SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease

 , ,
, ,
Abstract
:
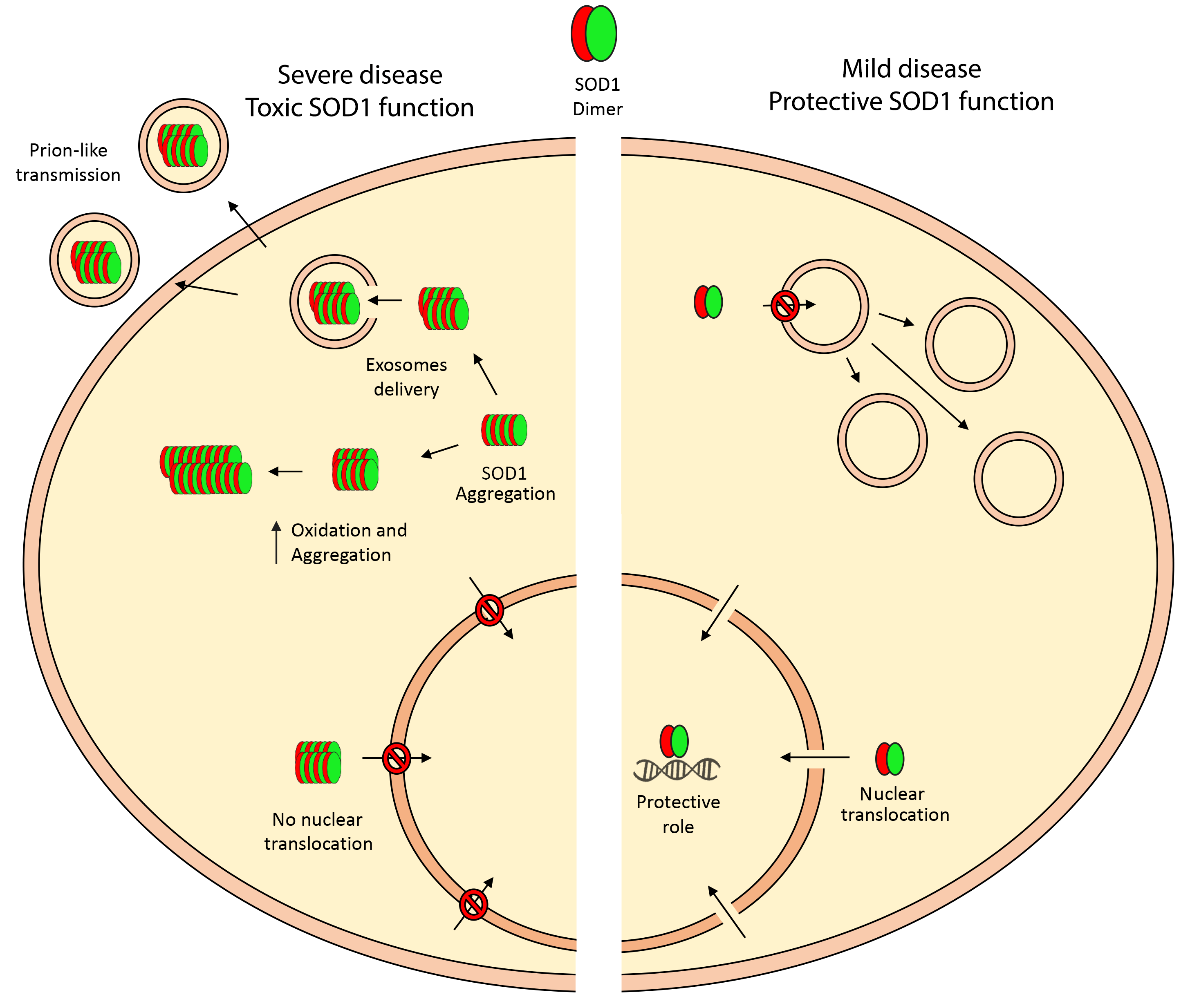{kind=link}
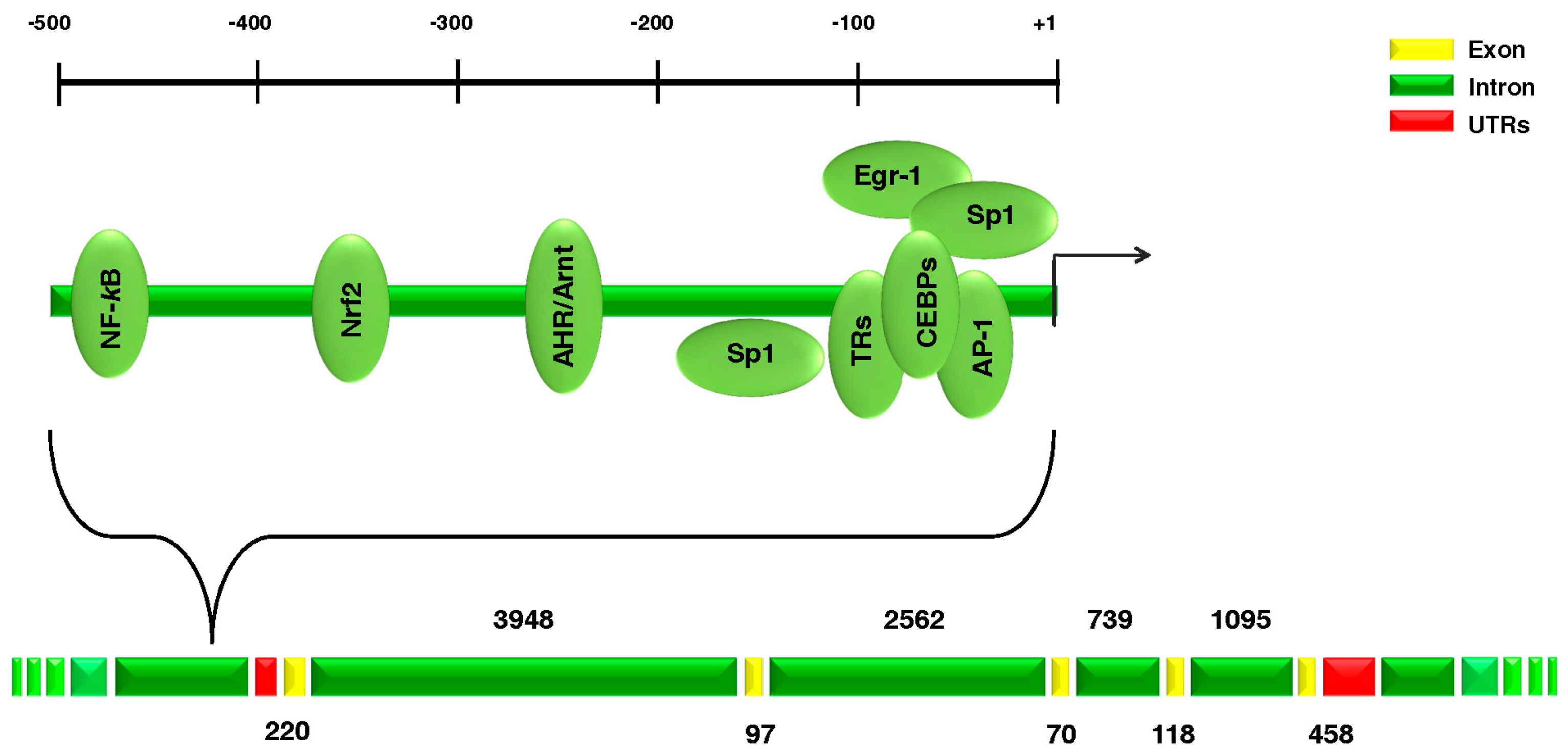{kind=link}
{kind=link}
1. Introduction
2. Superoxide Dismutase 1 (SOD1) Gene
3. SOD1 Protein
4. Transcriptional and Post-Transcriptional Regulation of SOD1
4.1. Trascriptional Regulation
4.2. Post-Trascriptional Regulation
5. Toxic Properties of SOD1
6. New Protective Function of Nuclear SOD1
7. Intercellular Transmission of SOD1
8. SOD1 as a Therapeutic Target in ALS Disease
9. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Brown, R.H., Jr.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 1602. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Traynor, B.J.; Collins, J.; Simeone, J.C.; Goldstein, L.A.; White, L.A. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, N.J.; Zhang, Y.J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.; et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.E. Capturing VCP. Another molecular piece in the ALS jigsaw puzzle. Neuron 2010, 68, 812–814. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.; Fisher, E.M.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136, 2342–2358. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.S.; Hart, P.J. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 3617–3622. [Google Scholar] [CrossRef] [PubMed]
- Reaume, A.G.; Elliott, J.L.; Hoffman, E.K.; Kowall, N.W.; Ferrante, R.J.; Siwek, D.F.; Wilcox, H.M.; Flood, D.G.; Beal, M.F.; Brown, R.H., Jr.; et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 1996, 13, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Tu, P.H.; Raju, P.; Robinson, K.A.; Gurney, M.E.; Trojanowski, J.Q.; Lee, V.M. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc. Natl. Acad. Sci. USA 1996, 93, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- McAlary, L.; Aquilina, J.A.; Yerbury, J.J. Susceptibility of mutant SOD1 to form a destabilized monomer predicts cellular aggregation and toxicity but not in vitro aggregation propensity. Front. Neurosci. 2016, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Doucette, P.A.; Whitson, L.J.; Cao, X.; Schirf, V.; Demeler, B.; Valentine, J.S.; Hansen, J.C.; Hart, P.J. Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant. Insights into the molecular basis for dimer stability. J. Biol. Chem. 2004, 279, 54558–54566. [Google Scholar] [CrossRef] [PubMed]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [PubMed]
- Seo, S.J.; Kang, S.S.; Cho, G.; Rho, H.M.; Jung, G. C/EBPα and C/EBPβ play similar roles in the transcription of the human Cu/Zn SOD gene. Gene 1997, 203, 11–15. [Google Scholar] [CrossRef]
- Hour, T.C.; Lai, Y.L.; Kuan, C.I.; Chou, C.K.; Wang, J.M.; Tu, H.Y.; Hu, H.T.; Lin, C.S.; Wu, W.J.; Pu, Y.S.; et al. Transcriptional up-regulation of SOD1 by CEBPD: A potential target for cisplatin resistant human urothelial carcinoma cells. Biochem. Pharmacol. 2010, 80, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Afonso, V.; Santos, G.; Collin, P.; Khatib, A.M.; Mitrovic, D.R.; Lomri, N.; Leitman, D.C.; Lomri, A. Tumor necrosis factor-α down-regulates human Cu/Zn superoxide dismutase 1 promoter via JNK/AP-1 signaling pathway. Free Radic. Biol. Med. 2006, 41, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Minc, E.; de Coppet, P.; Masson, P.; Thiery, L.; Dutertre, S.; Amor-Guéret, M.; Jaulin, C. The human copper-zinc superoxide dismutase gene (SOD1) proximal promoter is regulated by Sp1, Egr-1, and WT1 via non-canonical binding sites. J. Biol. Chem. 1999, 274, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, S.; Aquilano, K.; Rotilio, G.; Ciriolo, M.R. Glutathione and copper, zinc superoxide dismutase are modulated by overexpression of neuronal nitric oxide synthase. Int. J. Biochem. Cell Biol. 2008, 40, 2660–2670. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.S.; Chang, M.S.; Rho, H.M. Transcriptional activation of the human Cu/Zn superoxide dismutase gene by 2,3,7,8-tetrachlorodibenzo-p-dioxin through the xenobiotic-responsive element. Mol. Genet. Genom. 2001, 266, 133–141. [Google Scholar]
- O’Dea, E.; Hoffmann, A. The regulatory logic of the NF-kappaB signaling system. Cold Spring Harb. Perspect. Biol. 2010, 2, a000216. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Salinas, M.; Martín, D.; Perona, R.; Cuadrado, A. Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-κB. J. Neurosci. 2004, 24, 7324–7334. [Google Scholar] [CrossRef] [PubMed]
- Santos, G.M.; Afonso, V.; Barra, G.B.; Togashi, M.; Webb, P.; Neves, F.A.; Lomri, N.; Lomri, A. Negative regulation of superoxide dismutase-1 promoter by thyroid hormone. Mol. Pharmacol. 2006, 70, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kong, A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 2009, 48, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Park, E.Y.; Rho, H.M. The transcriptional activation of the human copper/zinc superoxide dismutase gene by 2,3,7,8-tetrachlorodibenzo-p-dioxin through two different regulator sites, the antioxidant responsive element and xenobiotic responsive element. Mol. Cell. Biochem. 2002, 240, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Dreger, H.; Westphal, K.; Wilck, N.; Baumann, G.; Stangl, V.; Stangl, K.; Meiners, S. Protection of vascular cells from oxidative stress by proteasome inhibition depends on Nrf2. Cardiovasc. Res. 2010, 85, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Dell’Orco, M.; Milani, P.; Arrigoni, L.; Pansarasa, O.; Sardone, V.; Maffioli, E.; Polveraccio, F.; Bordoni, M.; Diamanti, L.; Ceroni, M.; et al. Hydrogen peroxide-mediated induction of SOD1 gene transcription is independent from Nrf2 in a cellular model of neurodegeneration. Biochim. Biophys. Acta 2016, 1859, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Andreassi, C.; Riccio, A. To localize or not to localize: MRNA fate is in 3′UTR ends. Trends Cell Biol. 2009, 19, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Kilk, A.; Laan, M.; Torp, A. Human CuZn superoxide dismutase enzymatic activity in cells is regulated by the length of the mRNA. FEBS Lett. 1995, 362, 323–327. [Google Scholar] [CrossRef]
- Pascale, A.; Amadio, M.; Quattrone, A. Defining a neuron: Neuronal ELAV proteins. Cell. Mol. Life Sci. 2008, 65, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Milani, P.; Amadio, M.; Laforenza, U.; Dell’Orco, M.; Diamanti, L.; Sardone, V.; Gagliardi, S.; Govoni, S.; Ceroni, M.; Pascale, A.; et al. Posttranscriptional regulation of SOD1 gene expression under oxidative stress: Potential role of ELAV proteins in sporadic ALS. Neurobiol. Dis. 2013, 60, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Cuanalo-Contreras, K.; Mukherjee, A.; Soto, C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013, 2013, 638083. [Google Scholar] [CrossRef] [PubMed]
- Gruzman, A.; Wood, W.L.; Alpert, E.; Prasad, M.D.; Miller, R.G.; Rothstein, J.D.; Bowser, R.; Hamilton, R.; Wood, T.D.; Cleveland, D.W.; et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 12524–12529. [Google Scholar] [CrossRef] [PubMed]
- Ezzi, S.A.; Urushitani, M.; Julien, J.P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.; Andersen, P.M.; Marklund, S.L.; Brannstrom, T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011, 121, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; et al. Mutation of SOD1 in ALS: A gain of a loss of function. Hum. Mol. Genet. 2007, 16, 1604–1618. [Google Scholar] [CrossRef] [PubMed]
- Guareschi, S.; Cova, E.; Cereda, C.; Ceroni, M.; Donetti, E.; Bosco, D.A.; Trotti, D.; Pasinelli, P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 2012, 109, 5074–5079. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, S.; Cova, E.; Davin, A.; Guareschi, S.; Abel, K.; Alvisi, E.; Laforenza, U.; Ghidoni, R.; Cashman, J.R.; Ceroni, M.; et al. SOD1 mRNA expression in sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2010, 39, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Cereda, C.; Cova, E.; Di Poto, C.; Galli, A.; Mazzini, G.; Corato, M.; Ceroni, M. Effect of nitric oxide on lymphocytes from sporadic amyotrophic lateral sclerosis patients: Toxic or protective role? Neurol. Sci. 2006, 27, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Cova, E.; Cereda, C.; Galli, A.; Curti, D.; Finotti, C.; Di Poto, C.; Corato, M.; Mazzini, G.; Ceroni, M. Modified expression of Bcl-2 and SOD1 proteins in lymphocytes from sporadic ALS patients. Neurosci. Lett. 2006, 399, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Cereda, C.; Leoni, E.; Milani, P.; Pansarasa, O.; Mazzini, G.; Guareschi, S.; Alvisi, E.; Ghiroldi, A.; Diamanti, L.; Bernuzzi, S.; et al. Altered intracellular localization of SOD1 in leukocytes from patients with sporadic amyotrophic lateral sclerosis. PLoS ONE 2013, 8, e75916. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.K.; Ziegler, Y.S.; McLeod, I.X.; Yates, J.R.; Nardulli, A.M. Effects of Cu/Zn superoxide dismutase on estrogen responsiveness and oxidative stress in human breast cancer cells. Mol. Endocrinol. 2008, 22, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Inoue, E.; Tano, K.; Yoshii, H.; Nakamura, J.; Tada, S.; Watanabe, M.; Seki, M.; Enomoto, T. SOD1 is essential for the viability of DT40 cells and nuclear SOD1 functions as a guardian of genomic DNA. J. Nucleic Acids 2010, 5, 795946. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.; Jonsson, P.A.; Andersen, P.M.; Bergemalm, D.; Graffmo, K.S.; Hultdin, M.; Jacobsson, J.; Rosquist, R.; Marklund, S.L.; Brännström, T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE 2010, 5, e11552. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.K.; Li, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef] [PubMed]
- Münch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad Sci. USA 2011, 108, 3548–3553. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Pokrishevsky, E.; Silverman, J.M.; Cashman, N.R. Exosome-dependent and independent mechanisms are involved in prion-like transmission of propagated Cu/Zn superoxide dismutase misfolding. Prion 2014, 8, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Cleveland, D.W. Prion-like spread of protein aggregates in neurodegeneration. J. Exp. Med. 2012, 209, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Andaloussi, S.E.; Mäger, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, C.; Keller, S.; Altevogt, P.; Costa, J. Evidence for secretion of Cu, Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 2007, 428, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Basso, M.; Pozzi, S.; Tortarolo, M.; Fiordaliso, F.; Bisighini, C.; Pasetto, L.; Spaltro, G.; Lidonnici, D.; Gensano, F.; Battaglia, E.; et al. Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: Implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J. Biol. Chem. 2013, 288, 15699–15711. [Google Scholar] [CrossRef] [PubMed]
- Bellingham, S.A.; Guo, B.B.; Coleman, B.M.; Hill, A.F. Exosomes. Vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front. Physiol. 2012, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [PubMed]
- Fratta, P. Antisense makes sense for amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 416–417. [Google Scholar] [CrossRef]
- Rossor, A.M.; Reilly, M.M.; Sleigh, J.N. Antisense oligonucleotides and other genetic therapies made simple. Pract. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Miller, T.M.; Yamanaka, K.; Monia, B.P.; Condon, T.P.; Hung, G.; Lobsiger, C.S.; Ward, C.M.; McAlonis-Downes, M.; Wei, H.; et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Investig. 2006, 116, 2290–2296. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef]
- Chen, L.; Watson, C.; Morsch, M.; Cole, N.J.; Chung, R.S.; Saunders, D.N.; Yerbury, J.J.; Vine, K.L. Improving the delivery of SOD1 antisense oligonucleotides to motor neurons using calcium phosphate-lipid nanoparticles. Front. Neurosci. 2017, 11, 476. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M.; Kaspar, B.K.; Kops, G.J.; Yamanaka, K.; Christian, L.J.; Gage, F.H.; Cleveland, D.W. Virus-delivered small RNA silencing sustains strength in amyotrophic lateral sclerosis. Ann. Neurol. 2005, 57, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Ralph, G.S.; Radcliffe, P.A.; Day, D.M.; Carthy, J.M.; Leroux, M.A.; Lee, D.C.; Wong, L.F.; Bilsland, L.G.; Greensmith, L.; Kingsman, S.M.; et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 2005, 11, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Borel, F.; Gernoux, G.; Cardozo, B.; Metterville, J.P.; Toro Cabreja, G.C.; Song, L.; Su, Q.; Gao, G.P.; Elmallah, M.K.; Brown, R.H., Jr.; et al. Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1(G93A) Mice and Nonhuman Primates. Hum. Gene Ther. 2016, 27, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Biferi, M.G.; Cohen-Tannoudji, M.; Cappelletto, A.; Giroux, B.; Roda, M.; Astord, S.; Marais, T.; Bos, C.; Voit, T.; Ferry, A.; et al. A New AAV10-U7-Mediated Gene Therapy Prolongs Survival and Restores Function in an ALS Mouse Model. Mol. Ther. 2017, 25, 2038–2052. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Adikaram, P.; Pandey, M.; Genis, A.; Simonds, W.F. Optimization of genome editing through CRISPR-Cas9 engineering. Bioengineered 2016, 7, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yi, F.; Fu, L.; Yang, J.; Wang, S.; Wang, Z.; Suzuki, K.; Sun, L.; Xu, X.; Yu, Y.; et al. CRISPR/Cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient iPSCs. Protein Cell 2017, 8, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Frakes, A.E.; Braun, L.; Ferraiuolo, L.; Guttridge, D.C.; Kaspar, B.K. Additive amelioration of ALS by co-targeting independent pathogenic mechanisms. Ann. Clin. Transl. Neurol. 2017, 4, 76–86. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345. https://doi.org/10.3390/ijms19051345
Pansarasa O, Bordoni M, Diamanti L, Sproviero D, Gagliardi S, Cereda C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. International Journal of Molecular Sciences. 2018; 19(5):1345. https://doi.org/10.3390/ijms19051345
Chicago/Turabian StylePansarasa, Orietta, Matteo Bordoni, Luca Diamanti, Daisy Sproviero, Stella Gagliardi, and Cristina Cereda. 2018. "SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease" International Journal of Molecular Sciences 19, no. 5: 1345. https://doi.org/10.3390/ijms19051345