Polarised VEGFA Signalling at Vascular Blood–Neural Barriers



Abstract
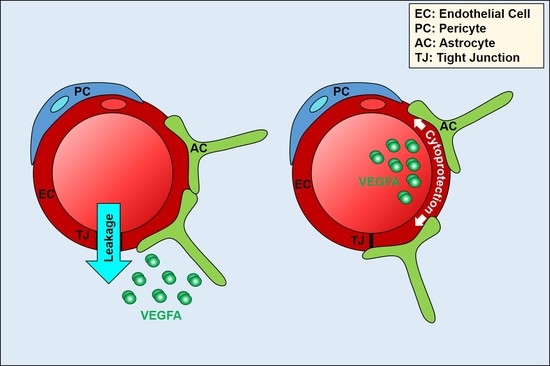
:
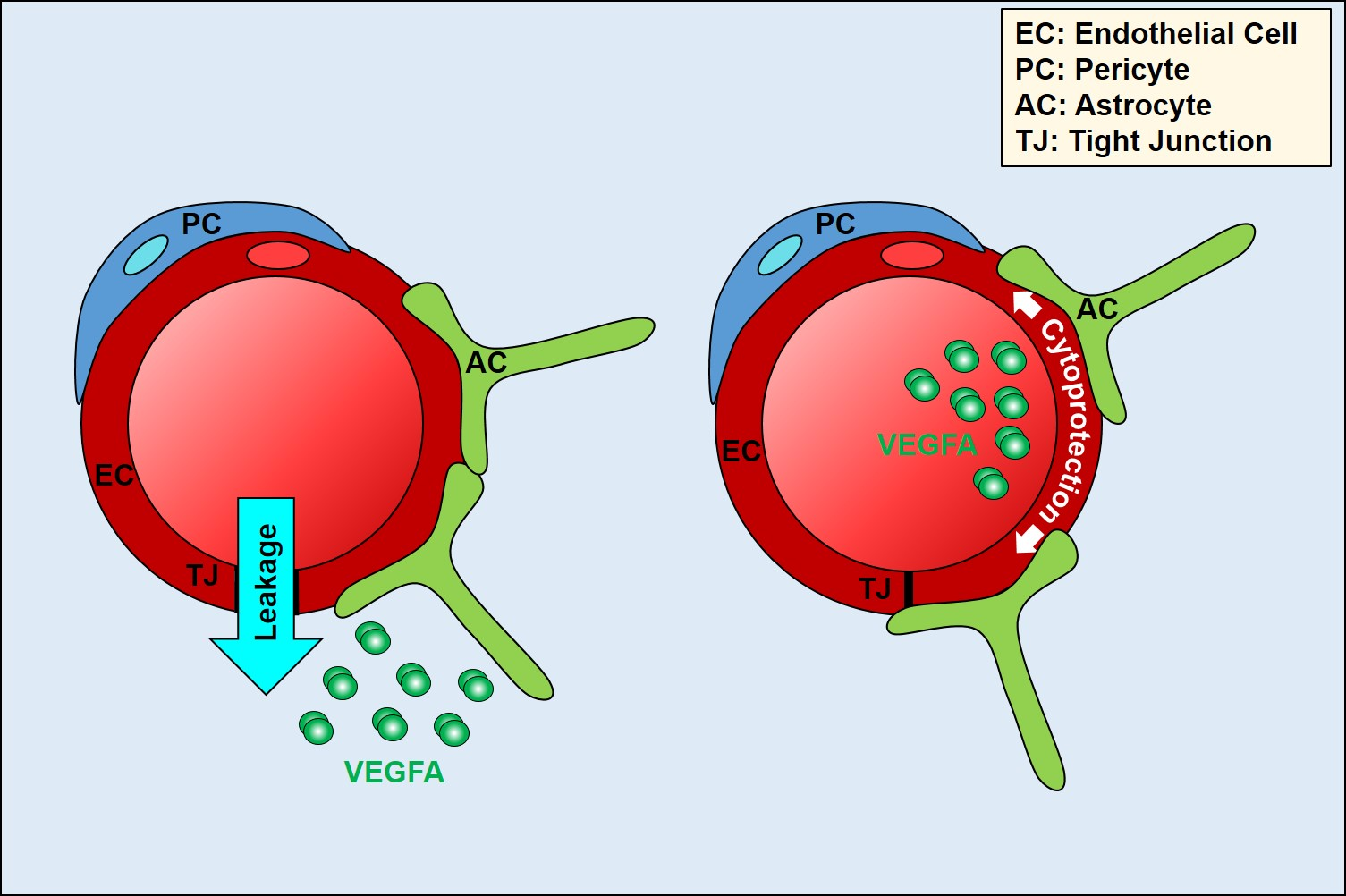{kind=link}
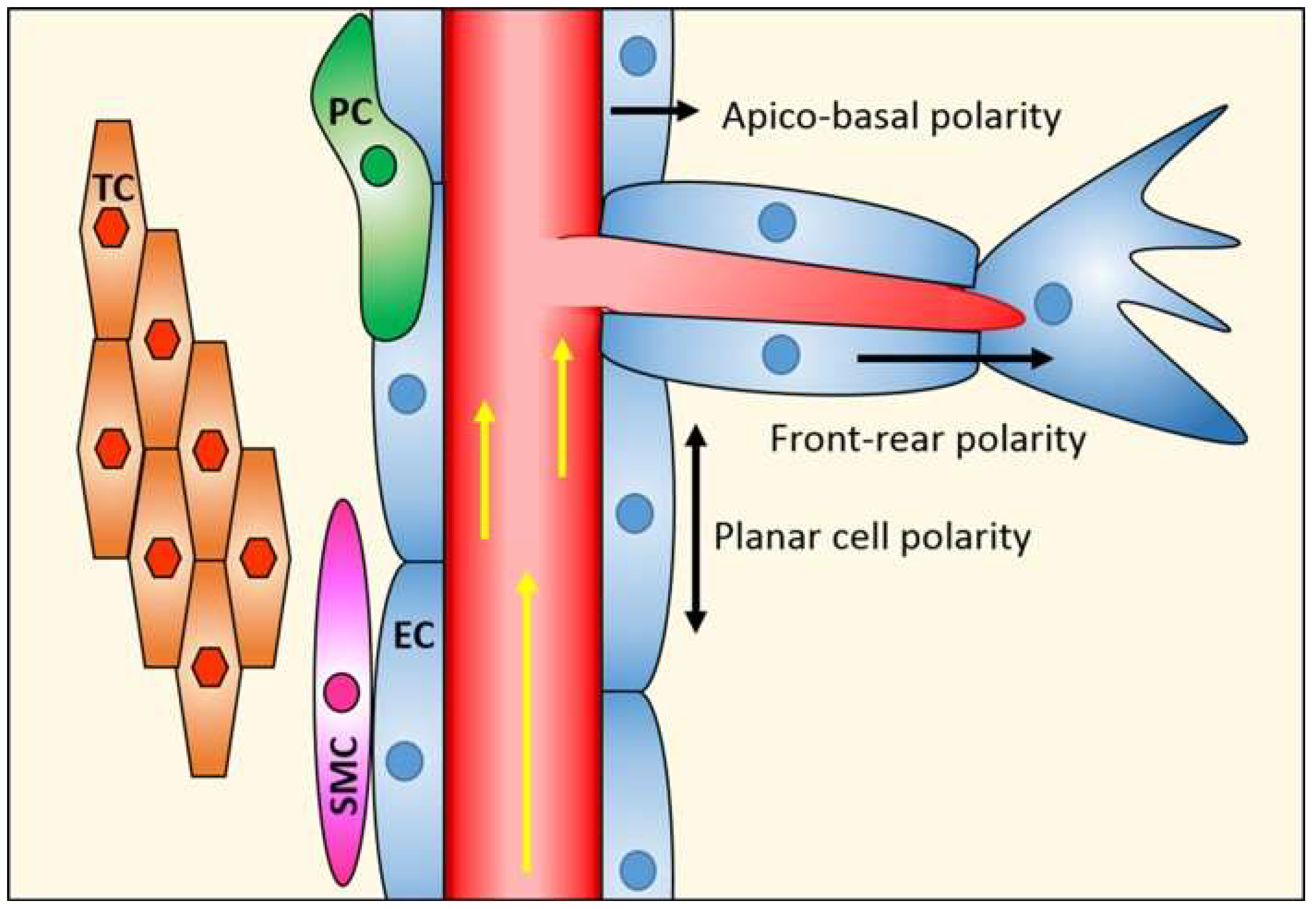{kind=link}
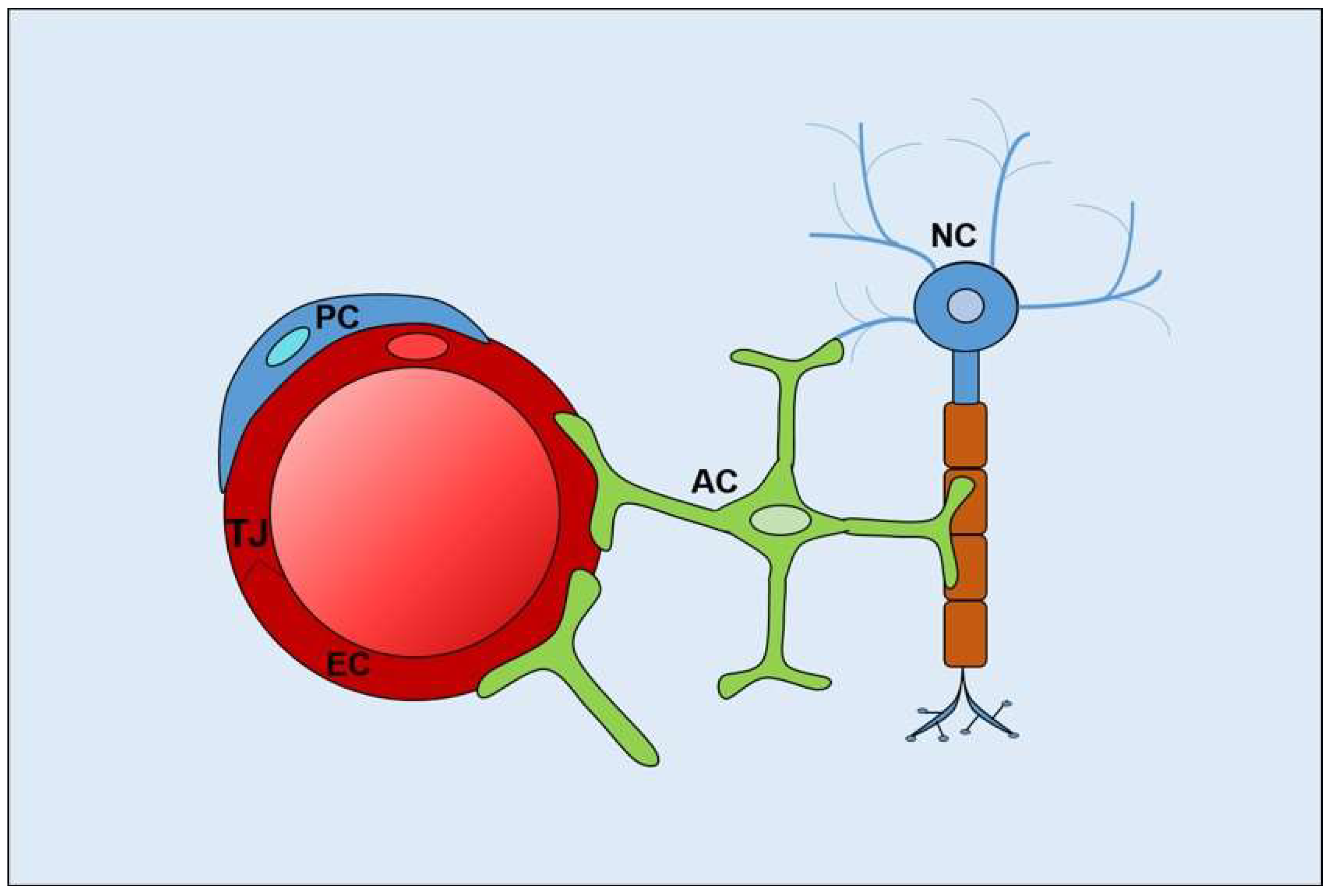{kind=link}
{kind=link}
1. Endothelial Cell Polarity
2. Polarity at Blood–Brain and Blood–Retinal Barriers
3. Vascular Endothelial Growth Factor Signalling in Endothelial Cells
4. Polarised VEGFA Signalling at Blood–Neural Barriers
5. Polarised Signalling at the BBB/BRB by Factors Other than VEGF
6. Conclusions and Future Perspectives
Acknowledgments
References
- Lee, J.L.; Streuli, C.H. Integrins and epithelial cell polarity. J. Cell Sci. 2014, 127, 3217–3225. [Google Scholar] [CrossRef] [PubMed]
- Ladoux, B.; Mège, R.M.; Trepat, X. Front-rear polarization by mechanical cues from single cells to tissues. Trends Cell Biol. 2016, 26, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Mack, N.A.; Georgiou, M. The interdependence of the Rho GTPases and apicobasal cell polarity. Small GTPases 2014, 5, e973768. [Google Scholar] [CrossRef] [PubMed]
- Ebnet, K.; Kummer, D.; Steinbacher, T.; Singh, A.; Nakayama, M.; Matis, M. Regulation of cell polarity by cell adhesion receptors. Semin. Cell Dev. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- St Johnston, D.; Sanson, B. Epithelial polarity and morphogenesis. Curr. Opin. Cell Biol. 2011, 23, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Boulan, E.; Macara, I.G. Organization and execution of the epithelial polarity programme. Nat. Rev. Mol. Cell Biol. 2014, 15, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Lizama, C.O.; Zovein, A.C. Polarizing pathways balancing endothelial polarity. Exp. Cell Res. 2013, 319, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Bautch, V.L. Ups and downs of guided vessel sprouting: The role of polarity. Physiology 2011, 26, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Carman, C.V.; Martinelli, R. T Lymphocyte-Endothelial Interactions: Emerging Understanding of Trafficking and Antigen-Specific Immunity. Front. Immunol. 2015, 6, 603. [Google Scholar] [CrossRef] [PubMed]
- Wacker, A.; Gerhardt, H.; Phng, L.K. Tissue guidance without filopodia. Commun. Integr. Biol. 2014, 7, e28820. [Google Scholar] [CrossRef] [PubMed]
- Halaoui, R.; McCaffrey, L. Rewiring cell polarity signaling in cancer. Oncogene 2015, 34, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D. Establishing and transducing cell polarity: Common themes and variations. Curr. Opin. Cell Biol. 2017, 51, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Worzfeld, T.; Schwaninger, M. Apicobasal polarity of brain endothelial cells. J. Cereb. Blood Flow Metab. 2016, 36, 340–362. [Google Scholar] [CrossRef] [PubMed]
- Bendayan, R.; Ronaldson, P.T.; Gingras, D.; Bendayan, M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J. Histochem. Cytochem. 2006, 54, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.; Melendez, A.J. Role of cell adhesion molecules and immune-cell migration in the initiation, onset and development of atherosclerosis. Cell Adh. Migr. 2007, 1, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; Oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.L.; Fine, R.E.; Sandra, A. Receptor-mediated endocytosis of transferrin at the blood-brain barrier. J. Cell Sci. 1993, 104, 521–532. [Google Scholar] [PubMed]
- Lopes da Silva, M.; Cutler, D.F. von Willebrand factor multimerization and the polarity of secretory pathways in endothelial cells. Blood 2016, 128, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R. The blood-brain barrier in health and disease. Ann. Neurol. 2012, 72, 648–672. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Matter, K. Tight junctions as regulators of tissue remodelling. Curr. Opin. Cell Biol. 2016, 42, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Tewes, B.J.; Galla, H.J. Lipid polarity in brain capillary endothelial cells. Endothelium 2001, 8, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.R.; Daneman, R.; Dziegielewska, K.M.; Liddelow, S.A. Transporters of the blood-brain and blood-CSF interfaces in development and in the adult. Mol. Aspects Med. 2013, 34, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, E.; Demeule, M.; Ghitescu, L.; Béliveau, R. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem. J. 1997, 326 Pt 2, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Bernacki, J.; Dobrowolska, A.; Nierwińska, K.; Małecki, A. Physiology and pharmacological role of the blood-brain barrier. Pharmacol. Rep. 2008, 60, 600–622. [Google Scholar] [PubMed]
- Gao, B.; Stieger, B.; Noé, B.; Fritschy, J.M.; Meier, P.J. Localization of the organic anion transporting polypeptide 2 (Oatp2) in capillary endothelium and choroid plexus epithelium of rat brain. J. Histochem. Cytochem. 1999, 47, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Zovein, A.C.; Luque, A.; Turlo, K.A.; Hofmann, J.J.; Yee, K.M.; Becker, M.S.; Fassler, R.; Mellman, I.; Lane, T.F.; Iruela-Arispe, M.L. β1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev. Cell 2010, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Ngok, S.P.; Geyer, R.; Liu, M.; Kourtidis, A.; Agrawal, S.; Wu, C.; Seerapu, H.R.; Lewis-Tuffin, L.J.; Moodie, K.L.; Huveldt, D.; et al. VEGF and Angiopoietin-1 exert opposing effects on cell junctions by regulating the Rho GEF Syx. J. Cell Biol. 2012, 199, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M.; et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Rigor, R.R.; Pivetti, C.D.; Wu, M.H.; Yuan, S.Y. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc. Res. 2010, 87, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Persidsky, Y.; Heilman, D.; Haorah, J.; Zelivyanskaya, M.; Persidsky, R.; Weber, G.A.; Shimokawa, H.; Kaibuchi, K.; Ikezu, T. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE). Blood 2006, 107, 4770–4780. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Sladojevic, N.; Keep, R.F.; Andjelkovic, A.V. Relocalization of junctional adhesion molecule A during inflammatory stimulation of brain endothelial cells. Mol. Cell. Biol. 2012, 32, 3414–3427. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.; Mahan, R.D.; Davis, G.E. Cdc42- and Rac1-mediated endothelial lumen formation requires Pak2, Pak4 and Par3, and PKC-dependent signaling. J. Cell Sci. 2008, 121 Pt 7, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 2014, 509, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.; Silver, D.L. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.H.; Chan, J.P.; Cazenave-Gassiot, A.; Poh, R.W.; Foo, J.C.; Galam, D.L.; Ghosh, S.; Nguyen, L.N.; Barathi, V.A.; Yeo, S.W.; et al. Mfsd2a Is a Transporter for the Essential ω-3 Fatty Acid Docosahexaenoic Acid (DHA) in Eye and Is Important for Photoreceptor Cell Development. J. Biol. Chem. 2016, 291, 10501–10514. [Google Scholar] [CrossRef] [PubMed]
- Betsholtz, C. Physiology: Double function at the blood-brain barrier. Nature 2014, 509, 432–433. [Google Scholar] [CrossRef] [PubMed]
- Quek, D.Q.; Nguyen, L.N.; Fan, H.; Silver, D.L. Structural Insights into the Transport Mechanism of the Human Sodium-dependent Lysophosphatidylcholine Transporter MFSD2A. J. Biol. Chem. 2016, 291, 9383–9394. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Kenny, B.A.; Prise, V.; Glenn, J.; Sarker, M.H.; Hudson, N.; Brandt, M.; Lopez, F.J.; Gale, D.; Luthert, P.J.; et al. Lipoprotein-associated phospholipase A2 (Lp-PLA2) as a therapeutic target to prevent retinal vasopermeability during diabetes. Proc. Natl. Acad. Sci. USA 2016, 113, 7213–7218. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L. VEGF receptor signal transduction—A brief update. Vasc. Pharmacol. 2016, 86, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Andersson, C.; Roomans, G.M.; Ito, N.; Claesson-Welsh, L. Signaling properties of VEGF receptor-1 and -2 homo- and heterodimers. Int. J. Biochem. Cell Biol. 2001, 33, 315–324. [Google Scholar] [CrossRef]
- Matsumoto, K.; Ema, M. Roles of VEGF-A signalling in development, regeneration, and tumours. J. Biochem. 2014, 156, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signaling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Nakayama, A.; van Lessen, M.; Yamamoto, H.; Hoffmann, S.; Drexler, H.C.; Itoh, N.; Hirose, T.; Breier, G.; Vestweber, D.; et al. Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat. Cell Biol. 2013, 15, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.M.; Ziyad, S.; Briot, A.; Der, A.; Iruela-Arispe, M.L. A ligand-independent VEGFR2 signaling pathway limits angiogenic responses in diabetes. Sci. Signal 2014, 7, ra1. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Zarkada, G.; Nurmi, H.; Jakobsson, L.; Heinolainen, K.; Tvorogov, D.; Zheng, W.; Franco, C.A.; Murtomäki, A.; Aranda, E.; et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat. Cell Biol. 2011, 13, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Galvagni, F.; Pennacchini, S.; Salameh, A.; Rocchigiani, M.; Neri, F.; Orlandini, M.; Petraglia, F.; Gotta, S.; Sardone, G.L.; Matteucci, G.; et al. Endothelial cell adhesion to the extracellular matrix induces c-Src-dependent VEGFR-3 phosphorylation without the activation of the receptor intrinsic kinase activity. Circ. Res. 2010, 106, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Dixelius, J.; Makinen, T.; Wirzenius, M.; Karkkainen, M.J.; Wernstedt, C.; Alitalo, K.; Claesson-Welsh, L. Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. J. Biol. Chem. 2003, 278, 40973–40979. [Google Scholar] [CrossRef] [PubMed]
- Mac Gabhann, F.; Popel, A.S. Dimerization of VEGF receptors and implications for signal transduction: A computational study. Biophys. Chem. 2007, 128, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Imoukhuede, P.I.; Popel, A.S. Quantification and cell-to-cell variation of vascular endothelial growth factor receptors. Exp. Cell Res. 2011, 317, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Cudmore, M.J.; Hewett, P.W.; Ahmad, S.; Wang, K.Q.; Cai, M.; Al-Ani, B.; Fujisawa, T.; Ma, B.; Sissaoui, S.; Ramma, W.; et al. The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis. Nat. Commun. 2012, 3, 972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witmer, A.N.; Vrensen, G.F.; Van Noorden, C.J.; Schlingemann, R.O. Vascular endothelial growth factors and angiogenesis in eye disease. Prog. Retina Eye Res. 2003, 22, 1–29. [Google Scholar] [CrossRef]
- Nagy, J.A.; Benjamin, L.; Zeng, H.; Dvorak, A.M.; Dvorak, H.F. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 2008, 11, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Venema, V.J.; Venema, R.C.; Tsai, N.; Behzadian, M.A.; Caldwell, R.B. VEGF-induced permeability increase is mediated by caveolae. Investig. Ophthalmol. Vis. Sci. 1999, 40, 157–167. [Google Scholar]
- Bates, D.O. Vascular endothelial growth factors and vascular permeability. Cardiovasc. Res. 2010, 87, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Turowski, P. Leakage at the Blood-Brain Barrier, in Blood-Brain Barrier and Inflammation. In Progress in Inflammation Research; Lyck, R., Enzmann, G., Eds.; Springer: Basel, Switzerland, 2017; pp. 81–102. [Google Scholar]
- Hofman, P.; Blaauwgeers, H.G.; Tolentino, M.J.; Adamis, A.P.; Nunes Cardozo, B.J.; Vrensen, G.F.; Schlingemann, R.O. VEGF-A induced hyperpermeability of blood-retinal barrier endothelium in vivo is predominantly associated with pinocytotic vesicular transport and not with formation of fenestrations. Vascular endothelial growth factor-A. Curr. Eye Res. 2000, 21, 637–645. [Google Scholar] [CrossRef]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Goddard, L.M.; Iruela-Arispe, M.L. Cellular and molecular regulation of vascular permeability. Thromb. Haemost. 2013, 109, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Hudson, N.; Powner, M.B.; Sarker, M.H.; Burgoyne, T.; Campbell, M.; Ockrim, Z.K.; Martinelli, R.; Futter, C.E.; Grant, M.B.; Fraser, P.A.; et al. Differential apicobasal VEGF signaling at vascular blood neural barriers. Dev. Cell 2014, 30, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Issbrücker, K.; Marti, H.H.; Hippenstiel, S.; Springmann, G.; Voswinckel, R.; Gaumann, A.; Breier, G.; Drexler, H.C.; Suttorp, N.; Clauss, M. p38 MAP kinase—A molecular switch between VEGF-induced angiogenesis and vascular hyperpermeability. FASEB J. 2003, 17, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L.; Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.H.; Yuan, S.Y.; Granger, H.J. The protein kinase MEK1/2 mediate vascular endothelial growth factor- and histamine-induced hyperpermeability in porcine coronary venules. J. Physiol. 2005, 563 Pt 1, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Breslin, J.W.; Zhu, J.; Yuan, S.Y.; Wu, M.H. Rho and ROCK signaling in VEGF-induced microvascular endothelial hyperpermeability. Microcirculation 2006, 13, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Huang, Q.; Yuan, Y.; Granger, H.J. VEGF induces NO-dependent hyperpermeability in coronary venules. Am. J. Physiol. 1996, 271, H2735–H2739. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Runkle, E.A.; Antonetti, D.A. The blood-retinal barrier: Structure and functional significance. Methods Mol. Biol. 2011, 686, 133–148. [Google Scholar] [PubMed]
- Sarker, M.H.; Hu, D.E.; Fraser, P.A. Regulation of cerebromicrovascular permeability by lysophosphatidic acid. Microcirculation 2010, 17, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Sarker, M.H.; Easton, A.S.; Fraser, P.A. Regulation of cerebral microvascular permeability by histamine in the anaesthetized rat. J. Physiol. 1998, 507 Pt 3, 909–918. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Mallet, C.; Keramidas, M.; Lamandé, N.; Gasc, J.M.; Dupuis-Girod, S.; Plauchu, H.; Feige, J.J.; Bailly, S. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ. Res. 2008, 102, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, P.L.; MacNaughton, D.E.; Clements, R.T.; Minnear, F.L.; Vincent, P.A. p38 MAPK activation by TGF-beta1 increases MLC phosphorylation and endothelial monolayer permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L146–L154. [Google Scholar] [CrossRef] [PubMed]
- Apodaca, G.; Gallo, L.I.; Bryant, D.M. Role of membrane traffic in the generation of epithelial cell asymmetry. Nat. Cell Biol. 2012, 14, 1235–1243. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dragoni, S.; Turowski, P. Polarised VEGFA Signalling at Vascular Blood–Neural Barriers. Int. J. Mol. Sci. 2018, 19, 1378. https://doi.org/10.3390/ijms19051378
Dragoni S, Turowski P. Polarised VEGFA Signalling at Vascular Blood–Neural Barriers. International Journal of Molecular Sciences. 2018; 19(5):1378. https://doi.org/10.3390/ijms19051378
Chicago/Turabian StyleDragoni, Silvia, and Patric Turowski. 2018. "Polarised VEGFA Signalling at Vascular Blood–Neural Barriers" International Journal of Molecular Sciences 19, no. 5: 1378. https://doi.org/10.3390/ijms19051378
APA StyleDragoni, S., & Turowski, P. (2018). Polarised VEGFA Signalling at Vascular Blood–Neural Barriers. International Journal of Molecular Sciences, 19(5), 1378. https://doi.org/10.3390/ijms19051378