An Integrated Approach of Proteomics and Computational Genetic Modification Effectiveness Analysis to Uncover the Mechanisms of Flood Tolerance in Soybeans



Abstract

1. Introduction
2. Results
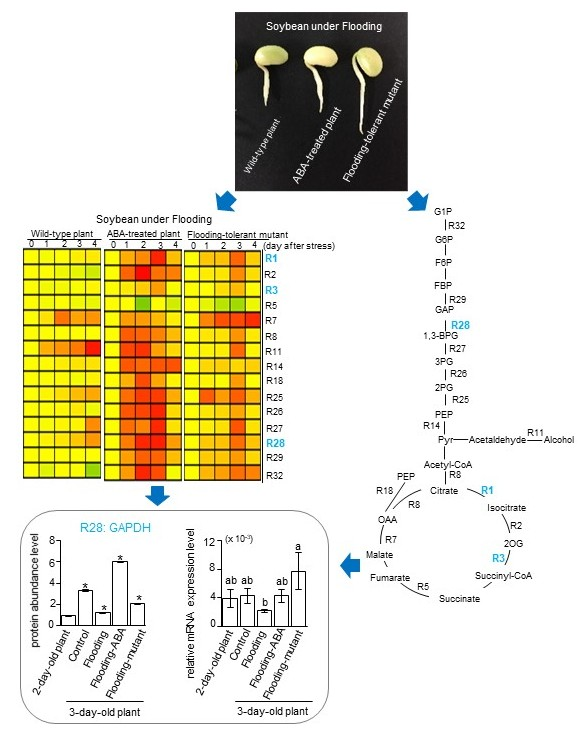
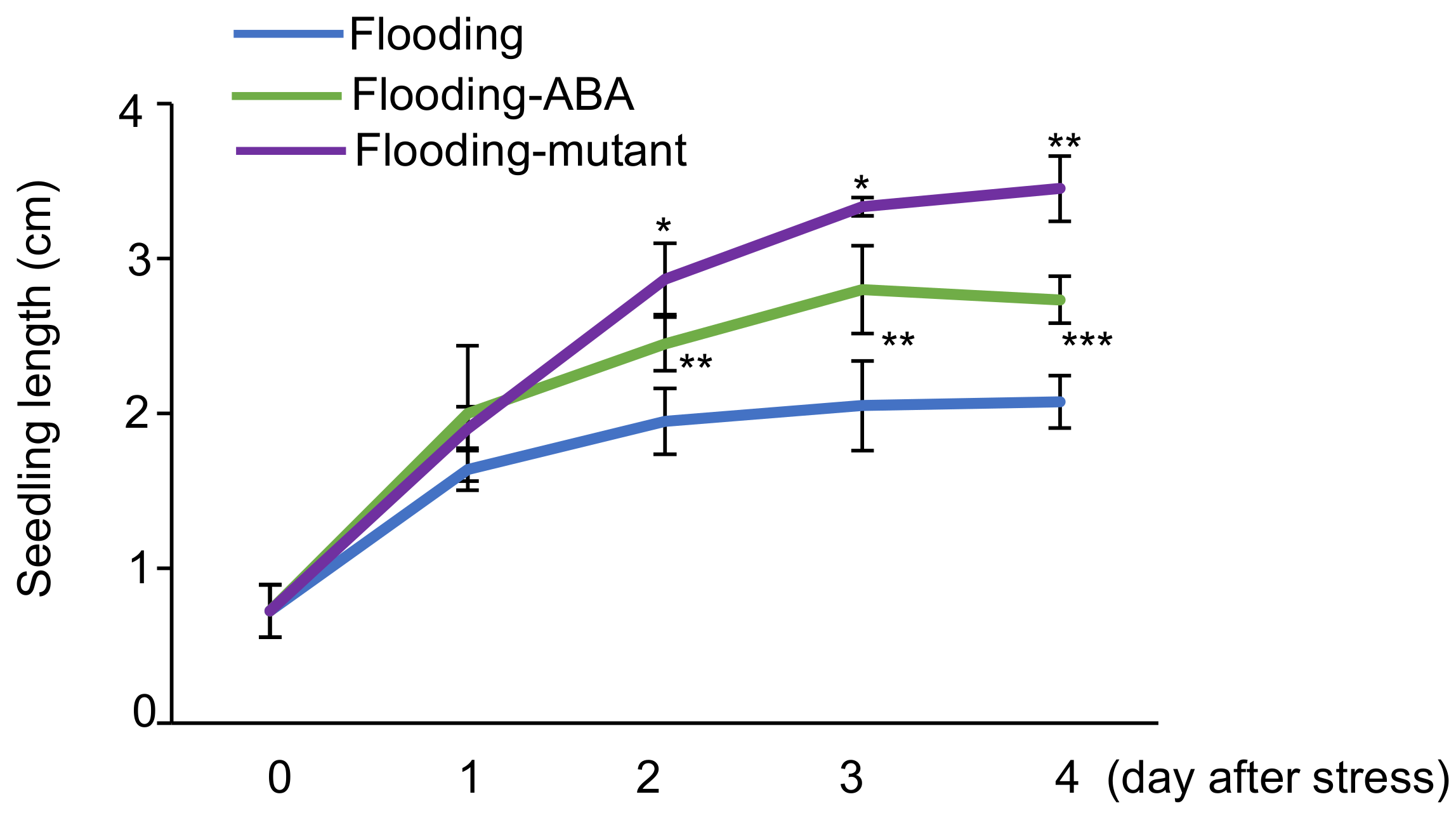
2.1. Effects of Flooding on Soybean Morphology
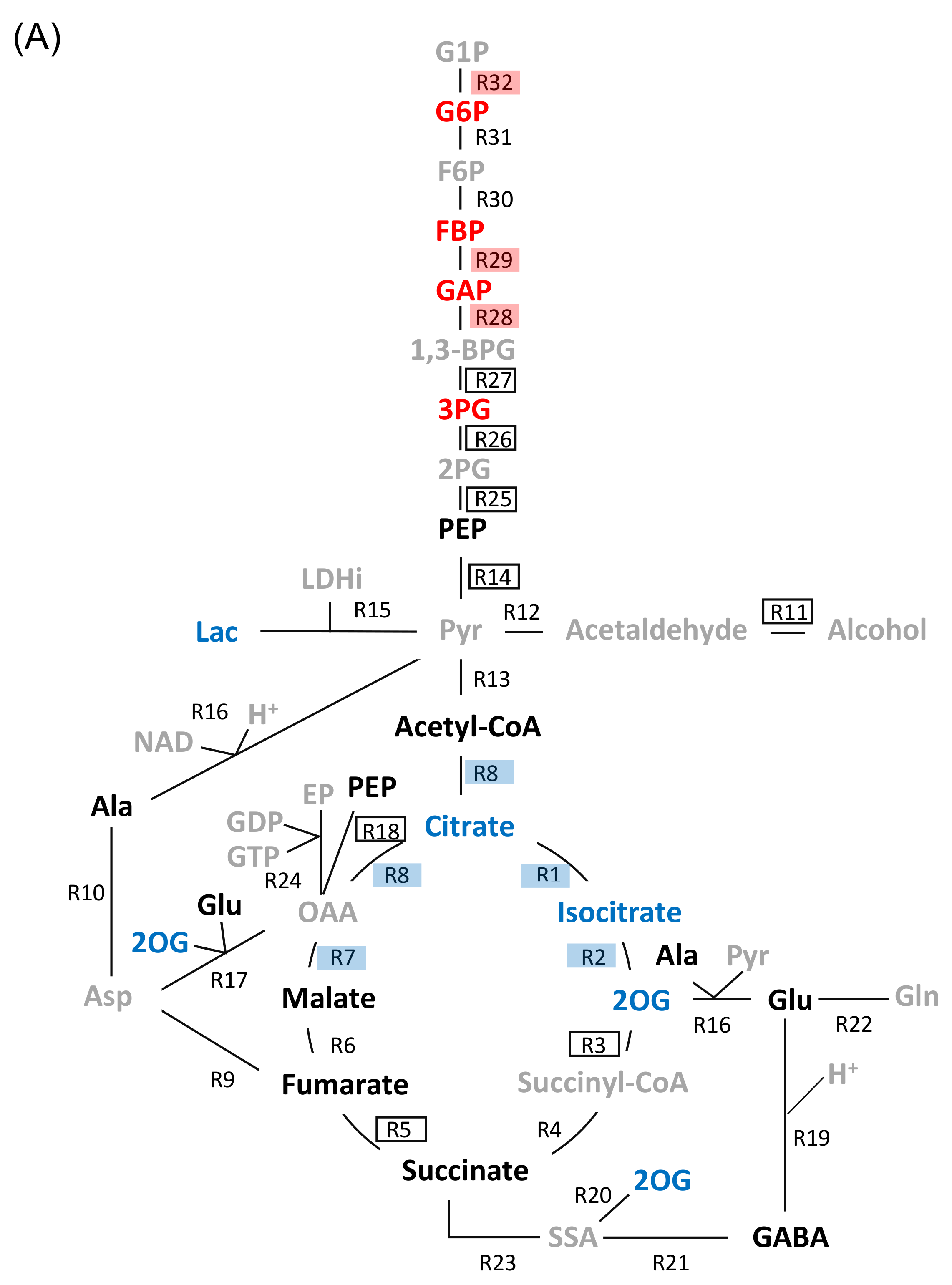
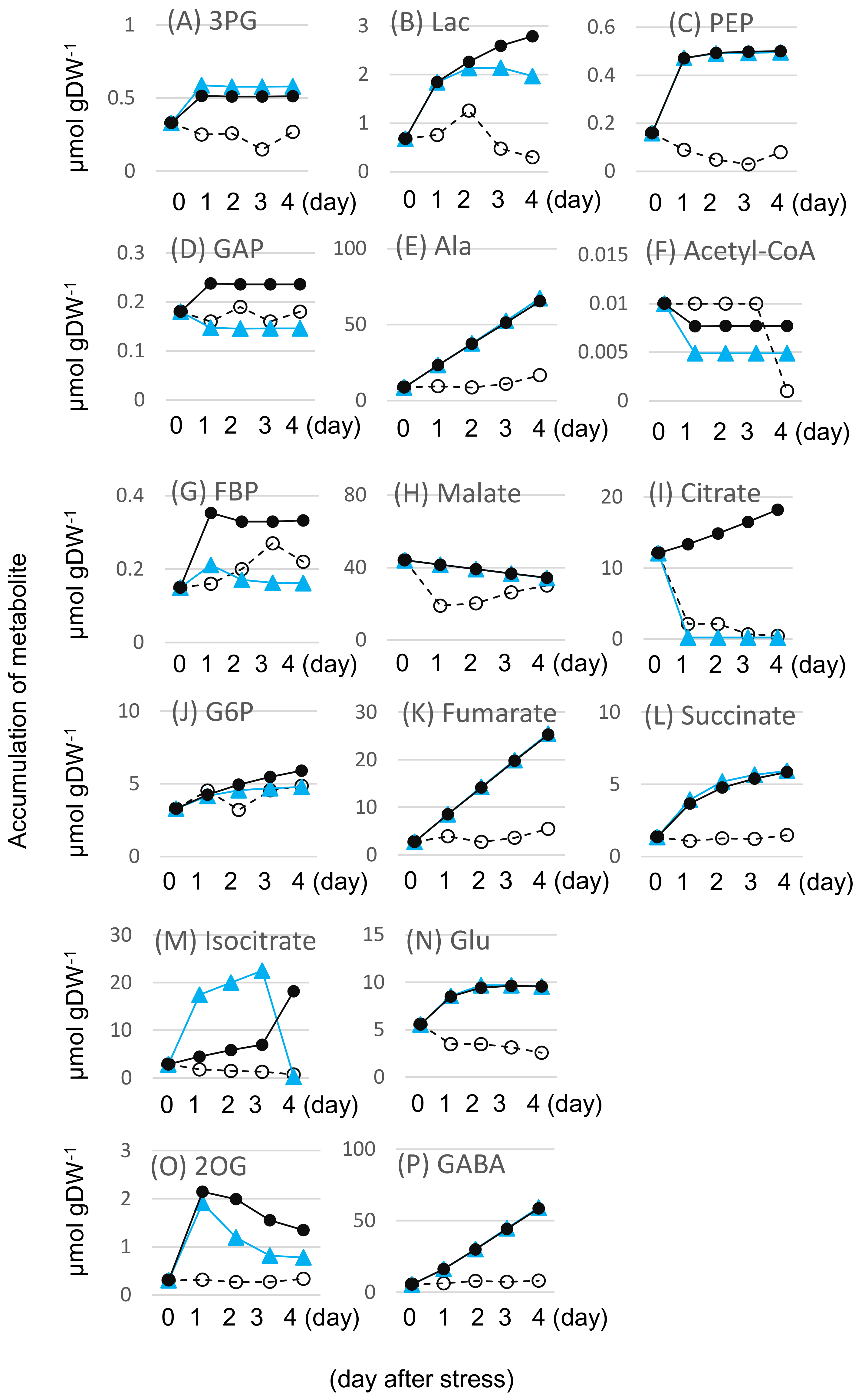
2.2. Effects of Flooding on the Primary Metabolism in Soybeans
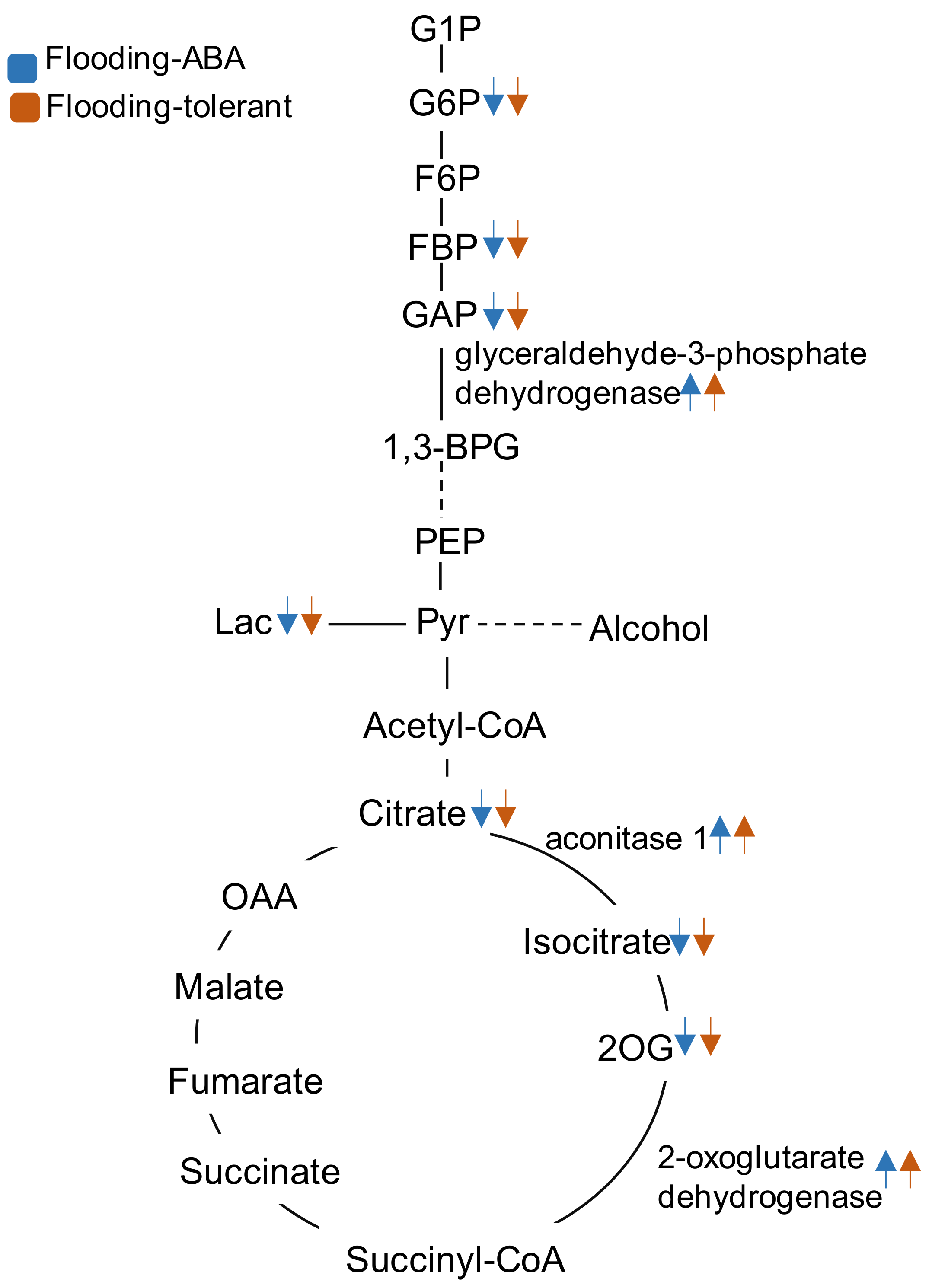
2.3. Genetic Modification Effectiveness Analysis Related to Glycolysis, Fermentation, and Tricarboxylic Acid Cycle in Soybeans
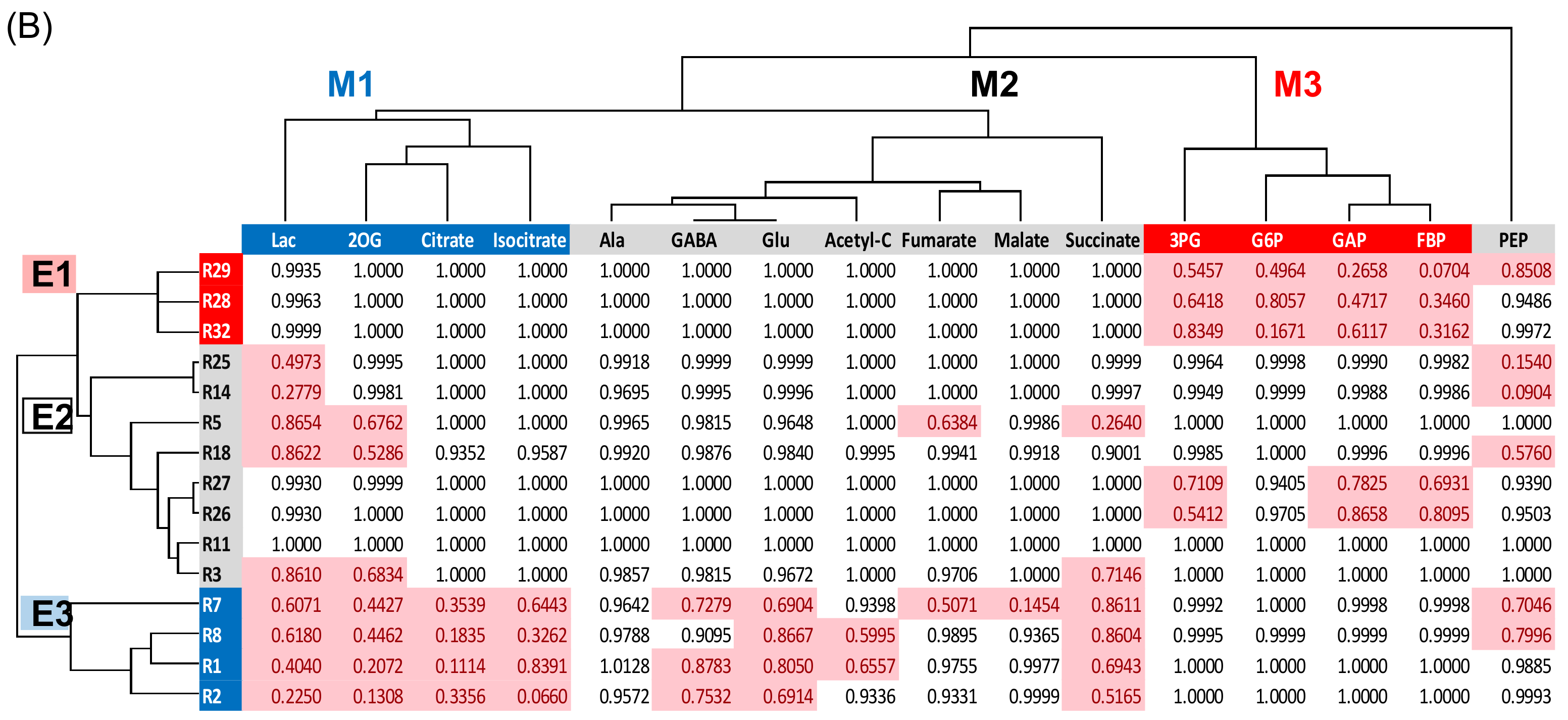
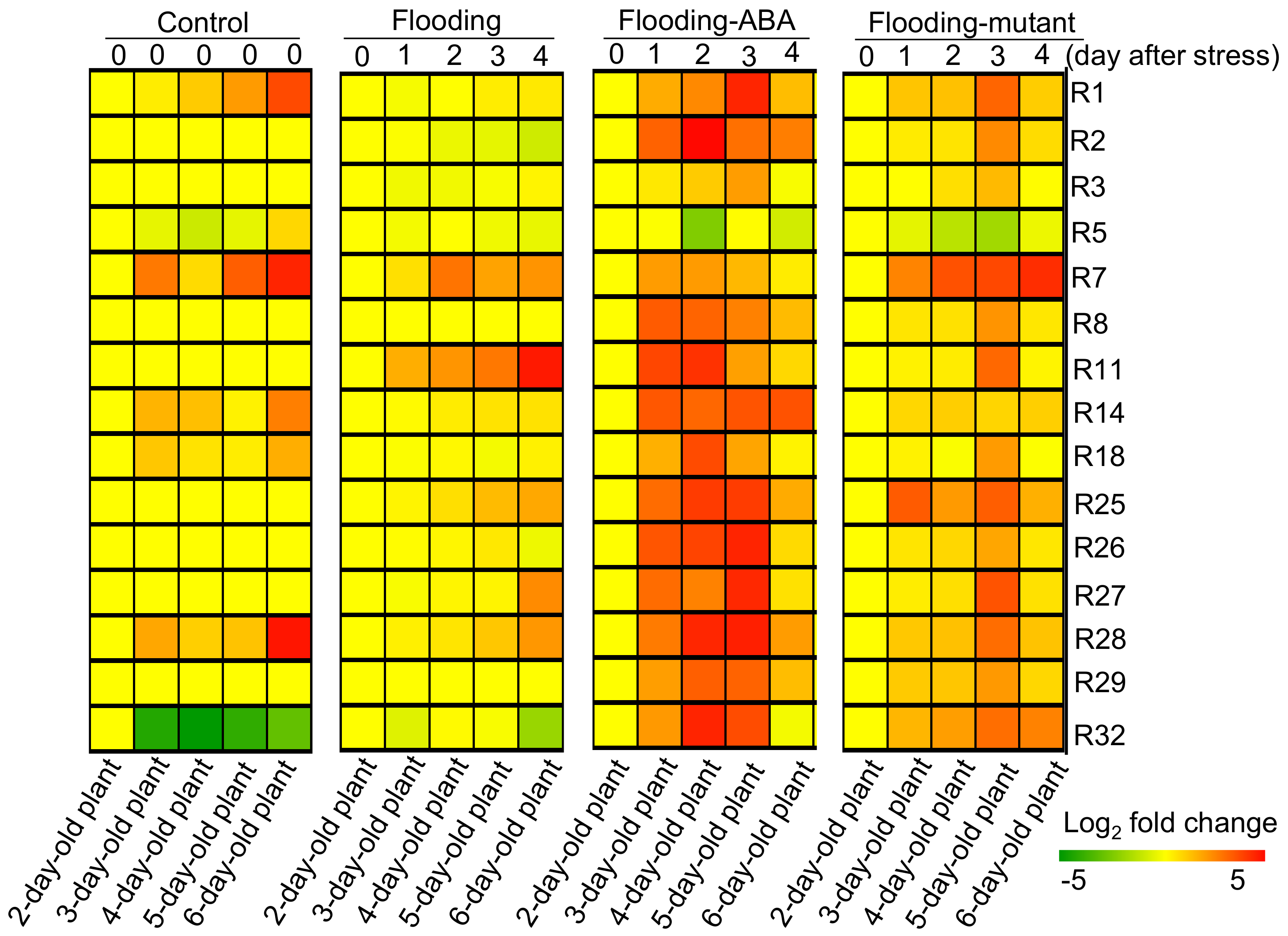
2.4. Cluster Analysis of Proteins Related to Glycolysis, Fermentation, and Tricarboxylic Acid Cycle in Soybeans
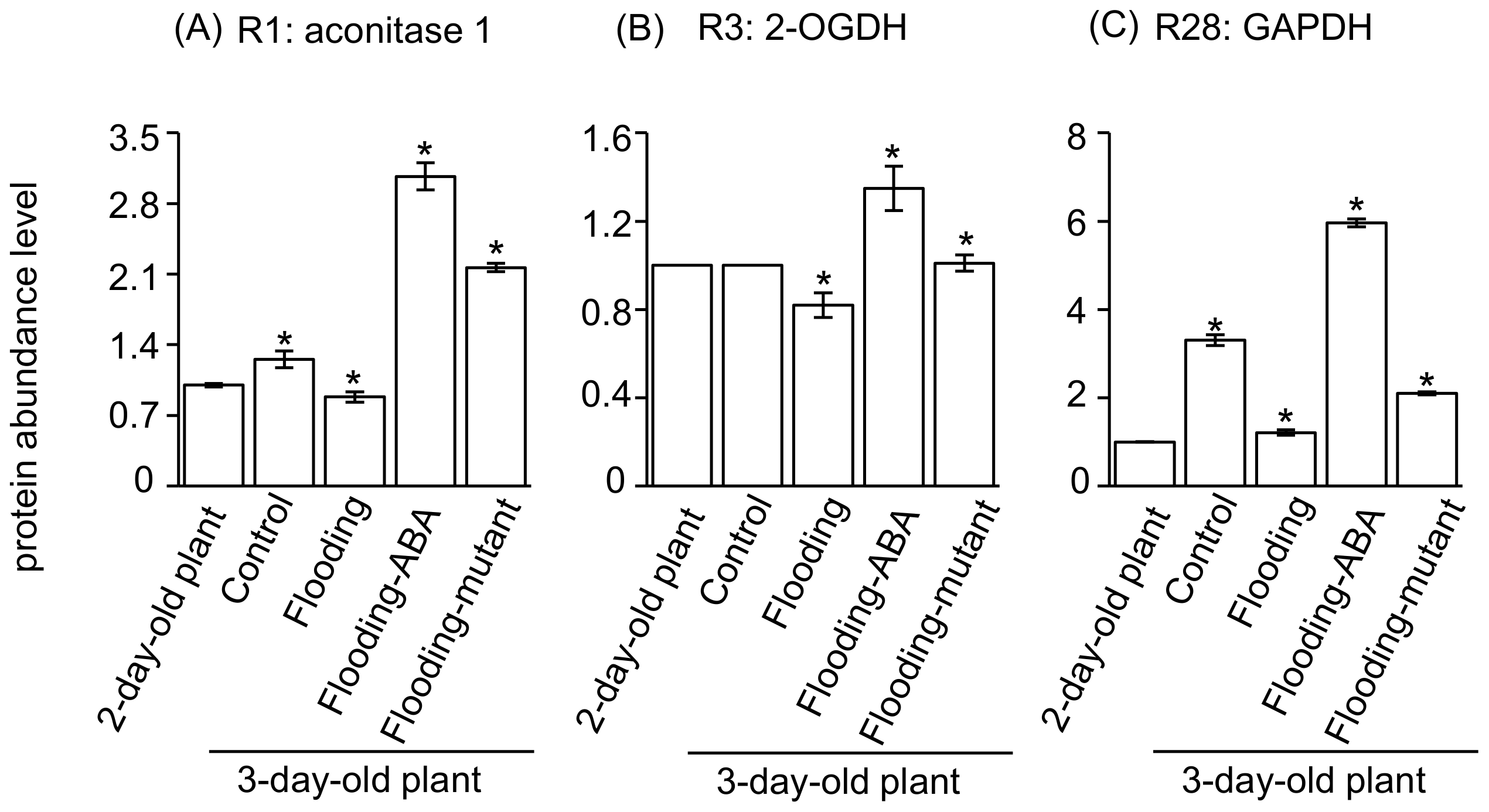
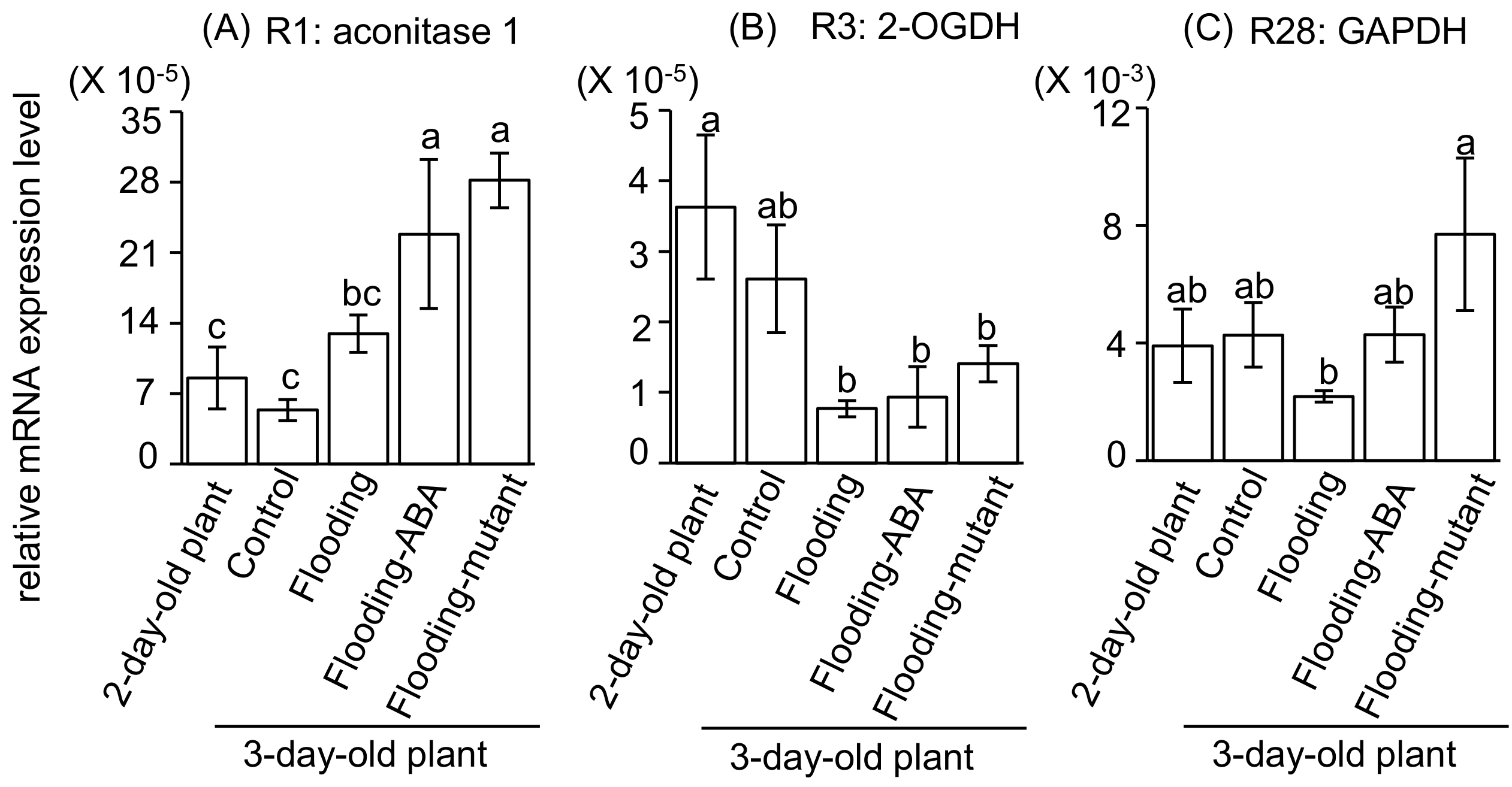
2.5. Integration of Protein Level and Transcript Level
3. Discussion
3.1. Glyceraldehyde-3-Phosphate Dehydrogenase in Glycolysis Mediate Flooding Tolerance in Soybeans
3.2. Aconitase 1 and 2-Oxoglutarate Dehydrogenase in Tricarboxylic Acid Cycle Regulate Flooding Tolerance in Soybeans
3.3. Proteomic Analysis and Metabolic Models as a Complementary Strategy to Screen Flood-Tolerance Factors in Glycolysis
4. Materials and Methods
4.1. Plant Materials and Treatments
4.2. Extraction of Proteins
4.3. Enrichment and Digestion of Proteins
4.4. Mass Spectrometry Analysis
4.5. Protein Identification from Acquired Mass Spectrometry Data
4.6. Analysis of Differential Protein Abundance
4.7. Functional Categorization
4.8. Cluster Analysis of Protein Level
4.9. Metabolic Network Modeling
4.10. RNA Extraction and Quantitative Reverse Transcription-Polymerase Chain Reaction
4.11. Statistical Analysis
4.12. Accession Codes
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| ABA | Abscisic Acid |
| LC | Liquid Chromatography |
| GAPDH | Glyceraldehyde-3-Phosphate Dehydrogenase |
| MS | Mass Spectrometry |
| qRT-PCR | Quantitative Reverse Transcription-Polymerase Chain Reaction |
References
- Kim, E.H.; Ro, H.M.; Kim, S.L.; Kim, H.S.; Chung, I.M. Analysis of isoflavone, phenolic, soyasapogenol, and tocopherol compounds in soybean [Glycine max (L.) Merrill] germplasms of different seed weights and origins. J. Agric. Food Chem. 2012, 60, 6045–6055. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, A.; Ueda, Y.; Takase, H.; Yazaki, K. Do soybeans select specific species of bradyrhizobium during growth? Commun. Integr. Biol. 2015, 8, e992734. [Google Scholar] [CrossRef] [PubMed]
- Li, M.W.; Xin, D.; Gao, Y.; Li, K.P.; Fan, K.; Muñoz, N.B.; Yung, W.S.; Lam, H.M. Using genomic information to improve soybean adaptability to climate change. J. Exp. Bot. 2017, 68, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Oh, M.; Sakata, K.; Komatsu, S. Gel-free/label-free proteomic analysis of root tip of soybean over time under flooding and drought stresses. J. Proteom. 2016, 130, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Komatsu, S. Proteomic analysis of calcium effects on soybean root tip under flooding and drought stresses. Plant Cell Physiol. 2017, 58, 1405–1420. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Komatsu, S. Gel-free/label-free proteomic analysis of endoplasmic reticulum proteins in soybean root tips under flooding and drought stresses. J. Proteom. Res. 2016, 15, 2211–2227. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Komatsu, S. Characterization of proteins in soybean roots under flooding and drought stresses. J. Proteom. 2015, 114, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Oh, M.; Komatsu, S. Characterization of S-adenosylmethionine synthetases in soybean under flooding and drought stresses. Biol. Plant. 2016, 60, 269–278. [Google Scholar] [CrossRef]
- Khatoon, A.; Rehman, S.; Hiraga, S.; Makino, T.; Komatsu, S. Organ-specific proteomics analysis for identification of response mechanism in soybean seedlings under flooding stress. J. Proteom. 2012, 75, 5706–5723. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Yamamoto, R.; Nanjo, Y.; Mikami, Y.; Yunokawa, H.; Sakata, K. A comprehensive analysis of the soybean genes and proteins expressed under flooding stress using transcriptome and proteome techniques. J. Proteom. Res. 2009, 8, 4766–4778. [Google Scholar] [CrossRef] [PubMed]
- Salavati, A.; Khatoon, A.; Nanjo, Y.; Komatsu, S. Analysis of proteomic changes in roots of soybean seedlings during recovery after flooding. J. Proteom. 2012, 75, 878–893. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.N.; Sakata, K.; Hiraga, S.; Komatsu, S. Quantitative proteomics reveals that peroxidases play key roles in post-flooding recovery in soybean roots. J. Proteom. Res. 2014, 13, 5812–5828. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Nanjo, Y.; Nishimura, M. Proteomic analysis of the flooding tolerance mechanism in mutant soybean. J. Proteom. 2013, 79, 231–250. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Han, C.; Nanjo, Y.; Altaf-Un-Nahar, M.; Wang, K.; He, D.; Yang, P. Label-free quantitative proteomic analysis of abscisic acid effect in early-stage soybean under flooding. J. Proteom. Res. 2013, 12, 4769–4784. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Nishimura, M.; Hajika, M.; Komatsu, S. Quantitative proteomics reveals the flooding-tolerance mechanism in mutant and abscisic acid-treated soybean. J. Proteom. Res. 2016, 15, 2008–2025. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, T.; Shinozaki, K. Research on plant abiotic stress responses in the post-genome era: Past, present and future. Plant. J. 2010, 61, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Shirasaka, N.; Sakata, K. ‘Omics’ techniques for identifying flooding-response mechanisms in soybean. J. Proteom. 2013, 93, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Sakata, K.; Ohyanagi, H.; Nobori, H.; Nakamura, T.; Hashiguchi, A.; Nanjo, Y.; Mikami, Y.; Yunokawa, H.; Komatsu, S. Soybean proteome database: A data resource for plant differential omics. J. Proteom. Res. 2009, 8, 3539–3548. [Google Scholar] [CrossRef] [PubMed]
- Shinto, H.; Tashiro, Y.; Yamashita, M.; Kobayashi, G.; Sekiguchi, T.; Hanai, T.; Kuriya, Y.; Okamoto, M.; Sonomoto, K. Kinetic modeling and sensitivity analysis of acetone-butanol-ethanol production. J. Biotechnol. 2007, 131, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Sakata, K.; Saito, T.; Ohyanagi, H.; Okumura, J.; Ishige, K.; Suzuki, H.; Nakamura, T.; Komatsu, S. Loss of variation of state detected in soybean metabolic and human myelomonocytic leukaemia cell transcriptional networks under external stimuli. Sci. Rep. 2016, 6, 35946. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Yamamoto, R.; Hiraga, S.; Nakayama, N.; Okazaki, K.; Takahashi, H.; Uchimiya, H.; Komatsu, S. Evaluation of metabolite alteration under flooding stress in soybeans. JARQ-Jpn. Agric. Res. Q. 2012, 46, 237–248. [Google Scholar] [CrossRef]
- Walley, J.W.; Sartor, R.C.; Shen, Z.; Schmitz, R.J.; Wu, K.J.; Urich, M.A.; Nery, J.R.; Smith, L.G.; Schnable, J.C.; Ecker, J.R.; et al. Integration of omic networks in a developmental atlas of maize. Science 2016, 353, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Duan, X.; Wang, B.; Hong, B.; Ho, T.H.D.; Wu, R. Expression of a late embryogenesis abundant protein gene, HVA1, from barley confers tolerance to water deficit and salt stress in transgenic rice. Plant Physiol. 1996, 110, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Nanjo, Y.; Skultety, L.; Ashraf, Y.; Komatsu, S. Comparative proteomic analysis of early-stage soybean seedlings responses to flooding by using gel and gel-free techniques. J. Proteom. Res. 2010, 9, 3989–4002. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, W.; Hashiguchi, A.; Nishimura, M.; Tian, J.; Komatsu, S. Metabolic profiles of flooding-tolerant mechanism in early-stage soybean responding to initial stress. Plant. Mol. Biol. 2017, 94, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Plaxton, W.C. The organization and regulation of plant glycolysis. Annu. Rev. Plant Physiol. Plant. Mol. Biol. 1996, 47, 185–214. [Google Scholar] [CrossRef] [PubMed]
- Velasco, R.; Salamini, F.; Bartels, D. Dehydration and ABA increase mRNA levels and enzyme activity of cytosolic GAPDH in the resurrection plant Craterostigma plantagineum. Plant. Mol. Biol. 1994, 26, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kwon, H.B.; Peng, H.P.; Shih, M.C. Stress responses and metabolic regulation of glyceraldehyde-3-phosphate dehydrogenase genes in Arabidopsis. Plant Physiol. 1993, 101, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Deng, R.; Guo, Z.; Yang, S.; Deng, X. Genome-wide identification and characterization of glyceraldehyde-3-phosphate dehydrogenase genes family in wheat (Triticum aestivum). BMC Genom. 2016, 17, 240. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.I.; Lim, H.M.; Siddiqui, Z.S.; Park, S.H.; Kim, A.R.; Kwon, T.R.; Lee, S.K.; Park, S.C.; Jeong, M.J.; Lee, G.S. Over-expression of PsGPD, a mushroom glyceraldehyde-3-phosphate dehydrogenase gene, enhances salt tolerance in rice plants. Biotechnol. Lett. 2014, 36, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Bertomeu, J.; Cascales-Miñana, B.; Mulet, J.M.; Baroja-Fernández, E.; Pozueta-Romero, J.; Kuhn, J.M.; Segura, J.; Ros, R. Plastidial glyceraldehyde-3-phosphate dehydrogenase deficiency leads to altered root development and affects the sugar and amino acid balance in Arabidopsis. Plant Physiol. 2009, 151, 541–558. [Google Scholar] [CrossRef] [PubMed]
- Sweetlove, L.J.; Heazlewood, J.L.; Herald, V.; Holtzapffel, R.; Day, D.A.; Leaver, C.J.; Millar, A.H. The impact of oxidative stress on Arabidopsis mitochondria. Plant J. 2002, 32, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Yamamoto, A.; Nakamura, T.; Nouri, M.Z.; Nanjo, Y.; Nishizawa, K.; Furukawa, K. Comprehensive analysis of mitochondria in roots and hypocotyls of soybean under flooding stress using proteomics and metabolomics techniques. J. Proteom. Res. 2011, 10, 3993–4004. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.H.; Komatsu, S. Involvement of reactive oxygen species and mitochondrial proteins in biophoton emission in roots of soybean plants under flooding stress. J. Proteom. Res. 2015, 14, 2219–2236. [Google Scholar] [CrossRef] [PubMed]
- Konert, G.; Trotta, A.; Kouvonen, P.; Rahikainen, M.; Durian, G.; Blokhina, O.; Fagerstedt, K.; Muth, D.; Corthals, G.L.; Kangasjärvi, S. Protein phosphatase 2A (PP2A) regulatory subunit B’γ interacts with cytoplasmic ACONITASE 3 and modulates the abundance of AOX1A and AOX1D in Arabidopsis thaliana. New Phytol. 2015, 205, 1250–1263. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.J.; Shah, J.K.; Brotman, Y.; Jahnke, K.; Willmitzer, L.; Kaiser, W.M.; Bauwe, H.; Igamberdiev, A.U. Inhibition of aconitase by nitric oxide leads to induction of the alternative oxidase and to a shift of metabolism towards biosynthesis of amino acids. J. Exp. Bot. 2012, 63, 1773–1784. [Google Scholar] [CrossRef] [PubMed]
- Navarre, D.A.; Wendehenne, D.; Durner, J.; Noad, R.; Klessig, D.F. Nitric oxide modulates the activity of tobacco aconitase. Plant Physiol. 2000, 122, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V.I.; Fernie, A.R. Metabolic control exerted by the 2-oxoglutarate dehydrogenase reaction: A cross-kingdom comparison of the crossroad between energy production and nitrogen assimilation. Biochem. J. 2009, 422, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Araújo, W.L.; Nunes-Nesi, A.; Trenkamp, S.; Bunik, V.I.; Fernie, A.R. Inhibition of 2-oxoglutarate dehydrogenase in potato tuber suggests the enzyme is limiting for respiration and confirms its importance in nitrogen assimilation. Plant Physiol. 2008, 148, 1782–1796. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Bryant, D.A. The tricarboxylic acid cycle in cyanobacteria. Science 2011, 334, 1551–1553. [Google Scholar] [CrossRef] [PubMed]
- Salavati, A.; Bushehri, A.A.; Taleei, A.; Hiraga, S.; Komatsu, S. A comparative proteomic analysis of the early response to compatible symbiotic bacteria in the roots of a supernodulating soybean variety. J. Proteom. 2012, 75, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Nanjo, Y.; Skultety, L.; Uvackova, L.; Klubicova, K.; Hajduch, M.; Komatsu, S. Mass spectrometry-based analysis of proteomic changes in the root tips of flooded soybean seedlings. J. Proteom. Res. 2012, 11, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; de Godoy, L.M.; Li, G.; Macek, B.; Mortensen, P.; Pesch, R.; Makarov, A.; Lange, O.; Horning, S.; Mann, M. Parts per million mass accuracy on an orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteom. 2005, 4, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wen, Z.; Washburn, M.P.; Florens, L. Effect of dynamic exclusion duration on spectral count based quantitative proteomics. Anal. Chem. 2009, 81, 6317–6326. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopholyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Brosch, M.; Yu, L.; Hubbard, T.; Choudhary, J. Accurate and sensitive peptide identification with mascot percolator. J. Proteom. Res. 2009, 8, 3176–3181. [Google Scholar] [CrossRef] [PubMed]
- Usadel, B.; Nagel, A.; Thimm, O.; Redestig, H.; Blaesing, O.E.; Palacios Rofas, N.; Selbig, J.; Hannemann, J.; Piques, M.C.; Steinhauser, D.; et al. Extension of the visualization tool mapman to allow statistical analysis of arrays, display of corresponding genes, and comparison with known databases. Plant Physiol. 2005, 138, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Usadel, B.; Poree, F.; Nagel, A.; Lohse, M.; Czedik-Eysenberg, A.; Stitt, M. A guide to using mapman to visualize and compare omics data in plants: A case study in the crop species, maize. Plant Cell Environ. 2009, 32, 1211–1229. [Google Scholar] [CrossRef] [PubMed]
- Sturn, A.; Quackenbush, J.; Trajanoski, Z. Genesis: Cluster analysis of microarray data. Bioinformatics 2002, 18, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, I.; Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G.; Schomburg, D. BRENDA, the enzyme database: Updates and major new developments. Nucleic Acids Res. 2004, 32, D431–D433. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, T.; Okamoto, M. Winbest-kit: Windows-based biochemical reaction simulator for metabolic pathways. J. Bioinform. Comput. Biol. 2006, 4, 621–638. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Côté, R.G.; Csordas, A.; Dianes, J.A.; Fabregat, A.; Foster, J.M.; Griss, J.; Alpi, E.; Birim, M.; Contell, J.; et al. The Proteomics Identifications (PRIDE) Database and Associated Tools: Status in 2013. Nucleic Acids Res. 2013, 41, D1063–D1069. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Hiraga, S.; Hajika, M.; Nishimura, M.; Komatsu, S. Transcriptomic Analysis Reveals the Flooding Tolerant Mechanism in Flooding Tolerant Line and Abscisic Acid Treated Soybean. Plant Mol. Biol. 2017, 93, 479–496. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Protein ID | Description | Ratio (Control Plant) | Ratio (Flooded Wile-Type Soybean) | Functional Category | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2-day-old | 3-day-old | 4-day-old | 5-day-old | 6-day-old | 1-day-flooded | 2-day-flooded | 3-day-flooded | 4-day-flooded | |||||
| 1 | R1a/b | Glyma11g08550.1 | aconitase 1 | 1 | 1.26 | 2.01 | 3.88 | 11.61 | 0.88 | 1.00 | 1.27 | 1.34 | TCA cycle |
| 2 | R2 | Glyma10g06590.1 | isocitrate dehydrogenase V | 1 | 1.00 | 1.00 | 1.00 | 1.00 | 0.96 | 0.77 | 0.71 | 0.51 | TCA cycle |
| 3 | R3a/b | Glyma18g52430.2 | 2-oxoglutarate dehydrogenase | 1 | 1.00 | 1.00 | 1.00 | 1.00 | 0.82 | 0.83 | 0.90 | 1.16 | TCA cycle |
| 4 | R5a/b | Glyma11g07250.1 | succinate dehydrogenase 1-1 | 1 | 0.72 | 0.49 | 0.71 | 1.70 | 0.86 | 1.00 | 0.81 | 0.74 | TCA cycle |
| 5 | R7a/b | Glyma06g34190.1 | malate dehydrogenase | 1 | 6.11 | 1.61 | 8.88 | 19.44 | 1.51 | 6.55 | 3.48 | 4.28 | TCA cycle |
| 6 | R8c | Glyma15g42301.1 | citrate synthase family protein | 1 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | TCA cycle |
| 7 | R11a/b | Glyma06g12780.1 | alcohol dehydrogenase 1 | 1 | 1.00 | 1.00 | 1.00 | 1.00 | 2.99 | 4.20 | 6.21 | 22.61 | fermentation |
| 8 | R14 | Glyma05g09310.2 | pyruvate kinase family protein | 1 | 2.74 | 2.36 | 1.19 | 5.66 | 1.06 | 1.29 | 1.46 | 1.47 | glycolysis |
| 9 | R18 | Glyma13g36670.1 | phosphoenolpyruvate carboxylase 1 | 1 | 2.12 | 1.45 | 1.26 | 2.98 | 0.96 | 1.09 | 0.86 | 1.20 | glycolysis |
| 10 | R25a/b | Glyma19g37520.1 | enolase | 1 | 1.00 | 1.00 | 1.00 | 1.00 | 1.16 | 1.53 | 2.49 | 3.25 | glycolysis |
| 11 | R26a/b | Glyma18g45121.1 | phosphoglycerate mutase | 1 | 1.00 | 1.00 | 1.00 | 1.00 | 1.03 | 1.12 | 1.35 | 0.79 | glycolysis |
| 12 | R27a/b | Glyma15g41550.1 | phosphoglycerate kinase | 1 | 1.00 | 1.00 | 1.00 | 1.00 | 0.91 | 1.14 | 1.16 | 4.82 | glycolysis |
| 13 | R28a/b | Glyma04g36860.1 | glyceraldehyde-3-phosphate dehydrogenase | 1 | 3.30 | 1.92 | 2.23 | 23.93 | 1.21 | 1.41 | 2.14 | 4.08 | glycolysis |
| 14 | R29a/b | Glyma03g34950.1 | aldolase superfamily protein | 1 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | glycolysis |
| 15 | R32a/b | Glyma20g32030.1 | phosphoglucomutase | 1 | 0.05 | 0.02 | 0.06 | 0.12 | 0.64 | 1.07 | 0.91 | 0.25 | glycolysis |
| Reaction | Protein ID | Description | Ratio (Control Plant) | Ratio (Flooded Wile-Type Soybean with ABA Treatment) | Ratio (Flooded soybean of flooding-Tolerant Mutant) | Functional Category | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2-day-old | 1-day-flooded | 2-day-flooded | 3-day-flooded | 4-day-flooded | 1-day-flooded | 2-day-flooded | 3-day-flooded | 4-day-flooded | |||||
| 1 | R1a/b | Glyma11g08550.1 | aconitase 1 | 1 | 3.07 | 4.97 | 19.17 | 2.45 | 2.17 | 2.31 | 8.04 | 1.95 | TCA cycle |
| 2 | R2 | Glyma14g39160.2 | cytosolic NADP dependent isocitrate dehydrogenase | 1 | 8.37 | 28.68 | 6.89 | 5.77 | 1.30 | 1.44 | 4.86 | 1.60 | TCA cycle |
| 3 | R3a/b | Glyma18g52430.2 | 2-oxoglutarate dehydrogenase | 1 | 1.35 | 2.00 | 3.75 | 0.91 | 1.01 | 1.53 | 2.52 | 1.03 | TCA cycle |
| 4 | R5a/b | Glyma01g38200.1 | succinate dehydrogenase 1-1 | 1 | 0.97 | 0.18 | 1.04 | 0.53 | 0.70 | 0.39 | 0.28 | 0.78 | TCA cycle |
| 5 | R7a/b | Glyma06g34190.1 | malate dehydrogenase | 1 | 3.84 | 3.88 | 2.62 | 1.28 | 5.32 | 10.52 | 11.90 | 17.32 | TCA cycle |
| 6 | R8a/b | Glyma09g04000.2 | ATP citrate lyase B1 | 1 | 9.24 | 8.11 | 5.52 | 2.51 | 1.42 | 1.50 | 4.25 | 1.37 | TCA cycle |
| 7 | R11a/b | Glyma09g08150.1 | aldehyde dehydrogenase 7B4 | 1 | 12.26 | 16.00 | 3.60 | 1.69 | 1.19 | 1.28 | 7.70 | 1.16 | fermentation |
| 8 | R14 | Glyma05g09310.2 | pyruvate kinase family protein | 1 | 9.79 | 7.88 | 10.14 | 10.41 | 1.71 | 1.93 | 1.75 | 1.91 | glycolysis |
| 9 | R18 | Glyma06g33380.1 | phosphoenolpyruvate carboxylase 3 | 1 | 2.90 | 11.44 | 3.38 | 1.17 | 1.19 | 0.93 | 3.86 | 0.97 | glycolysis |
| 10 | R25a/b | Glyma03g34830.1 | enolase | 1 | 7.34 | 14.18 | 14.10 | 3.11 | 9.18 | 3.92 | 8.87 | 2.89 | glycolysis |
| 11 | R26a/b | Glyma09g40690.1 | phosphoglycerate mutase 2,3 bisphosphoglycerate | 1 | 9.98 | 12.68 | 19.37 | 1.65 | 1.42 | 1.72 | 3.35 | 1.37 | glycolysis |
| 12 | R27a/b | Glyma08g17600.1 | phosphoglycerate kinase | 1 | 7.29 | 5.40 | 18.41 | 1.50 | 1.26 | 1.54 | 10.29 | 1.49 | glycolysis |
| 13 | R28a/b | Glyma04g36860.1 | glyceraldehyde-3-phosphate dehydrogenase | 1 | 5.96 | 19.08 | 20.78 | 3.83 | 2.10 | 2.31 | 7.22 | 2.23 | glycolysis |
| 14 | R29a/b | Glyma03g34950.1 | aldolase superfamily protein | 1 | 3.70 | 8.76 | 8.26 | 2.42 | 2.05 | 2.16 | 3.99 | 1.71 | glycolysis |
| 15 | R32a/b | Glyma05g34790.1 | phosphoglucomutase | 1 | 3.97 | 19.52 | 11.35 | 0.86 | 2.69 | 3.70 | 7.08 | 5.45 | glycolysis |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Sakata, K.; Komatsu, S. An Integrated Approach of Proteomics and Computational Genetic Modification Effectiveness Analysis to Uncover the Mechanisms of Flood Tolerance in Soybeans. Int. J. Mol. Sci. 2018, 19, 1301. https://doi.org/10.3390/ijms19051301
Wang X, Sakata K, Komatsu S. An Integrated Approach of Proteomics and Computational Genetic Modification Effectiveness Analysis to Uncover the Mechanisms of Flood Tolerance in Soybeans. International Journal of Molecular Sciences. 2018; 19(5):1301. https://doi.org/10.3390/ijms19051301
Chicago/Turabian StyleWang, Xin, Katsumi Sakata, and Setsuko Komatsu. 2018. "An Integrated Approach of Proteomics and Computational Genetic Modification Effectiveness Analysis to Uncover the Mechanisms of Flood Tolerance in Soybeans" International Journal of Molecular Sciences 19, no. 5: 1301. https://doi.org/10.3390/ijms19051301
APA StyleWang, X., Sakata, K., & Komatsu, S. (2018). An Integrated Approach of Proteomics and Computational Genetic Modification Effectiveness Analysis to Uncover the Mechanisms of Flood Tolerance in Soybeans. International Journal of Molecular Sciences, 19(5), 1301. https://doi.org/10.3390/ijms19051301