DNA Methyltransferases, DNA Methylation, and Age-Associated Cognitive Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
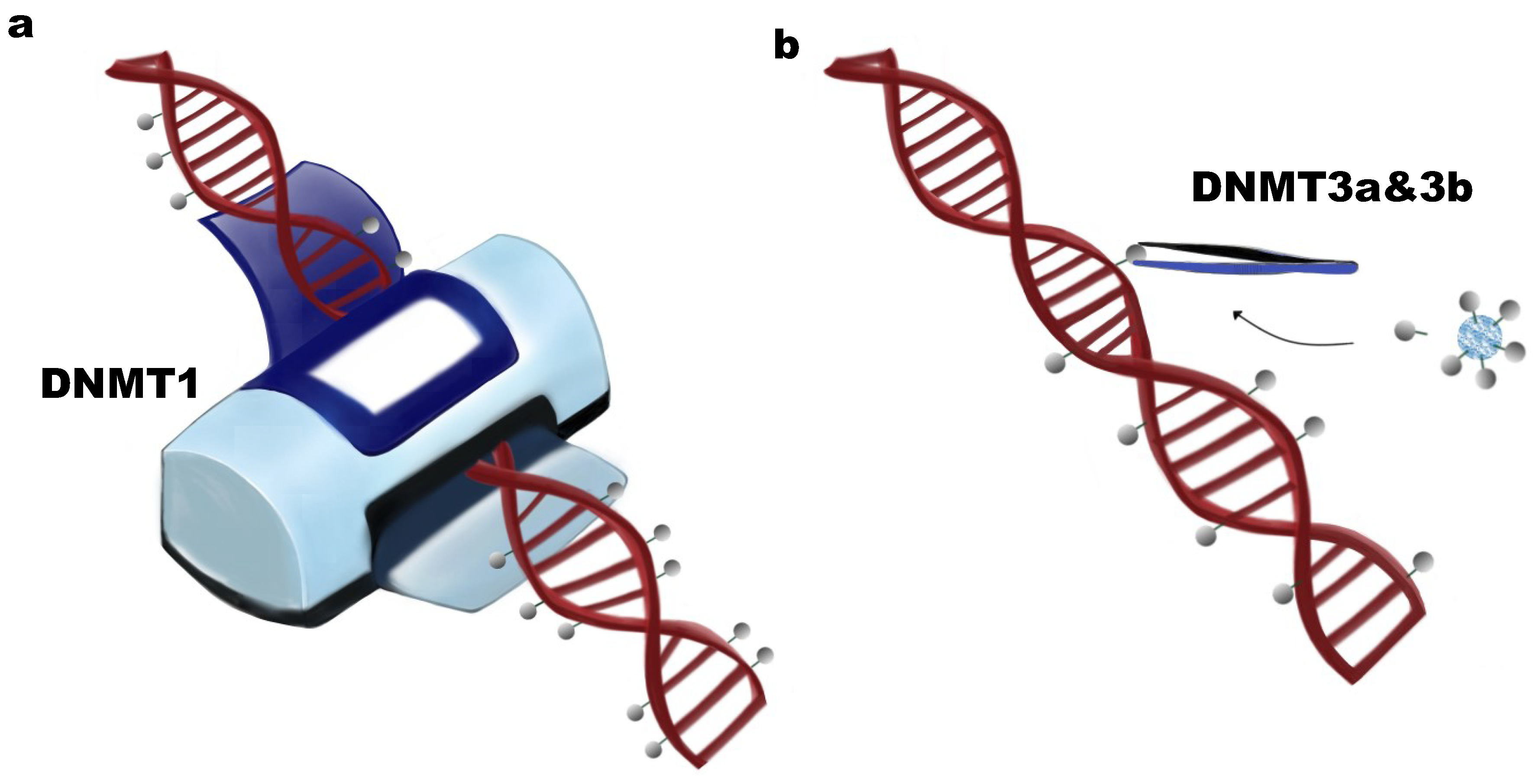
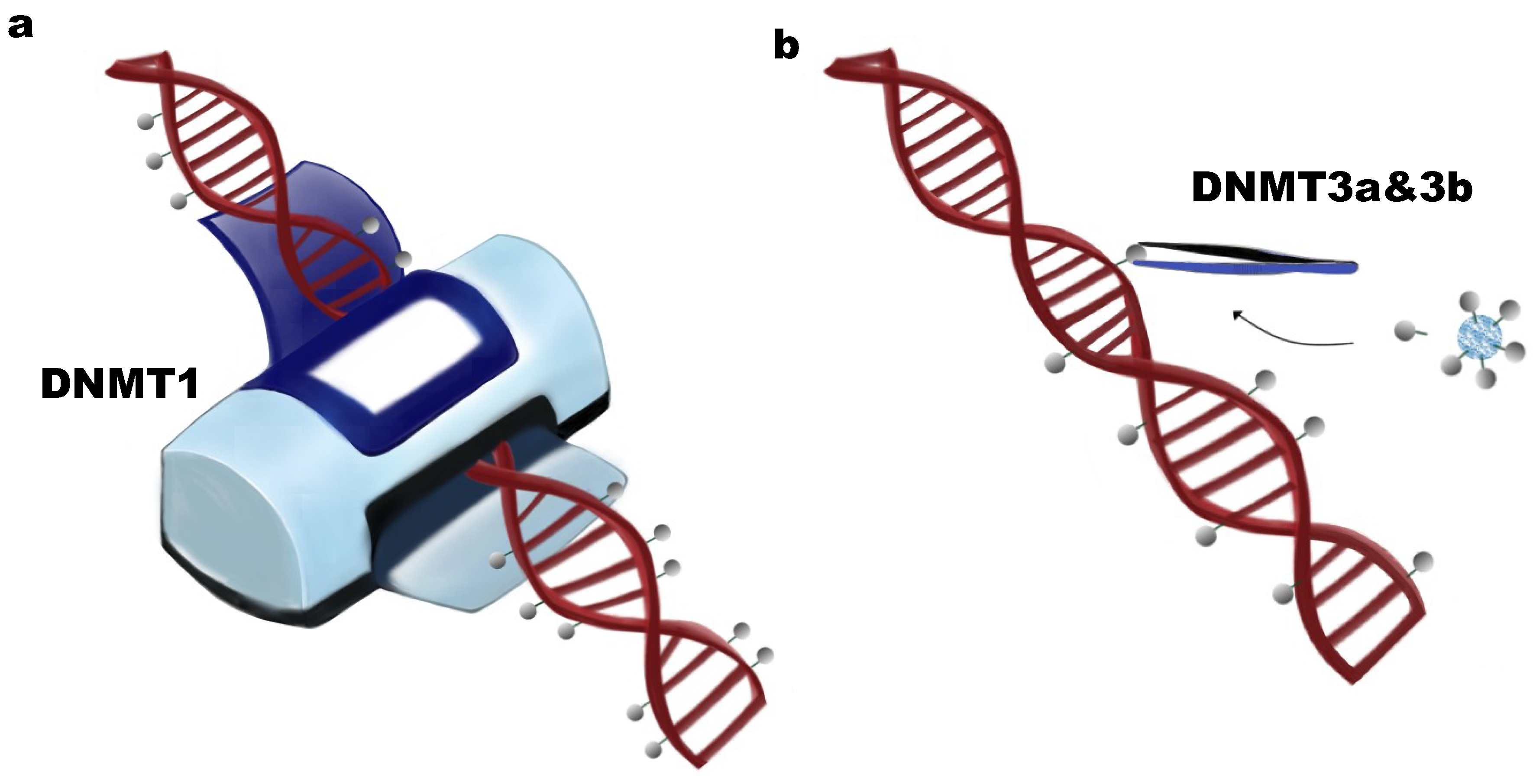
:1. DNA Methylation and DNA Methyltransferases
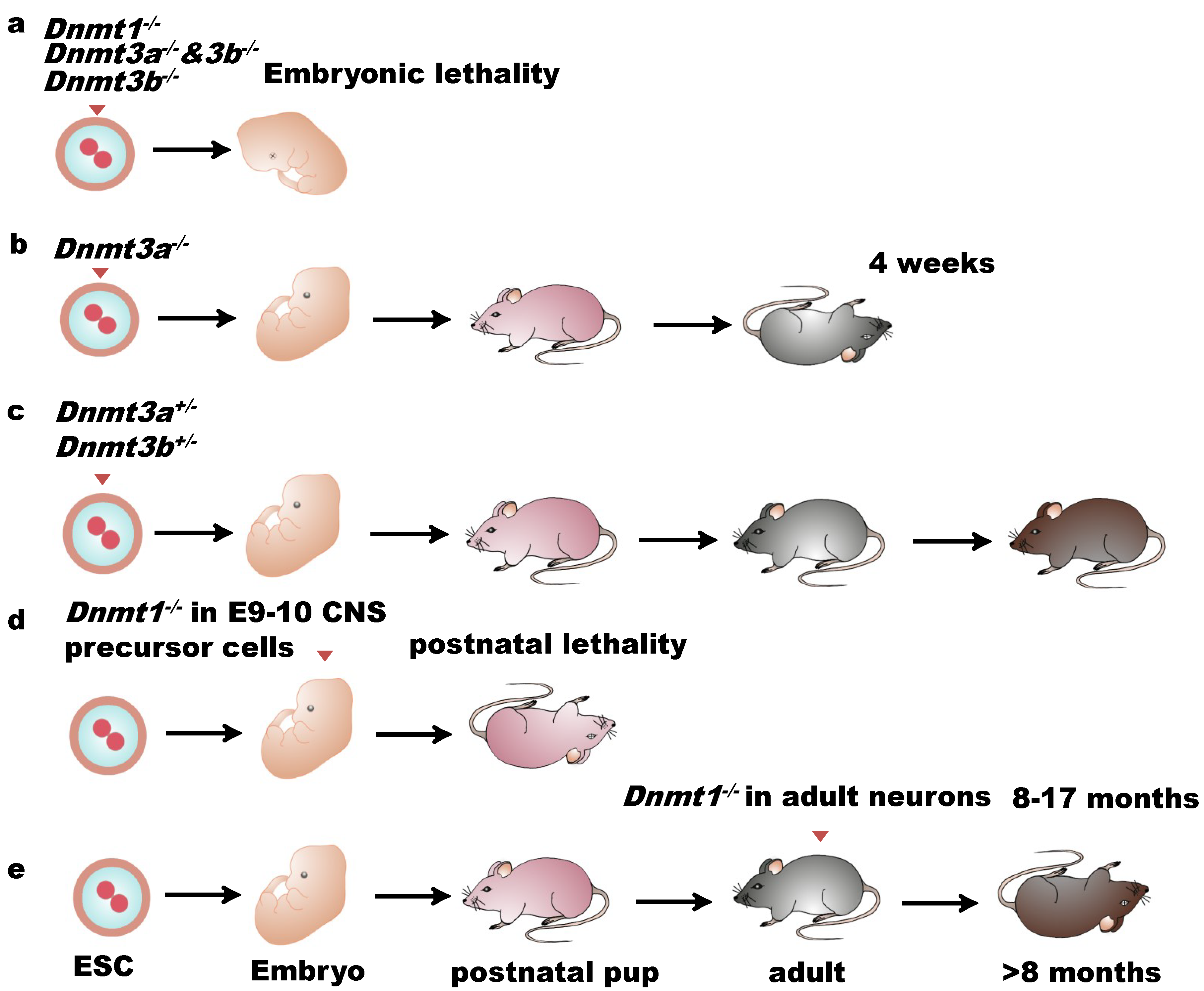
2. DNA Methylation, DNA Methyltransferases, and Mammalian CNS Development
3. DNA Methylation, DNA Methyltransferases, Mammalian CNS Ageing, and Alzheimer´s Disease
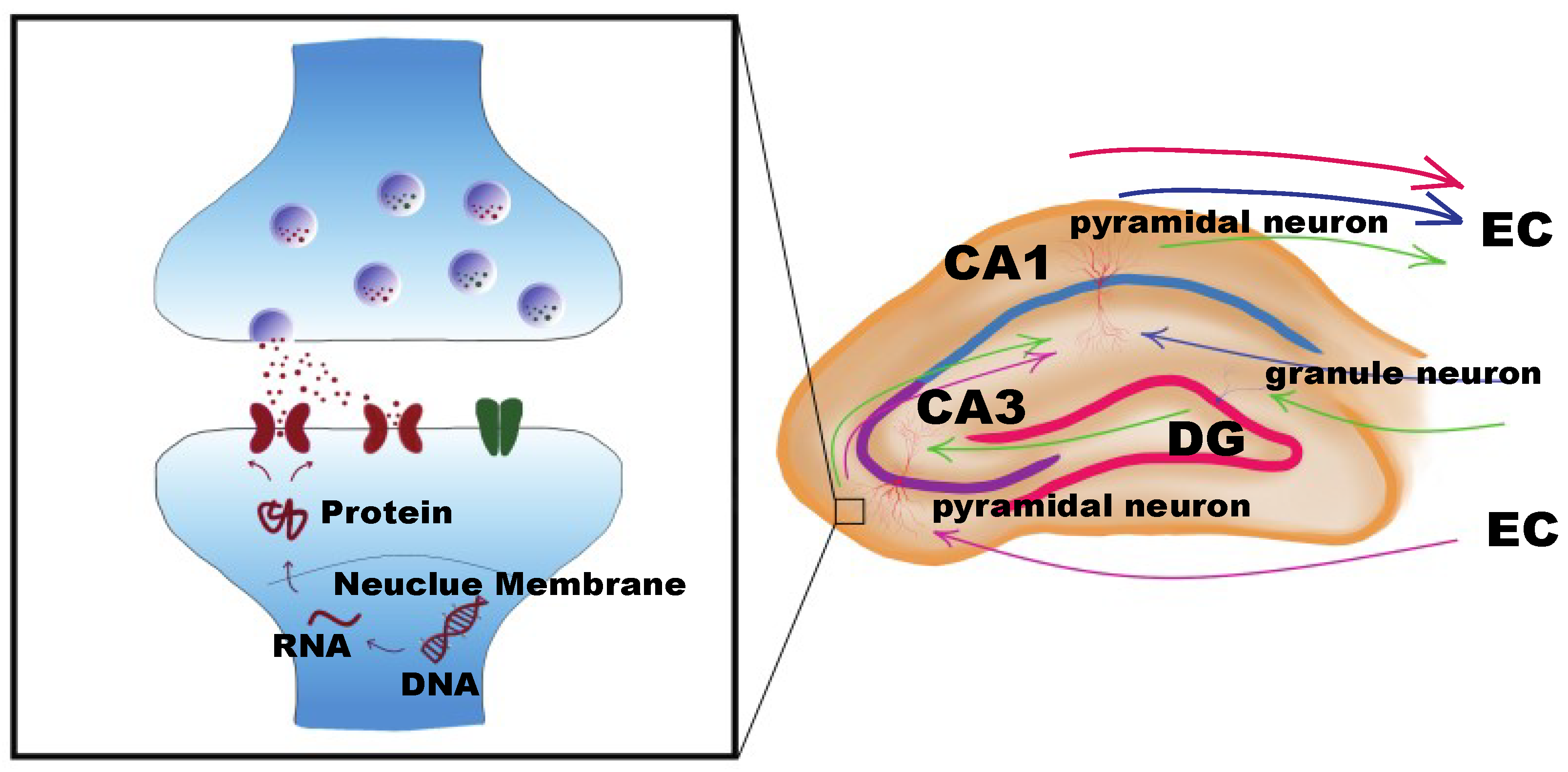
4. Mechanisms of DNA Methyltransferases in Regulating Neuronal Synaptic Plasticity-Related Gene Transcription
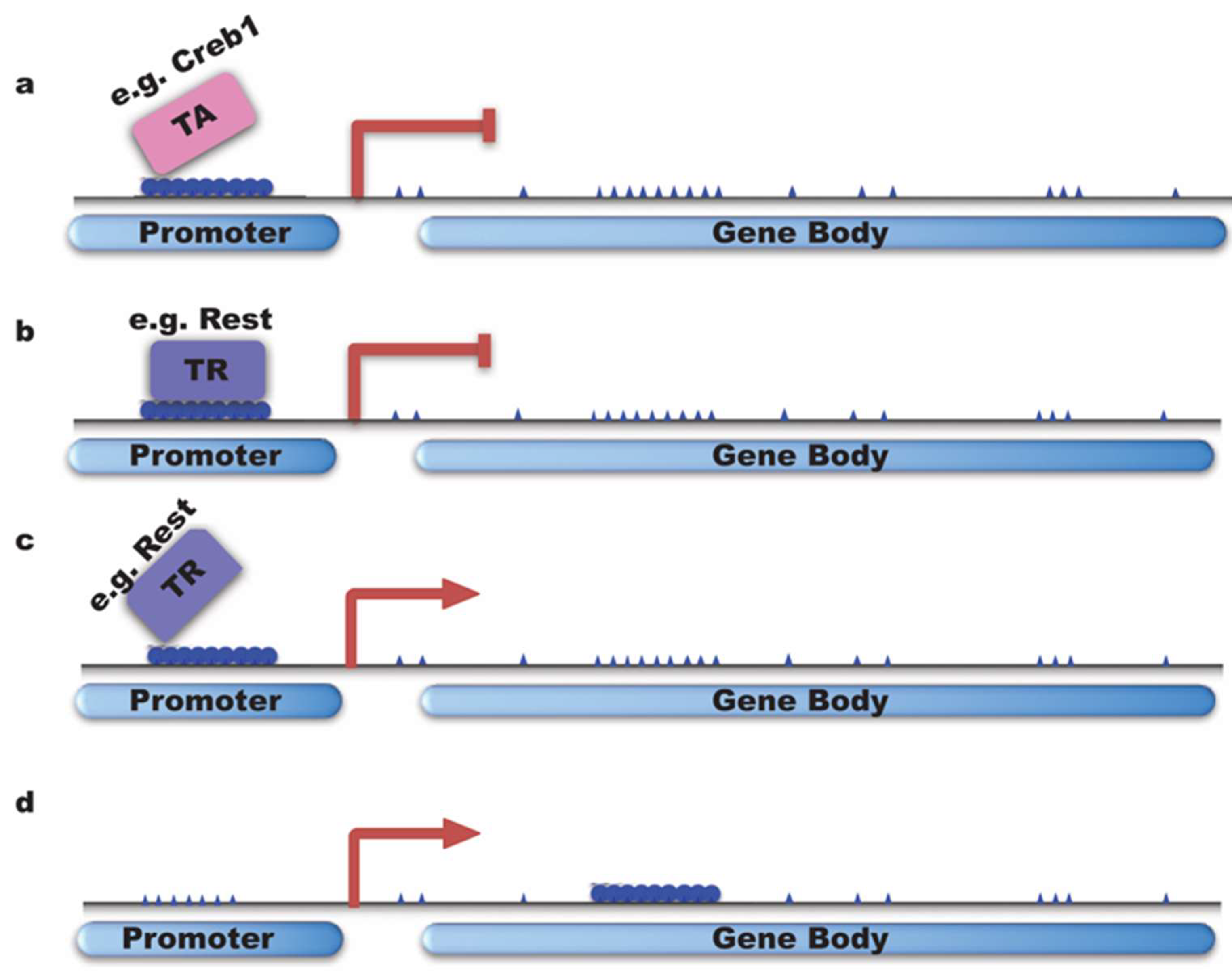
4.1. DNMTs Regulate Neuronal Gene Transcription by Fine-Tuning the Methylation Pattern on DNA Elements Such as Promoter, Enhancer, and Gene Body
4.1.1. DNMTs Inhibit Gene Transcription
4.1.2. DNMTs Activate/Enhance Gene Transcription
4.2. DNMTs Regulate Neuronal Gene Transcription by Coordinating the Function Of Methyl-DNA-Binding Proteins and Histones
4.2.1. DNMTs Associates with Methyl-DNA-Binding Protein in Neuronal Gene Regulation
4.2.2. DNMTs Associate with Histone Modification in Neuronal Gene Regulation
5. Perspectives of DNA Methylation on Cognitive Ageing
Author contributions
Acknowledgments
Conflicts of Interest
References
- Waddington, C.H. The epigenotype. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Blanco, M.A.; Gu, L.; Sendinc, E.; Liu, J.; Aristizábal-Corrales, D.; Hsu, C.H.; Aravind, L.; He, C.; Shi, Y. DNA methylation on N6-adenine in C. elegans. Cell 2015, 161, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457. [Google Scholar] [CrossRef] [PubMed]
- Xu, X. DNA methylation and cognitive aging. Oncotarget 2015, 6, 13922–13932. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Xu, X. Reduction in DNA methyltransferases and alteration of DNA methylation pattern associate with mouse skin ageing. Exp. Dermatol. 2014, 23, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Laird, P.W. Cancer-epigenetics comes of age. Nat. Genet. 1999, 21, 163. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.-Z.; Blanco, M.A.; Greer, E.L.; He, C.; Shi, Y. DNA N6-methyladenine: A new epigenetic mark in eukaryotes? Nat. Rev. Mol. Cell Biol. 2015, 16, 705. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M.; et al. DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Varela-Rey, M.; Iruarrizaga-Lejarreta, M.; Lozano, J.J.; Aransay, A.M.; Fernandez, A.F.; Lavin, J.L.; Mósen-Ansorena, D.; Berdasco, M.; Turmaine, M.; Luka, Z.; et al. S-adenosylmethionine levels regulate the schwann cell DNA methylome. Neuron 2014, 81, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, B.; Bird, A. Mammalian methyltransferases and methyl-CpG-binding domains: Proteins involved in DNA methylation. Curr. Top. Microbiol. Immunol. 2000, 249, 55. [Google Scholar] [PubMed]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.S.; Gruenewald-Schneider, U.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Smith, C.; Harrison, D.J.; Andrews, R.; Bird, A.P. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010, 6, e1001134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoder, J.A.; Soman, N.S.; Verdine, G.L.; Bestor, T.H. DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J. Mol. Biol. 1997, 270, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Suetake, I.; Shinozaki, F.; Miyagawa, J.; Takeshima, H.; Tajima, S. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J. Biol. Chem. 2004, 279, 27816–27823. [Google Scholar] [CrossRef] [PubMed]
- Suetake, I.; Morimoto, Y.; Fuchikami, T.; Abe, K.; Tajima, S. Stimulation effect of Dnmt3L on the DNA methylation activity of Dnmt3a2. J. Biochem. 2006, 140, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Nimura, K.; Ishida, C.; Koriyama, H.; Hata, K.; Yamanaka, S.; Li, E.; Ura, K.; Kaneda, Y. Dnmt3a2 targets endogenous Dnmt3L to ES cell chromatin and induces regional DNA methylation. Genes Cells 2006, 11, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Li, E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 2002, 3, 662. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wu, J. Epigenetic regulation in mammalian preimplantation embryo development. Reprod. Biol. Endocrinol. 2009, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, X.; Liang, D.; Li, T.; Zhu, P.; Guo, H.; Wu, X.; Wen, L.; Gu, T.P.; Hu, B.; et al. Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell 2014, 15, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Yan, L.; Guo, H.; Li, L.; Hu, B.; Zhao, Y.; Yong, J.; Hu, Y.; Wang, X.; Wei, Y.; Wang, W. The transcriptome and DNA methylome landscapes of human primordial germ cells. Cell 2015, 161, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Howlett, S.K.; Reik, W. Methylation levels of maternal and paternal genomes during preimplantation development. Development 1991, 113, 119–127. [Google Scholar] [PubMed]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schübeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, K.D.; Connor, C.M.; Campan, M.; Long, T.I.; Weisenberger, D.J.; Biniszkiewicz, D.; Jaenisch, R.; Laird, P.W.; Akbarian, S. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS ONE 2007, 2, e895. [Google Scholar] [CrossRef] [PubMed]
- Numata, S.; Ye, T.; Hyde, T.M.; Guitart-Navarro, X.; Tao, R.; Wininger, M.; Colantuoni, C.; Weinberger, D.R.; Kleinman, J.E.; Lipska, B.K. DNA methylation signatures in development and aging of the human prefrontal cortex. Am. J. Hum. Genet. 2012, 90, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, T.M.; Bonder, M.J.; Stark, A.K.; Krueger, F.; von Meyenn, F.; Stegle, O.; Reik, W. Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 2017, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W. Epigenetic aging clocks in mice and men. Genome Biol. 2017, 18, 107. [Google Scholar] [CrossRef] [PubMed]
- Petkovich, D.A.; Podolskiy, D.I.; Lobanov, A.V.; Lee, S.-G.; Miller, R.A.; Gladyshev, V.N. Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab. 2017, 25, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J.; Marietta, C.; Goldman, D. DNA mismatch repair and DNA methylation in adult brain neurons. J. Neurosci. 1996, 16, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Numata, M.; Komura, J.-I.; Ono, T.; Bestor, T.H.; Kondo, H. Expression of DNA methyltransferase gene in mature and immature neurons as well as proliferating cells in mice. Differentiation 1994, 56, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Golshani, P.; Hutnick, L.; Schweizer, F.; Fan, G. Conditional Dnmt1 deletion in dorsal forebrain disrupts development of somatosensory barrel cortex and thalamocortical long-term potentiation. Thalamus Relat. Syst. 2005, 3, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Beard, C.; Chen, R.Z.; Csankovszki, G.; Sun, Y.; Siniaia, M.; Biniszkiewicz, D.; Bates, B.; Lee, P.P.; Kühn, R.; et al. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J. Neurosci. 2001, 21, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Mallela, R.K.; Hayes, A.; Dunham, N.R.; Hedden, M.E.; Enke, R.A.; Fariss, R.N.; Sternberg, H.; West, M.D.; Nasonkin, I.O. Dnmt1, Dnmt3a and Dnmt3b cooperate in photoreceptor and outer plexiform layer development in the mammalian retina. Exp. Eye Res. 2017, 159, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Smets, M.; Link, S.; Wolf, P.; Schneider, K.; Solis, V.; Ryan, J.; Meilinger, D.; Qin, W.; Leonhardt, H. DNMT1 mutations found in HSANIE patients affect interaction with UHRF1 and neuronal differentiation. Hum. Mol. Genet. 2017, 26, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.J.; Botuyan, M.V.; Wu, Y.; Ward, C.J.; Nicholson, G.A.; Hammans, S.; Hojo, K.; Yamanishi, H.; Karpf, A.R.; Wallace, D.C.; et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 2011, 43, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.J.; Bird, T.; Ertekin-Taner, N.; Lincoln, S.; Hjorth, R.; Wu, Y.; Kwok, J.; Mer, G.; Dyck, P.J.; Nicholson, G.A. DNMT1 mutation hot spot causes varied phenotypes of HSAN1 with dementia and hearing loss. Neurology 2013, 80, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases.Cloning and characterization of a family of novel mammalian DNA. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Wang, Z.; Okano, M.; Nogami, M.; Li, Y.; He, W.W.; Okumura, K.; Li, E. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene 1999, 236, 87–95. [Google Scholar] [CrossRef]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.J.; Xu, J.; Gu, Z.H.; Pan, C.M.; Lu, G.; Shen, Y.; Shi, J.Y.; Zhu, Y.M.; Tang, L.; Zhang, X.W.; Liang, W.X. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011, 43, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Tao, Q.; Peng, J.; Soo, H.M.; Wu, W.; Ying, J.; Fields, C.R.; Delmas, A.L.; Liu, X.; Qiu, J.; et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum. Mol. Genet. 2008, 17, 690–709. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.S.; Wijmenga, C.; Luo, P.; Stanek, A.M.; Canfield, T.K.; Weemaes, C.M.; Gartler, S.M. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 14412–14417. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Okano, M.; Williams, C.; Chen, T.; Georgopoulos, K.; Li, E. Roles for Dnmt3b in mammalian development: A mouse model for the ICF syndrome. Development 2006, 133, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Glisky, E.L. Changes in cognitive function in human aging. Brain Aging Model. Methods Mech. 2017, 19, 3–20. [Google Scholar]
- Baddeley, A. Working memory: Looking back and looking forward. Nat. Rev. Neurosci. 2003, 4, 829. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.A.; Galanter, E.; Pribram, K.H. Plans and the Structure of Behavior; Adams Bannister Cox: New York, NY, USA, 1986. [Google Scholar]
- Jonides, J.; Smith, E.E.; Koeppe, R.A.; Awh, E.; Minoshima, S.; Mintun, M.A. Spatial working-memory in humans as revealed by PET. Nature 1993, 363, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Forman, S.D.; Braver, T.S.; Casey, B.J.; Servan-Schreiber, D.; Noll, D.C. Activation of the prefrontal cortex in a nonspatial working memory task with functional MRI. Hum. Brain Mapp. 1994, 1, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Aggleton, J.P.; Hunt, P.R.; Rawlins, J.N.P. The effects of hippocampal lesions upon spatial and non-spatial tests of working memory. Behav. Brain Res. 1986, 19, 133–146. [Google Scholar] [CrossRef]
- Scoville, W.B.; Milner, B. Loss of recent memory after bilateral hippocampal lesions. J. Neurol. Neurosurg. Psychiatry 1957, 20, 11. [Google Scholar] [CrossRef] [PubMed]
- Lledo, P.-M.; Alonso, M.; Grubb, M.S. Adult neurogenesis and functional plasticity in neuronal circuits. Nat. Rev. Neurosci. 2006, 7, 179. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Gage, F.H. Neurogenesis in the adult hippocampus. In Neural Transplantation in Neurodegenerative Disease: Current Status and New Directions: Novartis Foundation Symposium 231; Novartis Foundation: London, UK, 2000; pp. 220–241. [Google Scholar]
- Shors, T.J. From stem cells to grandmother cells: How neurogenesis relates to learning and memory. Cell Stem Cell 2008, 3, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, I.; Medina, J.H. Memory formation: The sequence of biochemical events in the hippocampus and its connection to activity in other brain structures. Neurobiol. Learn. Mem. 1997, 68, 285–316. [Google Scholar] [CrossRef] [PubMed]
- Tsien, J.Z.; Huerta, P.T.; Tonegawa, S. The essential role of hippocampal CA1 NMDA receptor–dependent synaptic plasticity in spatial memory. Cell 1996, 87, 1327–1338. [Google Scholar] [CrossRef]
- Burger, C. Region-specific genetic alterations in the aging hippocampus: Implications for cognitive aging. Front. Aging Neurosci. 2010, 2, 140. [Google Scholar] [CrossRef] [PubMed]
- Geinisman, Y. Loss of axosomatic synapses in the dentate gyrus of aged rats. Brain Res. 1979, 168, 485–492. [Google Scholar] [CrossRef]
- Nicholson, D.A.; Yoshida, R.; Berry, R.W.; Gallagher, M.; Geinisman, Y. Reduction in size of perforated postsynaptic densities in hippocampal axospinous synapses and age-related spatial learning impairments. J. Neurosci. 2004, 24, 7648–7653. [Google Scholar] [CrossRef] [PubMed]
- Vanyushin, B.F.; Nemirovsky, L.E.; Klimenko, V.V.; Vasiliev, V.K.; Belozersky, A.N. The 5-methylcytosine in DNA of rats. Gerontology 1973, 19, 138–152. [Google Scholar] [CrossRef]
- Wilson, V.L.; Smith, R.A.; Ma, S.; Cutler, R.G. Genomic 5-methyldeoxycytidine decreases with age. J. Biol. Chem. 1987, 262, 9948–9951. [Google Scholar] [PubMed]
- Mugatroyd, C.; Wu, Y.; Bockmühl, Y.; Spengler, D. The Janus face of DNA methylation in aging. Aging (Albany NY) 2010, 2, 107. [Google Scholar] [CrossRef] [PubMed]
- Day, J.J.; Childs, D.; Guzman-Karlsson, M.C.; Kibe, M.; Moulden, J.; Song, E.; Tahir, A.; Sweatt, J.D. DNA methylation regulates associative reward learning. Nat. Neurosci. 2013, 16, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Day, J.J.; Sweatt, J.D. DNA methylation and memory formation. Nat. Neurosci. 2010, 13, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Gavin, C.F.; White, J.A.; Parrish, R.R.; Honasoge, A.; Yancey, C.R.; Rivera, I.M.; Rubio, M.D.; Rumbaugh, G.; Sweatt, J.D. Cortical DNA methylation maintains remote memory. Nat. Neurosci. 2010, 13, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Levenson, J.M.; Roth, T.L.; Lubin, F.D.; Miller, C.A.; Huang, I.C.; Desai, P.; Malone, L.M.; Sweatt, J.D. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006, 281, 15763–15773. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; van Groen, T.; Kadish, I.; Tollefsbol, T.O. DNA methylation impacts on learning and memory in aging. Neurobiol. Aging 2009, 30, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.M.M.; Hemstedt, T.J.; Bading, H. Rescue of aging-associated decline in DnmtDNMT3a2 expression restores cognitive abilities. Nat. Neurosci. 2012, 15, 1111. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Nabeshima, T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J. Pharmacol. Sci. 2003, 91, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Tunc-Ozcan, E.; Wert, S.L.; Lim, P.H.; Ferreira, A.; Redei, E.E. Hippocampus-dependent memory and allele-specific gene expression in adult offspring of alcohol-consuming dams after neonatal treatment with thyroxin or metformin. Mol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Adachi, M.; Na, E.S.; Monteggia, L.M. Selective role for DNMT3a in learning and memory. Neurobiol. Learn. Mem. 2014, 115, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Kenis, G.; Visser, P.J.; Scheltens, P.; Tsolaki, M.; Jones, R.W.; Kehoe, P.G.; Graff, C.; Girtler, N.G.; Wallin, Å.K.; et al. DNMT3A moderates cognitive decline in subjects with mild cognitive impairment: Replicated evidence from two mild cognitive impairment cohorts. Epigenomics 2015, 7, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Bey, K.; Wolfsgruber, S.; Karaca, I.; Wagner, H.; Lardenoije, R.; Becker, J.; Milz, E.; Kornhuber, J.; Peters, O.; Frölich, L.; et al. No association of the variant rs11887120 in DNMT3A with cognitive decline in individuals with mild cognitive impairment. Epigenomics 2016, 8, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.M.M.; Hemstedt, T.J.; Freitag, H.E.; Bading, H. Dnmt3a2: A hub for enhancing cognitive functions. Mol. Psychiatry 2016, 21, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Córdova-Palomera, A.; Fatjó-Vilas, M.; Kebir, O.; Gastó, C.; Krebs, M.O.; Fañanás, L. Polymorphic variation in the epigenetic gene DNMT3B modulates the environmental impact on cognitive ability: A twin study. Eur. Psychiatry 2015, 30, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Association, A. 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar]
- Irier, H.A.; Jin, P. Dynamics of DNA methylation in aging and Alzheimer’s disease. DNA Cell Biol. 2012, 31, S42–S48. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Jones, L.; Owen, M.J.; Williams, J. Alzheimer’s disease genetics: Current knowledge and future challenges. Int. J. Geriatr. Psychiatry 2011, 26, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.Y.; Choi, E.N.; Jo, S.A.; Oh, S.; Ahn, J.-H. Amyloid protein-mediated differential DNA methylation status regulates gene expression in Alzheimer’s disease model cell line. Biochem. Biophys. Res. Commun. 2011, 414, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol. Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; van den Hove, D.L. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Liu, Z.; Liu, W.; Jiang, L.; Zhang, R.; Cui, D.; Zhang, Q.; Xu, S. DNA Methylation and Tag SNPs of the BDNF Gene in Conversion of Amnestic Mild Cognitive Impairment into Alzheimer’s Disease: A Cross-Sectional Cohort Study. J. Alzheimer’s Dis. 2017, 58, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, J.D.; Ulland, T.K.; Colonna, M.; Holtzman, D.M. Elucidating the Role of TREM2 in Alzheimer’s Disease. Neuron 2017, 94, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Yoshino, Y.; Yamazaki, K.; Sao, T.; Mori, Y.; Ochi, S.; Yoshida, T.; Mori, T.; Iga, J.I.; Ueno, S.I. DNA methylation changes at TREM2 intron 1 and TREM2 mRNA expression in patients with Alzheimer’s disease. J. Psychiatr. Res. 2017, 92, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Boden, K.A.; Barber, I.S.; Clement, N.; Patel, T.; Guetta-Baranes, T.; Brookes, K.J.; Chappell, S.; Craigon, J.; Chapman, N.H.; Morgan, K.; et al. Methylation profiling RIN3 and MEF2C identifies epigenetic marks associated with sporadic early onset Alzheimer’s disease. J. Alzheimer’s Dis. Rep. 2017, 1, 97–108. [Google Scholar] [CrossRef]
- Roubroeks, J.A.Y.; Smith, R.G.; van den Hove, D.L.A.; Lunnon, K. Epigenetics and DNA methylomic profiling in Alzheimer’s disease and other neurodegenerative diseases. J. Neurochem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Steward, O.; Worley, P.F. Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activation. Neuron 2001, 30, 227–240. [Google Scholar] [CrossRef]
- Li, F.; Tsien, J.Z. Memory and the NMDA receptors. N. Engl. J. Med. 2009, 361, 302. [Google Scholar] [CrossRef] [PubMed]
- Plath, N.; Ohana, O.; Dammermann, B.; Errington, M.L.; Schmitz, D.; Gross, C.; Mao, X.; Engelsberg, A.; Mahlke, C.; Welzl, H.; et al. Arc/Arg3. 1 is essential for the consolidation of synaptic plasticity and memories. Neuron 2006, 52, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.D.; Kavalali, E.T.; Monteggia, L.M. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J. Neurosci. 2008, 28, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Lubin, F.D. Epigenetic gene regulation in the adult mammalian brain: Multiple roles in memory formation. Neurobiol. Learn. Mem. 2011, 96, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Halder, R.; Hennion, M.; Vidal, R.O.; Shomroni, O.; Rahman, R.U.; Rajput, A.; Centeno, T.P.; van Bebber, F.; Capece, V.; Vizcaino, J.C.; et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat. Neurosci. 2016, 19, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, G.; Kreutz, M.R. Neuronal DNA Methyltransferases: Epigenetic Mediators between Synaptic Activity and Gene Expression? Neuroscientist 2017, 1, 1073858417707457. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, K.; Bernardi, G. Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene 2004, 333, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Ioshikhes, I.P.; Zhang, M.Q. Large-scale human promoter mapping using CpG islands. Nat. Genet. 2000, 26, 61. [Google Scholar] [PubMed]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484. [Google Scholar] [CrossRef] [PubMed]
- Dyrvig, M.; Gøtzsche, C.R.; Woldbye, D.P.D.; Lichota, J. Epigenetic regulation of Dnmt3a and Arc gene expression after electroconvulsive stimulation in the rat. Mol. Cell. Neurosci. 2015, 67, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T. The role of methylation in gene expression. Nat. Educ. 2008, 1, 116. [Google Scholar]
- Zhang, D.; Wu, B.; Wang, P.; Wang, Y.; Lu, P.; Nechiporuk, T.; Floss, T.; Greally, J.M.; Zheng, D.; Zhou, B. Non-CpG methylation by DNMT3B facilitates REST binding and gene silencing in developing mouse hearts. Nucleic Acids Res. 2017, 45, 3102–3115. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Sabato, S.; Hellman, A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013, 14, R21. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J.; Handy, D.E. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease (2013 Grover Conference series). Pulm. Circ. 2014, 4, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchal, C.; Miotto, B. Emerging concept in DNA methylation: Role of transcription factors in shaping DNA methylation patterns. J. Cell. Physiol. 2015, 230, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Toperoff, G.; Rosenberg, M.; Hellman, A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2010, 20, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Meehan, R.R.; Bird, A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993, 21, 4886–4892. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Su, S.C.; Hrvatin, S.; Greben, A.W.; Renthal, W.; Boxer, L.D.; Nagy, M.A.; Hochbaum, D.R.; Kinde, B.; Gabel, H.W.; et al. Early-life gene expression in neurons modulates lasting epigenetic states. Cell 2017, 171, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.-H.; Johnson, C.A.; Laherty, C.D. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.H.; Zhang, Y.; Hendrich, B.; Johnson, C.A.; Turner, B.M.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D.; Bird, A. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet. 1999, 23, 58. [Google Scholar]
- Jia, Y.; Li, P.; Fang, L.; Zhu, H.; Xu, L.; Cheng, H.; Zhang, J.; Li, F.; Feng, Y.; Li, Y.; et al. Negative regulation of DNMT3A de novo DNA methylation by frequently overexpressed UHRF family proteins as a mechanism for widespread DNA hypomethylation in cancer. Cell Discov. 2016, 2, 16007. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, Q.; Li, P.; Zhao, Q.; Zhang, J.; Li, J.; Koseki, H.; Wong, J. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun. 2013, 4, 1563. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, V.; Bayam, E.; Cernilogar, F.M.; Bonapace, I.M.; Schulze, M.; Riemenschneider, M.J.; Schotta, G.; Götz, M. Loss of Uhrf1 in neural stem cells leads to activation of retroviral elements and delayed neurodegeneration. Genes Dev. 2016, 30, 2199–2212. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Hennekam, R.C. Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. A 2006, 140, 2603–2624. [Google Scholar] [CrossRef] [PubMed]
- Saffrey, M.J. Cellular changes in the enteric nervous system during ageing. Dev. Biol. 2013, 382, 344–355. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, D.; Xu, X. DNA Methyltransferases, DNA Methylation, and Age-Associated Cognitive Function. Int. J. Mol. Sci. 2018, 19, 1315. https://doi.org/10.3390/ijms19051315
Cui D, Xu X. DNA Methyltransferases, DNA Methylation, and Age-Associated Cognitive Function. International Journal of Molecular Sciences. 2018; 19(5):1315. https://doi.org/10.3390/ijms19051315
Chicago/Turabian StyleCui, Di, and Xiangru Xu. 2018. "DNA Methyltransferases, DNA Methylation, and Age-Associated Cognitive Function" International Journal of Molecular Sciences 19, no. 5: 1315. https://doi.org/10.3390/ijms19051315
APA StyleCui, D., & Xu, X. (2018). DNA Methyltransferases, DNA Methylation, and Age-Associated Cognitive Function. International Journal of Molecular Sciences, 19(5), 1315. https://doi.org/10.3390/ijms19051315