Computational Characterization of Small Molecules Binding to the Human XPF Active Site and Virtual Screening to Identify Potential New DNA Repair Inhibitors Targeting the ERCC1-XPF Endonuclease




Abstract
:1. Introduction
2. Results and Discussion
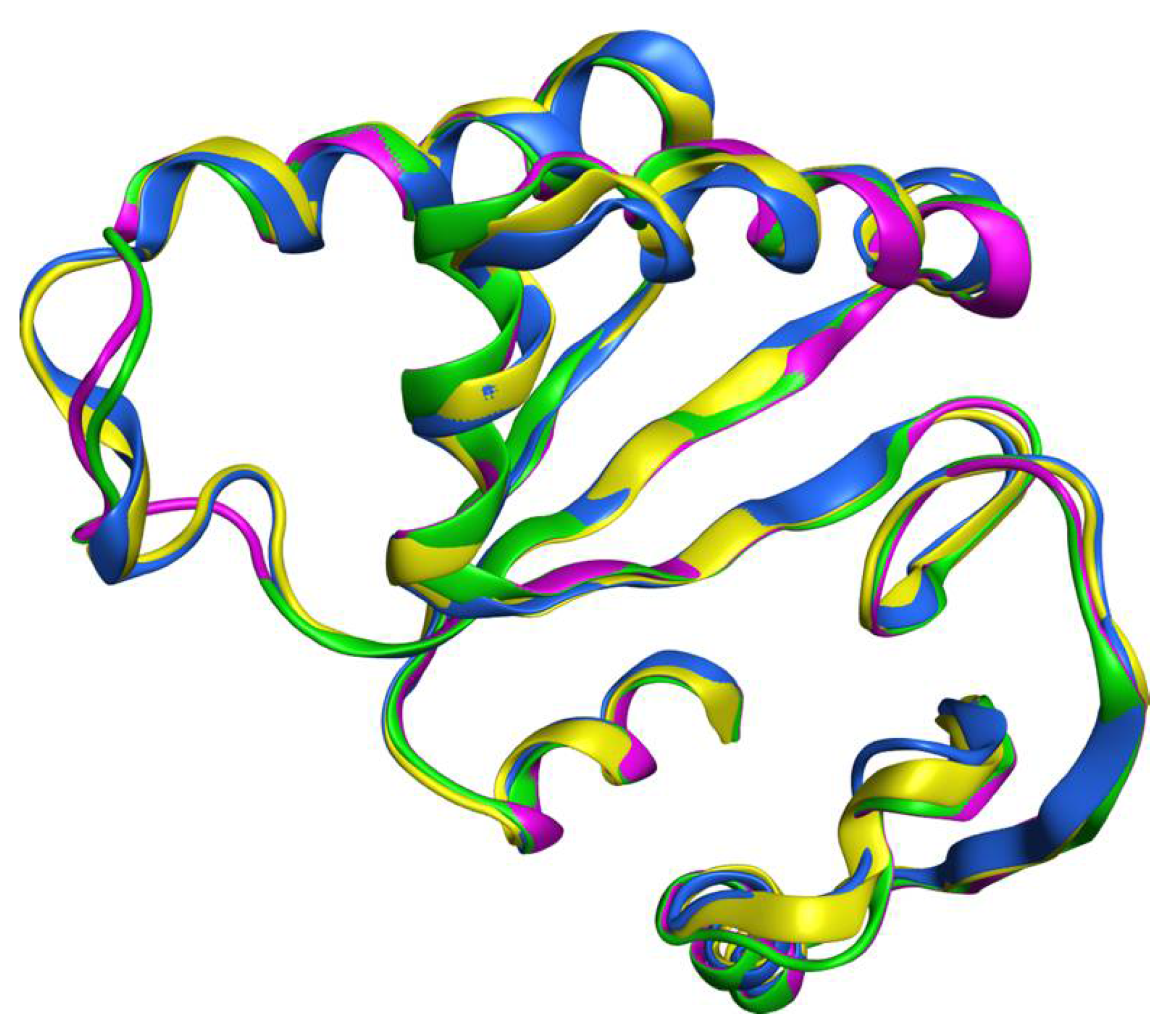
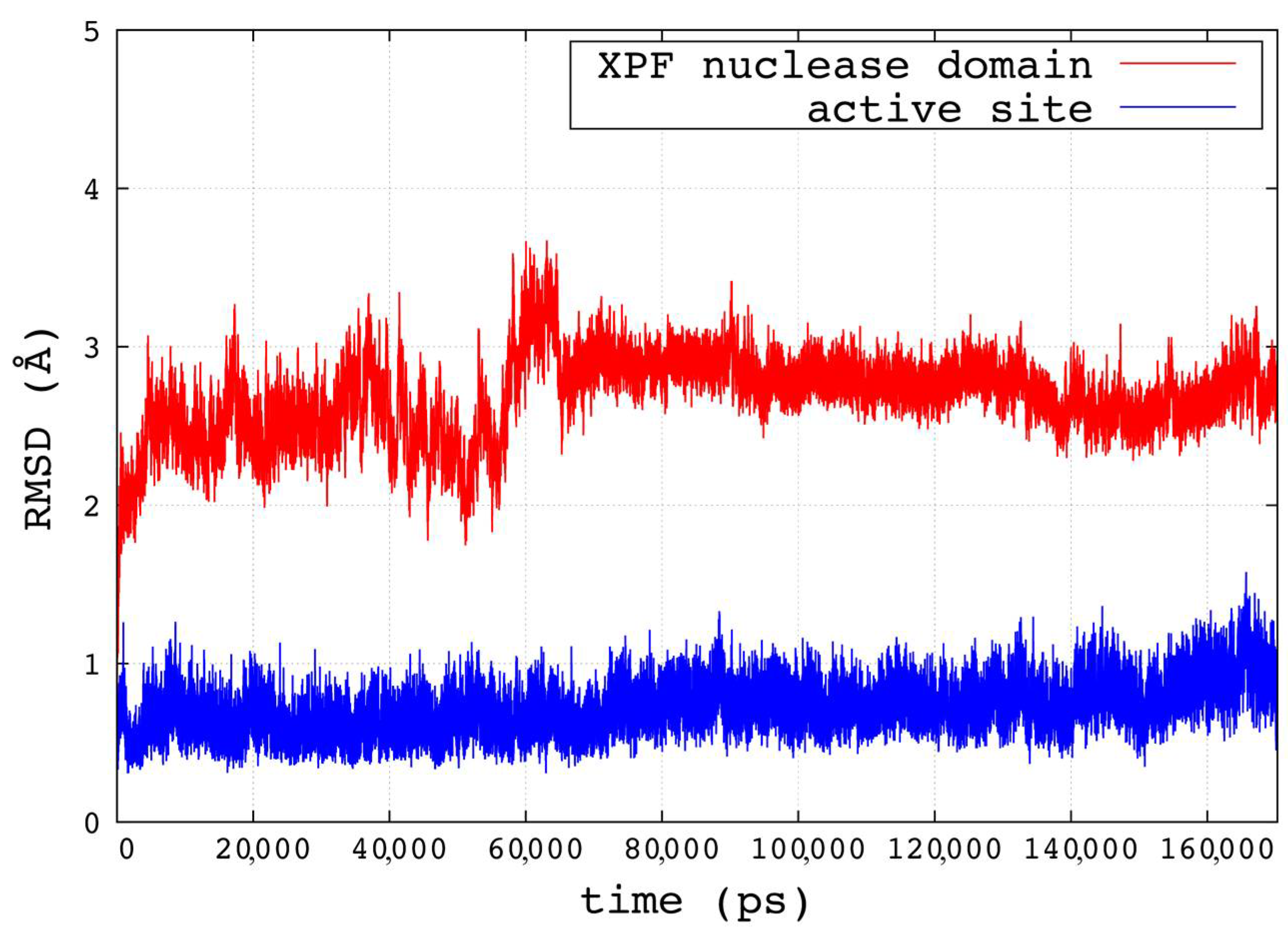
2.1. Homology Modeling and Molecular Dynamics Simulation
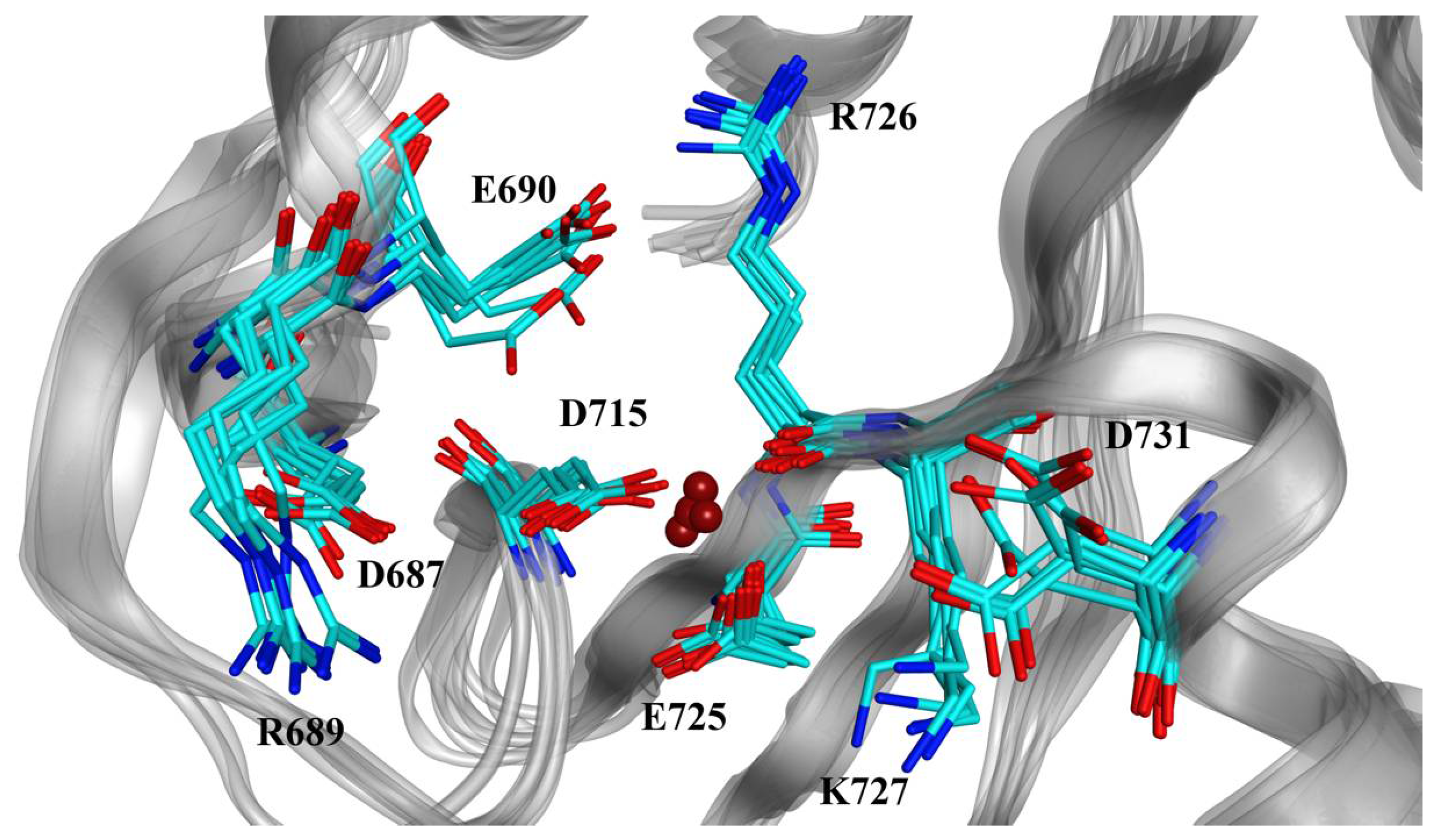
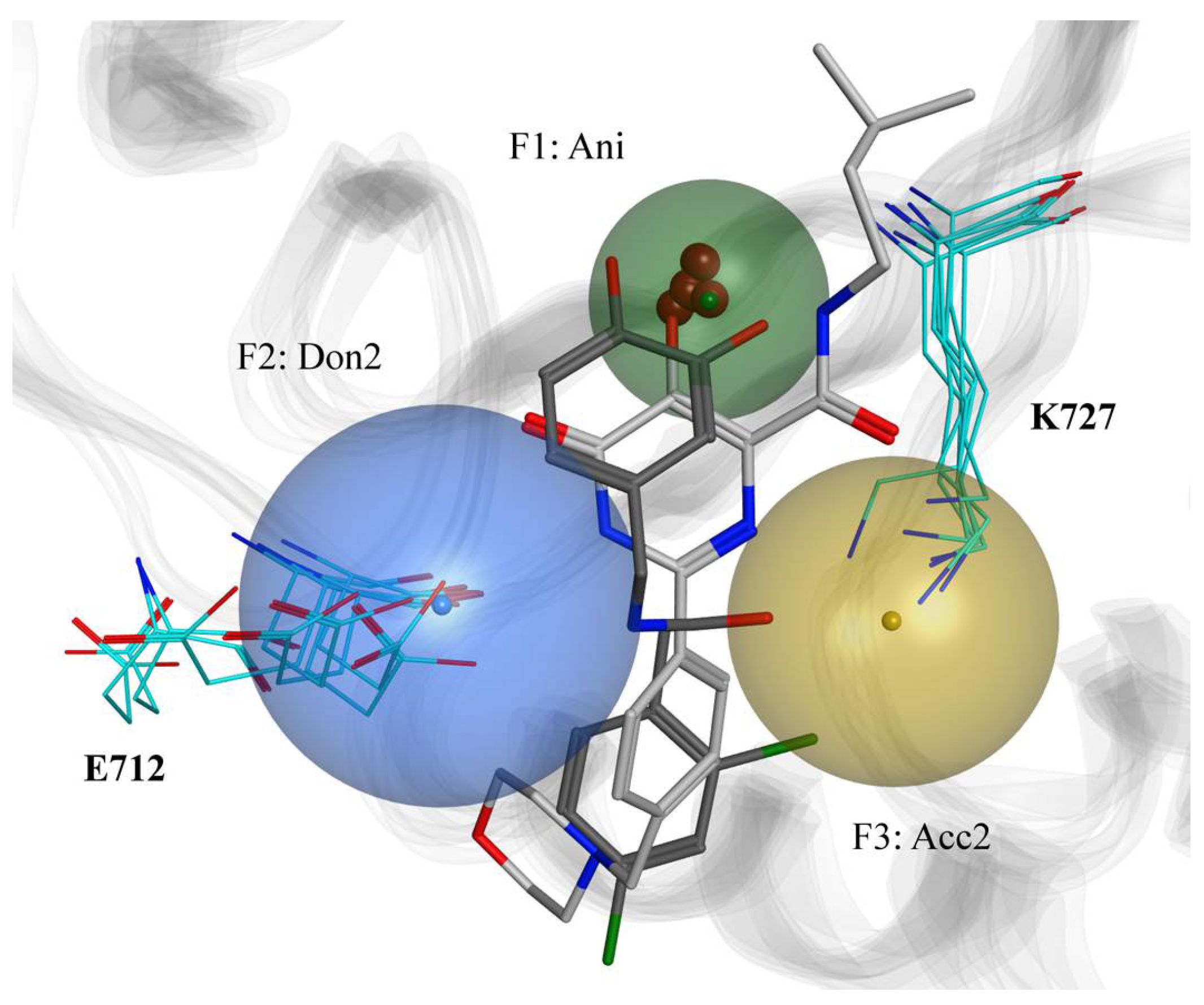
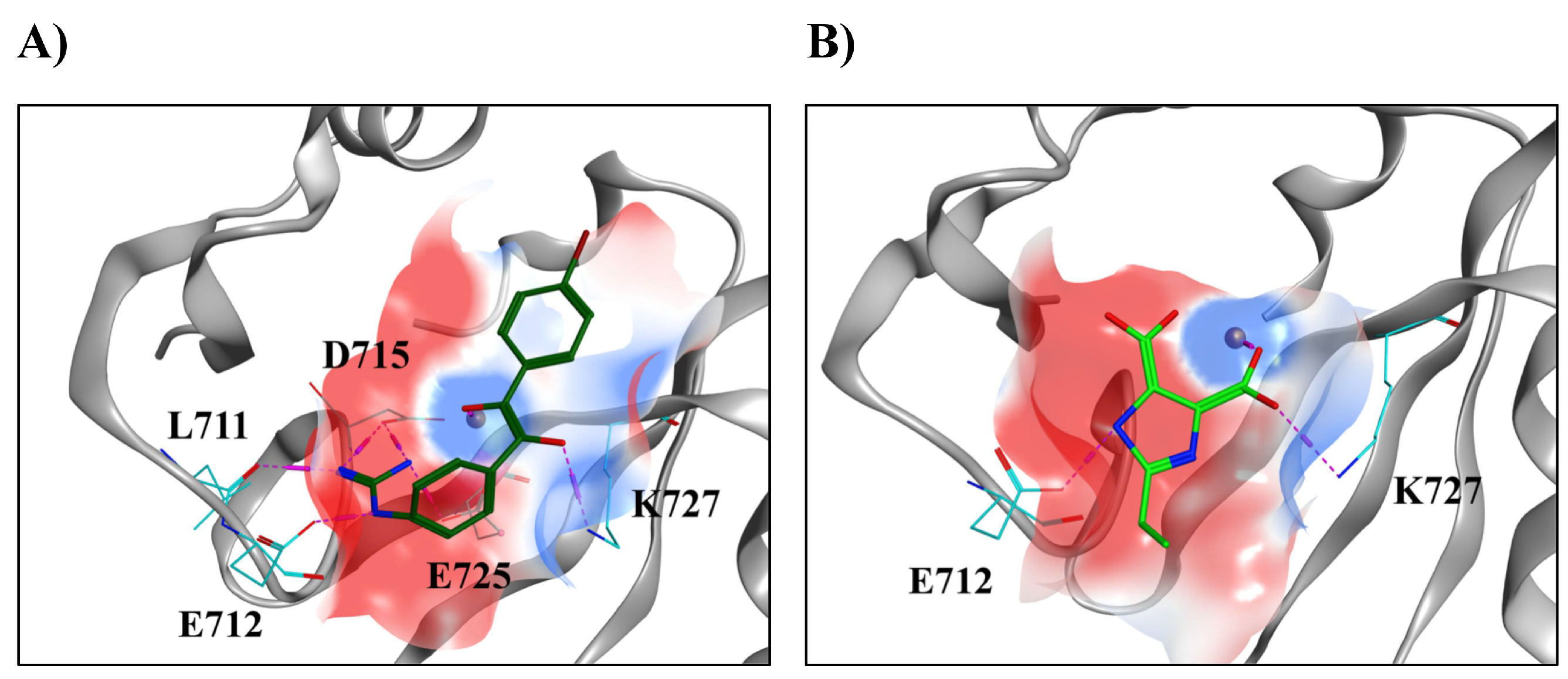
2.2. Modeling of Small Molecules Binding to the Human XPF Active Site
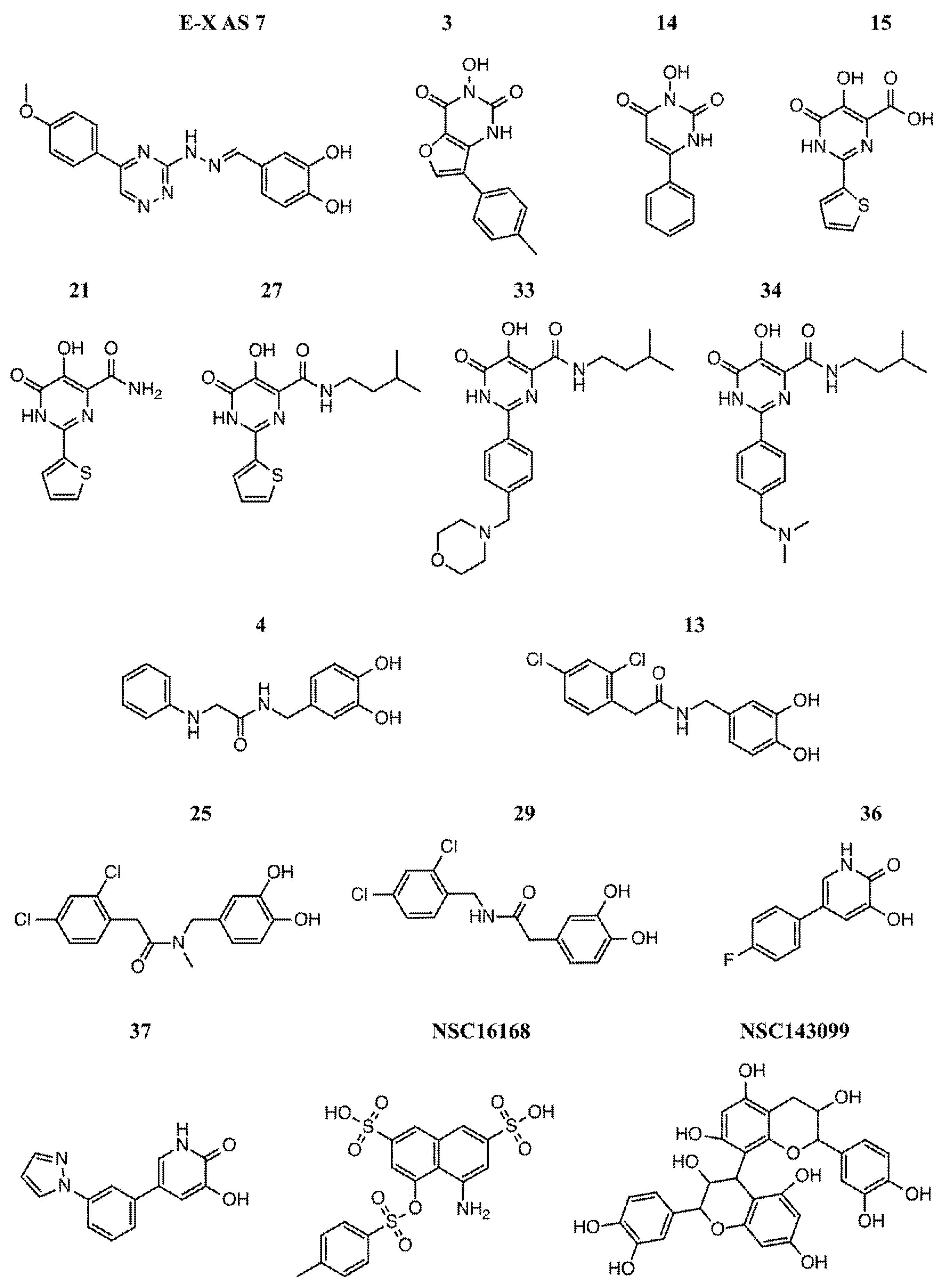
2.3. Virtual Screening
3. Materials and Methods
3.1. Homology Modeling of the Human XPF Nuclease Domain
3.2. Molecular Dynamics Simulation and Clustering of the Trajectory
3.3. Molecular Docking of Known Inhibitors and Pharmacophore Modeling
3.4. Virtual Screening of the ZINC15 Database
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Iyama, T.; Wilson, D.M. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Köberle, B.; Masters, J.R.W.; Hartley, J.A.; Wood, R.D. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr. Biol. 1999, 9, 273–278. [Google Scholar] [CrossRef]
- Mendoza, J.; Martínez, J.; Hernández, C.; Pérez-Montiel, D.; Castro, C.; Fabián-Morales, E.; Santibáñez, M.; González-Barrios, R.; Díaz-Chávez, J.; Andonegui, M.A.; et al. Association between ERCC1 and XPA expression and polymorphisms and the response to cisplatin in testicular germ cell tumours. Br. J. Cancer 2013, 109, 68–75. [Google Scholar] [CrossRef] [PubMed]
- O’Grady, S.; Finn, S.P.; Cuffe, S.; Richard, D.J.; O’Byrne, K.J.; Barr, M.P. The role of DNA repair pathways in cisplatin resistant lung cancer. Cancer Treat. Rev. 2014, 40, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Barakat, K.; Gajewski, M.; Tuszynski, J.A. DNA repair inhibitors: The next major step to improve cancer therapy. Curr. Top. Med. Chem. 2012, 12, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Nouspikel, T. DNA repair in mammalian cells: Nucleotide excision repair: Variations on versatility. Cell. Mol. Life Sci. 2009, 66, 994–1009. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Tuszynski, J.A.; Barakat, K.H. Modelling DNA Repair Pathways: Recent Advances and Future Directions. Curr. Pharm. Des. 2016, 22, 3527–3546. [Google Scholar] [CrossRef] [PubMed]
- McNeil, E.M.; Melton, D.W. DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res. 2012, 40, 9990–10004. [Google Scholar] [CrossRef] [PubMed]
- Barakat, K.H.; Jordheim, L.P.; Perez-Pineiro, R.; Wishart, D.; Dumontet, C.; Tuszynski, J.A. Virtual screening and biological evaluation of inhibitors targeting the XPA-ERCC1 interaction. PLoS ONE 2012, 7, e51329. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Tuszynski, J.A.; Barakat, K.H. New design of nucleotide excision repair (NER) inhibitors for combination cancer therapy. J. Mol. Graph. Model. 2016, 1, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Barakat, K.H.; Heinrich-Balard, L.; Matera, E.-L.; Cros-Perrial, E.; Bouledrak, K.; El Sabeh, R.; Perez-Pineiro, R.; Wishart, D.S.; Cohen, R.; et al. Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells. Mol. Pharmacol. 2013, 84, 12–24. [Google Scholar] [CrossRef] [PubMed]
- McNeil, E.M.; Astell, K.R.; Ritchie, A.-M.; Shave, S.; Houston, D.R.; Bakrania, P.; Jones, H.M.; Khurana, P.; Wallace, C.; Chapman, T.; et al. Inhibition of the ERCC1-XPF structure-specific endonuclease to overcome cancer chemoresistance. DNA Repair 2015, 31, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.M.; Wallace, C.; Gillen, K.J.; Bakrania, P.; Khurana, P.; Coombs, P.J.; Fox, S.; Bureau, E.A.; Brownlees, J.; Melton, D.W.; et al. N-Hydroxyimides and hydroxypyrimidinones as inhibitors of the DNA repair complex ERCC1-XPF. Bioorg. Med. Chem. Lett. 2015, 25, 4104–4108. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.M.; Gillen, K.J.; Wallace, C.; Lee, M.T.; Bakrania, P.; Khurana, P.; Coombs, P.J.; Stennett, L.; Fox, S.; Bureau, E.A.; et al. Catechols and 3-hydroxypyridones as inhibitors of the DNA repair complex ERCC1-XPF. Bioorg. Med. Chem. Lett. 2015, 25, 4097–4103. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Heyza, J.; Zhang, H.; Kalman-Maltese, V.; Tillison, K.; Floyd, A.M.; Chalfin, E.M.; Bepler, G.; Patrick, S.M. Identification of small molecule inhibitors of ERCC1-XPF that inhibit DNA repair and potentiate cisplatin efficacy in cancer cells. Oncotarget 2016, 7, 75104–75117. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.; Murray-Rust, J.; Lally, J.; Rudolf, J.; Fadden, A.; Knowles, P.P.; White, M.F.; McDonald, N.Q. Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J. 2005, 24, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Komori, K.; Ishino, Y.; Morikawa, K. X-ray and biochemical anatomy of an archaeal XPF/Rad1/Mus81 family nuclease: Similarity between its endonuclease domain and restriction enzymes. Structure 2003, 11, 445–457. [Google Scholar] [CrossRef]
- Ciccia, A.; McDonald, N.; West, S.C. Structural and Functional Relationships of the XPF/MUS81 Family of Proteins. Annu. Rev. Biochem. 2008, 77, 259–287. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G. Performance evaluation of amino acid substitution matrices. Proteins 1993, 17, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R. Comparison of methods for searching protein sequence databases. Protein Sci. 1995, 4, 1145–1160. [Google Scholar] [CrossRef] [PubMed]
- The Statistics of Sequence Similarity Scores—National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov/BLAST/tutorial (accessed on 8 December 2015).
- Enzlin, J.H.; Schärer, O.D. The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J. 2002, 21, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lee, J.Y.; Nowotny, M. Making and breaking nucleic acids: Two-Mg2+-ion catalysis and substrate specificity. Mol. Cell 2006, 22, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Gwon, G.H.; Jo, A.; Baek, K.; Jin, K.S.; Fu, Y.; Lee, J.-B.; Kim, Y.; Cho, Y. Crystal structures of the structure-selective nuclease Mus81-Eme1 bound to flap DNA substrates. EMBO J. 2014, 33, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2017. [Google Scholar]
- Kelly, K. 3D Bioinformatics and Comparative Modeling in MOE. Available online: https://www.chemcomp.com/journal/bio1999.htm (accessed on 8 December 2015).
- Kelly, K. Exhaustive and Iterative Clustering of the Protein Databank. Available online: https://www.chemcomp.com/journal/families.htm (accessed on 7 March 2018).
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [PubMed]
- Gonnet, G.H.; Cohen, M.A.; Benner, S.A. Exhaustive matching of the entire protein sequence database. Science 1992, 256, 1443–1445. [Google Scholar] [CrossRef] [PubMed]
- Dayhoff, M.O.; Schwartz, R.; Orcutt, B.C. A model of evolutionary change in proteins. In Atlas of Protein Sequence and Structure; Dayhoff, M.O., Ed.; National Biomedical Research Foundation: Silver Spring, MD, USA, 1978; pp. 345–352. [Google Scholar]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. Protonate 3D: Assignment of Macromolecular Protonation State and Geometry. Available online: http://www.ccl.net/cca/documents/proton/ (accessed on 31 March 2014).
- AmberTools 12 Reference Manual. Available online: http://ambermd.org/doc12/AmberTools12.pdf (accessed on 6 March 2016).
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, K.M. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Song, L.F.; Merz, K.M. Systematic parameterization of monovalent ions employing the nonbonded model. J. Chem. Theory Comput. 2015, 11, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baron, R.; McCammon, J.A. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput. Aided Mol. Des. 2008, 22, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef] [PubMed]
- Barakat, K.; Tuszynski, J. Relaxed complex scheme suggests novel inhibitors for the lyase activity of DNA polymerase beta. J. Mol. Graph. Model. 2011, 29, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Wolfson, H.J. Efficient detection of three-dimensional structural motifs in biological macromolecules by computer vision techniques. Proc. Natl. Acad. Sci. USA 1991, 88, 10495–10499. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Lin, A. Overview of Pharmacophore Applications in MOE. Available online: https://www.chemcomp.com/journal/ph4.htm (accessed on 20 July 2015).
- Oprea, T.I. Property distribution of drug-related chemical databases. J. Comput. Aided Mol. Des. 2000, 14, 251–264. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substitution Matrix | Top Templates | Others |
|---|---|---|
| BLOSUM50 | 2BGW, 2BHN | 1J22, 2ZIU, 4P0P |
| BLOSUM62 | 2BGW, 2BHN, 1J22 | 2ZIU, 2ZIX, 4P0P |
| Gonnet | 2BGW, 2BHN | 1J22 |
| PAM250 | 2BGW, 2BHN | 1J22 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentile, F.; Barakat, K.H.; Tuszynski, J.A. Computational Characterization of Small Molecules Binding to the Human XPF Active Site and Virtual Screening to Identify Potential New DNA Repair Inhibitors Targeting the ERCC1-XPF Endonuclease. Int. J. Mol. Sci. 2018, 19, 1328. https://doi.org/10.3390/ijms19051328
Gentile F, Barakat KH, Tuszynski JA. Computational Characterization of Small Molecules Binding to the Human XPF Active Site and Virtual Screening to Identify Potential New DNA Repair Inhibitors Targeting the ERCC1-XPF Endonuclease. International Journal of Molecular Sciences. 2018; 19(5):1328. https://doi.org/10.3390/ijms19051328
Chicago/Turabian StyleGentile, Francesco, Khaled H. Barakat, and Jack A. Tuszynski. 2018. "Computational Characterization of Small Molecules Binding to the Human XPF Active Site and Virtual Screening to Identify Potential New DNA Repair Inhibitors Targeting the ERCC1-XPF Endonuclease" International Journal of Molecular Sciences 19, no. 5: 1328. https://doi.org/10.3390/ijms19051328
APA StyleGentile, F., Barakat, K. H., & Tuszynski, J. A. (2018). Computational Characterization of Small Molecules Binding to the Human XPF Active Site and Virtual Screening to Identify Potential New DNA Repair Inhibitors Targeting the ERCC1-XPF Endonuclease. International Journal of Molecular Sciences, 19(5), 1328. https://doi.org/10.3390/ijms19051328