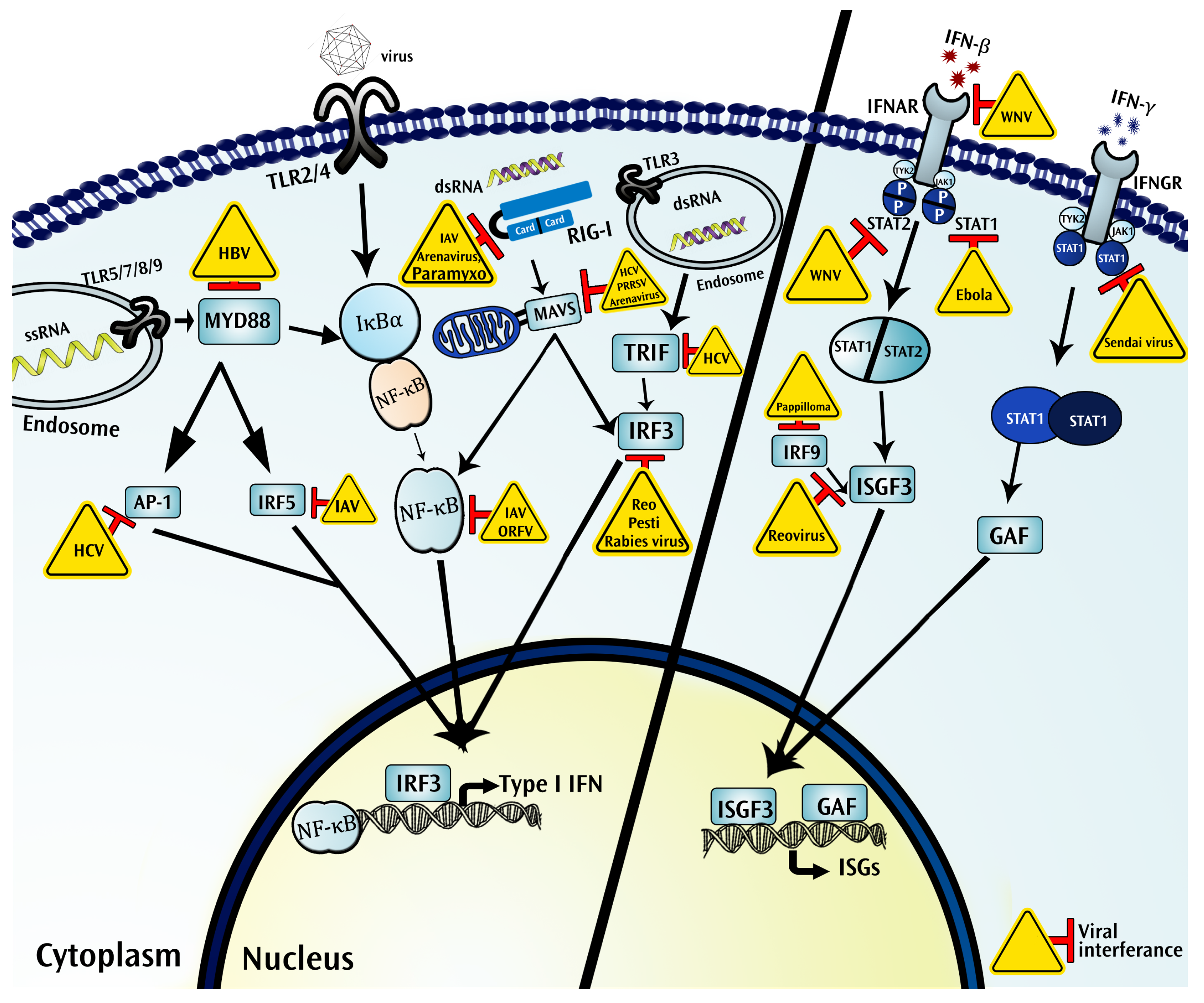
4.1. Pattern Recognition Receptors
4.1.1. Toll Like Receptors
4.1.2. RIG-I Like Receptors
4.1.3. NOD-Like Receptors (NLRs)
4.2. Interferons Function as Critical Components in the Immune Ecosystem of a Virus Infected Tissue
5. Adaptive Immune Response to Viral Infection
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AP1 | Activation Protein1 |
| APC | Antigen Presenting Cells |
| ATF2 | Activating transcription factor-2 |
| CARD | Caspase Activation and Recruitment Domains |
| CMV | Cytomegaloviruses |
| CTL | Cytotoxic T Lymphocytes |
| ds/ssRNA | Double/Single Stranded RNA |
| ER | Endoplasmic Reticulum |
| FMD | Foot-and-mouth disease |
| GAS | Gamma-Activated Sequence |
| HCV | Hepatitis C Virus |
| IAV | Influenza A Virus |
| IFN | Interferon |
| IFNAR | Interferon α Receptor |
| IFGAR | Interferon γ Receptor |
| IL | Interleukins |
| ISGF | IFN-Stimulated Gene Factor |
| JAK1 | Janus Kinase 1 |
| LCMV | Lymphocytic Choriomeningitis Virus |
| LGP2 | Laboratory of Genetics and Physiology 2 |
| MAPKs | Mitogen-activated Protein |
| MAVS | Mitochondrial Antiviral Signaling Protein |
| MDA5 | Melanoma Differentiation-associated Gene 5 |
| MHC | Major Histocompatibility Complex |
| miRNAs | microRNAs |
| MLV | Murine Leukemia virus |
| MYD88 | Myeloid Differentiation Primary Response 88 |
| ncRNA | Non-coding RNAs |
| NF-κB | Nuclear Factor κB |
| NK | Natural Killer |
| NLRP3 | NOD-, LRR- and pyrine domain-containg 3 |
| NOD | Nucleotide-binding Oligomerization |
| OAS | 2′-5′ Oligoadenylate Synthetase |
| PAMP | Pathogen Associated Molecular Patterns |
| PI3K | Phospho-Inositol 3 Kinase |
| PRR | Pattern Recognition Receptors |
| STING | Stimulator of interferon genes |
| RIG-I | Retinoic Acid Inducible Gene |
| RSV | Respiratory Syncytial Virus |
| STAT | Signal Transducers and Activators of Transcription |
| TCM | Central memory T cells |
| TEM | Effector memory T cells |
| TLR | Toll Like Receptors |
| TYK2 | Tyrosine Kinase 2 |
| WNV | West Nile virus |
References
- Doherty, P.C.; Turner, S.J. The virus-immunity ecosystem. In Infectious Diseases from Nature: Mechanisms of Viral Emergence and Persistence; Springer: Vienna, Austria, 2005; pp. 17–32. [Google Scholar]
- Labuda, M.; Nuttall, P.A.; Kozuch, O.; Eleckova, E.; Williams, T.; Zuffova, E.; Sabo, A. Non-viraemic transmission of tick-borne encephalitis virus: A mechanism for arbovirus survival in nature. Experientia 1993, 49, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Morse, S.S. The public health threat of emerging viral disease. J. Nutr. 1997, 127, S951–S957. [Google Scholar]
- Buttner, S.; Eisenberg, T.; Herker, E.; Carmona-Gutierrez, D.; Kroemer, G.; Madeo, F. Why yeast cells can undergo apoptosis: Death in times of peace, love, and war. J. Cell Biol. 2006, 175, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Galluzzi, L.; Panaretakis, T.; Tesniere, A.; Schlemmer, F.; Madeo, F.; Zitvogel, L.; Kroemer, G. Viral subversion of immunogenic cell death. Cell Cycle 2009, 8, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C. Immunobiology: The Immune System in Health and Disease, 6th ed.; Garland Science: New York, NY, USA, 2005; p. xxiii. 823p. [Google Scholar]
- Janeway, C.A., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Akira, S. Toll-like receptors and innate immunity. J. Mol. Med. 2006, 84, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Xagorari, A.; Chlichlia, K. Toll-like receptors and viruses: Induction of innate antiviral immune responses. Open Microbiol. J. 2008, 2, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptor and RIG-I-like receptor signaling. Ann. N. Y. Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef] [PubMed]
- Goraya, M.U.; Wang, S.; Munir, M.; Chen, J.L. Induction of innate immunity and its perturbation by influenza viruses. Protein Cell 2015, 6, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.T.; Chen, S.S. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell. Mol. Immunol. 2016, 13, 11–35. [Google Scholar] [CrossRef] [PubMed]
- Grinde, B. Herpesviruses: Latency and reactivation–viral strategies and host response. J. Oral Microbiol. 2013, 5, 22766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schotsaert, M.; Garcia-Sastre, A. A High-Resolution Look at Influenza Virus Antigenic Drift. J. Infect. Dis. 2016, 214, 982. [Google Scholar] [CrossRef] [PubMed]
- Voronin, Y.; Holte, S.; Overbaugh, J.; Emerman, M. Genetic drift of HIV populations in culture. PLoS Genet. 2009, 5, e1000431. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Tian, S.; Luo, M.; Xie, W.; Liu, T.; Duan, T.; Wu, Y.; Cui, J. Tetherin Suppresses Type I Interferon Signaling by Targeting MAVS for NDP52-Mediated Selective Autophagic Degradation in Human Cells. Mol. Cell 2017, 68, 308–322. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.C.; Cardenas, W.B.; Basler, C.F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J. Virol. 2009, 83, 3069–3077. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Liu, L.; Lei, X.; Zhou, Z.; He, B.; Wang, J. 3C Protease of Enterovirus D68 Inhibits Cellular Defense Mediated by Interferon Regulatory Factor 7. J. Virol. 2016, 90, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.; Haas, D.A.; Farrell, P.J.; Pichlmair, A.; Bowie, A.G. Poxvirus Protein MC132 from Molluscum Contagiosum Virus Inhibits NF-B Activation by Targeting p65 for Degradation. J. Virol. 2015, 89, 8406–8415. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.; Fernandez-Sesma, A.; Garcia-Sastre, A. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, A.E.; Mitchell, A.; Brownell, J.; Miklin, D.J.; Golden-Mason, L.; Polyak, S.J.; Gale, M.J., Jr.; Rosen, H.R. Hepatitis C virus core protein inhibits interferon production by a human plasmacytoid dendritic cell line and dysregulates interferon regulatory factor-7 and signal transducer and activator of transcription (STAT) 1 protein expression. PLoS ONE 2014, 9, e95627. [Google Scholar] [CrossRef] [PubMed]
- Harman, A.N.; Nasr, N.; Feetham, A.; Galoyan, A.; Alshehri, A.A.; Rambukwelle, D.; Botting, R.A.; Hiener, B.M.; Diefenbach, E.; Diefenbach, R.J.; et al. HIV Blocks Interferon Induction in Human Dendritic Cells and Macrophages by Dysregulation of TBK1. J. Virol. 2015, 89, 6575–6584. [Google Scholar] [CrossRef] [PubMed]
- Saez-Cirion, A.; Manel, N. Immune Responses to Retroviruses. Annu. Rev. Immunol. 2018, 36, 193–220. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, X.; Manel, N. Viral and cellular mechanisms of the innate immune sensing of HIV. Curr. Opin. Virol. 2015, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Finberg, R.W.; Kurt-Jones, E.A. Viruses and Toll-like receptors. Microbes Infect. 2004, 6, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Ishii, K.J.; Kumar, H.; Tanimoto, T.; Coban, C.; Uematsu, S.; Kawai, T.; Akira, S. Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J. Immunol. 2007, 179, 4711–4720. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.; Heit, A.; Guggemoos, S.; Krug, A.; Mages, J.; Schiemann, M.; Adler, H.; Drexler, I.; Haas, T.; Lang, R.; et al. Interferon-regulatory-factor 1 controls Toll-like receptor 9-mediated IFN-beta production in myeloid dendritic cells. Eur. J. Immunol. 2007, 37, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.J.; Kawagoe, T.; Koyama, S.; Matsui, K.; Kumar, H.; Kawai, T.; Uematsu, S.; Takeuchi, O.; Takeshita, F.; Coban, C.; et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 2008, 451, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Travers, P.; Walport, M.; Janeway, C. Janeway’s Immunobiology, 8th ed.; Garland Science: London, UK; Taylor & Francis: London, UK, 2012. [Google Scholar]
- Watanabe, Y. Fifty years of interference. Nat. Immunol. 2004, 5, 1193. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.C.; Jacques, D.A. Intracellular immunity: Finding the enemy within—How cells recognize and respond to intracellular pathogens. J. Leukocyte Biol. 2014, 96, 233–244. [Google Scholar] [CrossRef] [PubMed]
- De Bentzmann, S.; Plotkowski, C.; Puchelle, E. Receptors in the Pseudomonas aeruginosa adherence to injured and repairing airway epithelium. Am. J. Respir. Crit. Care Med. 1996, 154, S155. [Google Scholar] [CrossRef] [PubMed]
- Didierlaurent, A.; Goulding, J.; Hussell, T. The impact of successive infections on the lung microenvironment. Immunology 2007, 122, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.Y.; Phipps, S. Regulation of inducible BALT formation and contribution to immunity and pathology. Mucosal Immunol. 2010, 3, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Sckisel, G.D.; Tietze, J.K.; Zamora, A.E.; Hsiao, H.H.; Priest, S.O.; Wilkins, D.E.; Lanier, L.L.; Blazar, B.R.; Baumgarth, N.; Murphy, W.J. Influenza infection results in local expansion of memory CD8+ T cells with antigen non-specific phenotype and function. Clin. Exp. Immunol. 2014, 175, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Misiak, A.; Wilk, M.M.; Raverdeau, M.; Mills, K.H. IL-17-Producing Innate and Pathogen-Specific Tissue Resident Memory gammadelta T Cells Expand in the Lungs of Bordetella pertussis-Infected Mice. J. Immunol. 2017, 198, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, S.; Schlums, H.; Gallais Serezal, I.; Martini, E.; Chiang, S.C.; Marquardt, N.; Gibbs, A.; Detlofsson, E.; Introini, A.; Forkel, M.; et al. CD49a Expression Defines Tissue-Resident CD8+ T Cells Poised for Cytotoxic Function in Human Skin. Immunity 2017, 46, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Hamerman, J.A.; Ogasawara, K.; Lanier, L.L. NK cells in innate immunity. Curr. Opin. Immunol. 2005, 17, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Becknell, B.; Caligiuri, M.A. Natural killer cells in innate immunity and cancer. J. Immunother. 2008, 31, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Guerra, N. Oncogenic stress sensed by the immune system: Role of natural killer cell receptors. Nat. Rev. Immunol. 2009, 9, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Schuster, I.S.; Coudert, J.D.; Andoniou, C.E.; Degli-Esposti, M.A. “Natural Regulators”: NK Cells as Modulators of T Cell Immunity. Front. Immunol. 2016, 7, 235. [Google Scholar] [CrossRef] [PubMed]
- Deitz, S.B.; Dodd, D.A.; Cooper, S.; Parham, P.; Kirkegaard, K. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. USA 2000, 97, 13790–13795. [Google Scholar] [CrossRef] [PubMed]
- Moffat, K.; Knox, C.; Howell, G.; Clark, S.J.; Yang, H.; Belsham, G.J.; Ryan, M.; Wileman, T. Inhibition of the secretory pathway by foot-and-mouth disease virus 2BC protein is reproduced by coexpression of 2B with 2C, and the site of inhibition is determined by the subcellular location of 2C. J. Virol. 2007, 81, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.L.; Morris, C.R.; Solheim, J.C. Virus evasion of MHC class I molecule presentation. J. Immunol. 2003, 171, 4473–4478. [Google Scholar] [CrossRef] [PubMed]
- Swann, S.A.; Williams, M.; Story, C.M.; Bobbitt, K.R.; Fleis, R.; Collins, K.L. HIV-1 Nef blocks transport of MHC class I molecules to the cell surface via a PI 3-kinase-dependent pathway. Virology 2001, 282, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Halenius, A.; Gerke, C.; Hengel, H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: So many targets-but how many arrows in the quiver? Cell. Mol. Immunol. 2015, 12, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Goulder, P.J.; Walker, B.D. HIV and HLA class I: An evolving relationship. Immunity 2012, 37, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Apps, R.; Del Prete, G.Q.; Chatterjee, P.; Lara, A.; Brumme, Z.L.; Brockman, M.A.; Neil, S.; Pickering, S.; Schneider, D.K.; Piechocka-Trocha, A.; et al. HIV-1 Vpu Mediates HLA-C Downregulation. Cell Host Microbe 2016, 19, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cheng, M.; Tian, Z. Hepatitis B virus down-regulates expressions of MHC class I molecules on hepatoplastoma cell line. Cell. Mol. Immunol. 2006, 3, 373–378. [Google Scholar] [PubMed]
- Noriega, V.M.; Hesse, J.; Gardner, T.J.; Besold, K.; Plachter, B.; Tortorella, D. Human cytomegalovirus US3 modulates destruction of MHC class I molecules. Mol. Immunol. 2012, 51, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Roder, G.; Geironson, L.; Bressendorff, I.; Paulsson, K. Viral proteins interfering with antigen presentation target the major histocompatibility complex class I peptide-loading complex. J. Virol. 2008, 82, 8246–8252. [Google Scholar] [CrossRef] [PubMed]
- Rosato, P.C.; Leib, D.A. Neurons versus herpes simplex virus: The innate immune interactions that contribute to a host-pathogen standoff. Future Virol. 2015, 10, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.C.; Wang, S.Y.; et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.W.; Carvalho, A.; La Rocca, C.; Palma, C.; Rodrigues, F.; Silvestre, R.; Kleinnijenhuis, J.; Lachmandas, E.; Goncalves, L.G.; Belinha, A.; et al. Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep. 2016, 17, 2562–2571. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Elliott, J.M.; Keyel, P.A.; Yang, L.; Carrero, J.A.; Yokoyama, W.M. Cytokine-induced memory-like natural killer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Askenase, M.H.; Han, S.J.; Byrd, A.L.; Morais da Fonseca, D.; Bouladoux, N.; Wilhelm, C.; Konkel, J.E.; Hand, T.W.; Lacerda-Queiroz, N.; Su, X.Z.; et al. Bone-Marrow-Resident NK Cells Prime Monocytes for Regulatory Function during Infection. Immunity 2015, 42, 1130–1142. [Google Scholar] [CrossRef] [PubMed]
- Quintin, J.; Saeed, S.; Martens, J.H.A.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.J.; Wijmenga, C.; et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef] [PubMed]
- White, K.A.; Enjuanes, L.; Berkhout, B.; Enjuanes, L.; Berkhout, B.; Andrew, K. RNA virus replication, transcription and recombination. RNA Biol. 2011, 8, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009, 227, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Wang, Y.Y. Innate immune responses to DNA viruses. Protein Cell 2013, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.; Thomsen, A.R. Sensing of RNA Viruses: A Review of Innate Immune Receptors Involved in Recognizing RNA Virus Invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Hu, J.; Chen, J.L. lncRNAs regulate the innate immune response to viral infection. Wiley Interdiscip. Rev. RNA 2016, 7, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ouyang, J.; Wei, J.; Maarouf, M.; Chen, J.L. Involvement of Host Non-Coding RNAs in the Pathogenesis of the Influenza Virus. Int. J. Mol. Sci. 2016, 18, 39. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Zhu, X.; Chen, Y.; Wei, H.; Chen, Q.; Chi, X.; Qi, B.; Zhang, L.; Zhao, Y.; Gao, G.F.; et al. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe 2014, 16, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.K.; Kotenko, S.V.; Durbin, J.E. Interferon induction and function at the mucosal surface. Immunol. Rev. 2013, 255, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chi, X.; Wei, H.; Chen, Y.; Chen, Z.; Huang, S.; Chen, J.L. Influenza A virus-induced degradation of eukaryotic translation initiation factor 4B contributes to viral replication by suppressing IFITM3 protein expression. J. Virol. 2014, 88, 8375–8385. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, X.; Satoh, T.; Gentili, M.; Cerboni, S.; Conrad, C.; Hurbain, I.; El Marjou, A.; Lacabaratz, C.; Lelievre, J.D.; Manel, N. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity 2013, 39, 1132–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Oak, S.; Kodys, K.; Golenbock, D.T.; Finberg, R.W.; Kurt-Jones, E.; Szabo, G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 2004, 127, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Fiola, S.; Gosselin, D.; Takada, K.; Gosselin, J. TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J. Immunol. 2010, 185, 3620–3631. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.S.; Lum, K.K.; Sheng, X.; Song, B.; Cristea, I.M. Diverse mechanisms evolved by DNA viruses to inhibit early host defenses. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 452–481. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Desmet, C.J.; Ishii, K.J. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat. Rev. Immunol. 2012, 12, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.-I.; Okabayashi, T.; Yokosawa, N.; Fujii, N. Measles virus P protein suppresses Toll-like receptor signal through up-regulation of ubiquitin-modifying enzyme A20. FASEB J. 2008, 22, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- Breiman, A.; Grandvaux, N.; Lin, R.; Ottone, C.; Akira, S.; Yoneyama, M.; Fujita, T.; Hiscott, J.; Meurs, E.F. Inhibition of RIG-I-dependent signaling to the interferon pathway during hepatitis C virus expression and restoration of signaling by IKKepsilon. J. Virol. 2005, 79, 3969–3978. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Kumar, D.; Gumma, P.K.; Chowdhury, S.J.; Karra, V.K. Down regulation of TRIF, TLR3, and MAVS in HCV infected liver correlates with the outcome of infection. J. Med. Virol. 2017, 89, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Nagendraprabhu, P.; Khatiwada, S.; Chaulagain, S.; Delhon, G.; Rock, D.L. A parapoxviral virion protein targets the retinoblastoma protein to inhibit NF-kappaB signaling. PLoS Pathog. 2017, 13, e1006779. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Ishii, K.J.; Coban, C.; Akira, S. Innate immune response to viral infection. Cytokine 2008, 43, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Finberg, R.W.; Wang, J.P.; Kurt-Jones, E.A. Toll like receptors and viruses. Rev. Med. Virol. 2007, 17, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Rassa, J.C.; Meyers, J.L.; Zhang, Y.; Kudaravalli, R.; Ross, S.R. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. USA 2002, 99, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Bieback, K.; Lien, E.; Klagge, I.M.; Avota, E.; Schneider-Schaulies, J.; Duprex, W.P.; Wagner, H.; Kirschning, C.J.; Ter Meulen, V.; Schneider-Schaulies, S. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 2002, 76, 8729–8736. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Chan, M.; Zhou, S.; Wang, J.; Reed, G.; Bronson, R.; Arnold, M.M.; Knipe, D.M.; Finberg, R.W. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 2004, 101, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, U.; von dem Bussche, A.; Kirschning, C.; Miyake, K.; Sauerbruch, T.; Spengler, U. Cell activation by synthetic lipopeptides of the hepatitis C virus (HCV)—Core protein is mediated by toll like receptors (TLRs) 2 and 4. Immunol. Lett. 2002, 84, 89–95. [Google Scholar] [CrossRef]
- Cheng, G.; Zhong, J.; Chung, J.; Chisari, F.V. Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc. Natl. Acad. Sci. USA 2007, 104, 9035–9040. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Valentine, R.; Smith, G.L. Inhibition of the RNA polymerase III-mediated dsDNA-sensing pathway of innate immunity by vaccinia virus protein E3. J. Gen. Virol. 2010, 91, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.R.; Bruns, A.M.; Horvath, C.M. MDA5 and LGP2: Accomplices and Antagonists of Antiviral Signal Transduction. J. Virol. 2014, 88, 8194–8200. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic Sensing of Viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Gack, M.U. RIG-I-like receptor regulation in virus infection and immunity. Curr. Opin. Virol. 2015, 12, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chen, L.M.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007, 36, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Ly, H.; Liang, Y. The Z proteins of pathogenic but not nonpathogenic arenaviruses inhibit RIG-I-like receptor-dependent interferon production. J. Virol. 2015, 89, 2944–2955. [Google Scholar] [CrossRef] [PubMed]
- Biacchesi, S.; Merour, E.; Chevret, D.; Lamoureux, A.; Bernard, J.; Bremont, M. NV Proteins of Fish Novirhabdovirus Recruit Cellular PPM1Bb Protein Phosphatase and Antagonize RIG-I-Mediated IFN Induction. Sci. Rep. 2017, 7, 44025. [Google Scholar] [CrossRef] [PubMed]
- Childs, K.S.; Andrejeva, J.; Randall, R.E.; Goodbourn, S. Mechanism of mda-5 Inhibition by Paramyxovirus V Proteins. J. Virol. 2009, 83, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Childs, K.; Randall, R.; Goodbourn, S. Paramyxovirus V Proteins Interact with the RNA Helicase LGP2 to Inhibit RIG-I-Dependent Interferon Induction. J. Virol. 2012, 86, 3411–3421. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Xu, S.; Wang, J.; Luo, R.; Wang, D.; Xiao, S.; Fang, L.; Chen, H.; Jiang, Y. Porcine reproductive and respiratory syndrome virus 3C protease cleaves the mitochondrial antiviral signalling complex to antagonize IFN-beta expression. J. Gen. Virol. 2015, 96, 3049–3058. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Chen, S.O.; Chang, S.P.; Lee, Y.P.; Yu, C.K.; Chen, C.L.; Tseng, P.C.; Hsieh, C.Y.; Chen, S.H.; Lin, C.F. Enterovirus 71 Proteins 2A and 3D Antagonize the Antiviral Activity of Gamma Interferon via Signaling Attenuation. J. Virol. 2015, 89, 7028–7037. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Su, J.; Wang, H.; Chang, J.; Wang, S.; Zheng, W.; Cai, Y.; Wei, W.; Gordy, J.T.; Markham, R.; et al. Disruption of MDA5-Mediated Innate Immune Responses by the 3C Proteins of Coxsackievirus A16, Coxsackievirus A6, and Enterovirus D68. J. Virol. 2017, 91, e00546-17. [Google Scholar] [CrossRef] [PubMed]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.Y.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β Production through the NLRP3 Inflammasome by Hepatic Macrophages Links Hepatitis C Virus Infection with Liver Inflammation and Disease. PLoS Pathog. 2013, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-C.; Jin, Y.; Zhi, X.-Y.; Yan, D.; Sun, S.-Q. NLRP3 Inflammasome Activation by Viroporins of Animal Viruses. Viruses 2015, 7, 3380–3391. [Google Scholar] [CrossRef] [PubMed]
- Cheong, W.-C.; Kang, H.-R.; Yoon, H.; Kang, S.-J.; Ting, J.P.-Y.; Song, M.J. Influenza A Virus NS1 Protein Inhibits the NLRP3 Inflammasome. PLoS ONE 2015, 10, e0126456. [Google Scholar] [CrossRef] [PubMed]
- Komune, N.; Ichinohe, T.; Ito, M.; Yanagi, Y. Measles Virus V Protein Inhibits NLRP3 Inflammasome-Mediated Interleukin-1 Secretion. J. Virol. 2011, 85, 13019–13026. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Ma, Y.; Wang, L.; Chi, X.; Yan, R.; Wang, S.; Li, X.; Chen, X.; Shao, W.; Chen, J.L. Alpha/beta interferon receptor deficiency in mice significantly enhances susceptibility of the animals to pseudorabies virus infection. Vet. Microbiol. 2017, 203, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.C.; Young, H.A. Interferons: Success in anti-viral immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, S.; Chen, Q.; Chen, Y.; Chi, X.; Zhang, L.; Huang, S.; Gao, G.F.; Chen, J.L. Correction: Suppression of Interferon Lambda Signaling by SOCS-1 Results in Their Excessive Production during Influenza Virus Infection. PLoS Pathog. 2016, 12, e1005402. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, M.; Veron, L.; Stempor, P.; Ahringer, J.; Sarkies, P.; Miska, E.A. An Alternative STAT Signaling Pathway Acts in Viral Immunity in Caenorhabditis elegans. mBio 2017, 8, e00924-17. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O’Shea, J.J. Mechanisms of Jak/STAT Signaling in Immunity and Disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.; Dickensheets, H.L.; Spann, K.M.; Alston, M.A.; Luongo, C.; Dumoutier, L.; Huang, J.; Renauld, J.C.; Kotenko, S.V.; Roederer, M.; et al. Alpha and lambda interferon together mediate suppression of CD4 T cells induced by respiratory syncytial virus. J. Virol. 2006, 80, 5032–5040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W. Interferon-stimulated genes: Roles in viral pathogenesis. Curr. Opin. Virol. 2014, 6, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Zang, T.M.; Rihn, S.J.; Zhang, F.; Kueck, T.; Alim, M.; Schoggins, J.; Rice, C.M.; Wilson, S.J.; Bieniasz, P.D. Identification of Interferon-Stimulated Genes with Antiretroviral Activity. Cell Host Microbe 2016, 20, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chen, Y.; Zhang, Z.; Ouyang, J.; Wang, Y.; Yan, R.; Huang, S.; Gao, G.F.; Guo, G.; Chen, J.L. Robust expression of vault RNAs induced by influenza A virus plays a critical role in suppression of PKR-mediated innate immunity. Nucleic Acids Res. 2015, 43, 10321–10337. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.A.; Patton, J.T. Shutdown of interferon signaling by a viral-hijacked E3 ubiquitin ligase. Microb. Cell 2017, 4, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, L.N.; Benveniste, E.N. Viral Exploitation of Host SOCS Protein Functions. J. Virol. 2011, 85, 1912–1921. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Moss, B. Vaccinia Virus C9 Ankyrin Repeat/F-Box Protein Is a Newly Identified Antagonist of the Type I Interferon-Induced Antiviral State. J. Virol. 2018, 92, e00053-18. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.W.; Kim, D.; Jung, J.U.; Lee, H.R. KSHV-encoded viral interferon regulatory factor 4 (vIRF4) interacts with IRF7 and inhibits interferon alpha production. Biochem. Biophys. Res. Commun. 2017, 486, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Gottipati, K.; Holthauzen, L.M.; Ruggli, N.; Choi, K.H. Pestivirus Npro Directly Interacts with Interferon Regulatory Factor 3 Monomer and Dimer. J. Virol. 2016, 90, 7740–7747. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef] [PubMed]
- Ziehr, B.; Vincent, H.A.; Moorman, N.J. Human Cytomegalovirus pTRS1 and pIRS1 Antagonize Protein Kinase R to Facilitate Virus Replication. J. Virol. 2016, 90, 3839–3848. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, P.W.; Vaidya, S.A.; Cheng, G. The art of war: Innate and adaptive immune responses. Cell. Mol. Life Sci. 2003, 60, 2604–2621. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.L. How do natural killer T cells help B cells? Expert Rev. Vaccines 2009, 8, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N.; Gebhardt, T.; Carbone, F.R.; Heath, W.R. Memory T cell subsets, migration patterns, and tissue residence. Annu. Rev. Immunol. 2013, 31, 137–161. [Google Scholar] [CrossRef] [PubMed]
- Clem, A.S. Fundamentals of vaccine immunology. J. Glob. Infect. Dis. 2011, 3, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Okoye, A.A.; Picker, L.J. CD4+ T-cell depletion in HIV infection: Mechanisms of immunological failure. Immunol. Rev. 2013, 254, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E. The Immune Response in Measles: Virus Control, Clearance and Protective Immunity. Viruses 2016, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Hatton, O.L.; Harris-Arnold, A.; Schaffert, S.; Krams, S.M.; Martinez, O.M. The interplay between Epstein-Barr virus and B lymphocytes: Implications for infection, immunity, and disease. Immunol. Res. 2014, 58, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Gossmann, J.; Lohler, J.; Lehmann-Grube, F. Entry of antivirally active T lymphocytes into the thymus of virus-infected mice. J. Immunol. 1991, 146, 293–297. [Google Scholar] [PubMed]
- Yoshimura, F.K.; Wang, T.; Cankovic, M. Sequences between the enhancer and promoter in the long terminal repeat affect murine leukemia virus pathogenicity and replication in the thymus. J. Virol. 1999, 73, 4890–4898. [Google Scholar] [PubMed]
- Kennedy, P.T.F.; Litwin, S.; Dolman, G.E.; Bertoletti, A.; Mason, W.S. Immune Tolerant Chronic Hepatitis B: The Unrecognized Risks. Viruses 2017, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T. Immune Tolerant Hepatitis B: A Clinical Dilemma. Gastroenterol. Hepatol. 2011, 7, 511–516. [Google Scholar]
- Sukriti, S.; Pati, N.T.; Bose, S.; Hissar, S.S.; Sarin, S.K. Impaired antigen processing and presentation machinery is associated with immunotolerant state in chronic hepatitis B virus infection. J. Clin. Immunol. 2010, 30, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Carey, I.; D’Antiga, L.; Bansal, S.; Longhi, M.S.; Ma, Y.; Mesa, I.R.; Mieli-Vergani, G.; Vergani, D. Immune and viral profile from tolerance to hepatitis B surface antigen clearance: A longitudinal study of vertically hepatitis B virus-infected children on combined therapy. J. Virol. 2011, 85, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.N.; Kuo, C.F.; Ou, J.J. Mechanisms of Hepatitis B Virus Persistence. Trends Microbiol. 2018, 26, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Golovkina, T. Common threads in persistent viral infections. J. Virol. 2010, 84, 4116–4123. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, M.P.; Proenca, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Conrad, R.J.; Ott, M. Retrovirus Integration: Some Assembly Required? Cell Host Microbe 2016, 20, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Grebely, J.; Prins, M.; Hellard, M.; Cox, A.L.; Osburn, W.O.; Lauer, G.; Page, K.; Lloyd, A.R.; Dore, G.J. Hepatitis C virus clearance, reinfection, and persistence, with insights from studies of injecting drug users: Towards a vaccine. Lancet Infect. Dis. 2012, 12, 408–414. [Google Scholar] [CrossRef]
- Knipe, D.M.; Howley, P.M. Fields Virology, 6th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields Virology, 5th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Moser, J.M.; Lukacher, A.E. Immunity to polyoma virus infection and tumorigenesis. Viral Immunol. 2001, 14, 199–216. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maarouf, M.; Rai, K.R.; Goraya, M.U.; Chen, J.-L. Immune Ecosystem of Virus-Infected Host Tissues. Int. J. Mol. Sci. 2018, 19, 1379. https://doi.org/10.3390/ijms19051379
Maarouf M, Rai KR, Goraya MU, Chen J-L. Immune Ecosystem of Virus-Infected Host Tissues. International Journal of Molecular Sciences. 2018; 19(5):1379. https://doi.org/10.3390/ijms19051379
Chicago/Turabian StyleMaarouf, Mohamed, Kul Raj Rai, Mohsan Ullah Goraya, and Ji-Long Chen. 2018. "Immune Ecosystem of Virus-Infected Host Tissues" International Journal of Molecular Sciences 19, no. 5: 1379. https://doi.org/10.3390/ijms19051379
APA StyleMaarouf, M., Rai, K. R., Goraya, M. U., & Chen, J.-L. (2018). Immune Ecosystem of Virus-Infected Host Tissues. International Journal of Molecular Sciences, 19(5), 1379. https://doi.org/10.3390/ijms19051379