The Costimulatory Pathways and T Regulatory Cells in Ischemia-Reperfusion Injury: A Strong Arm in the Inflammatory Response?

 ,
, 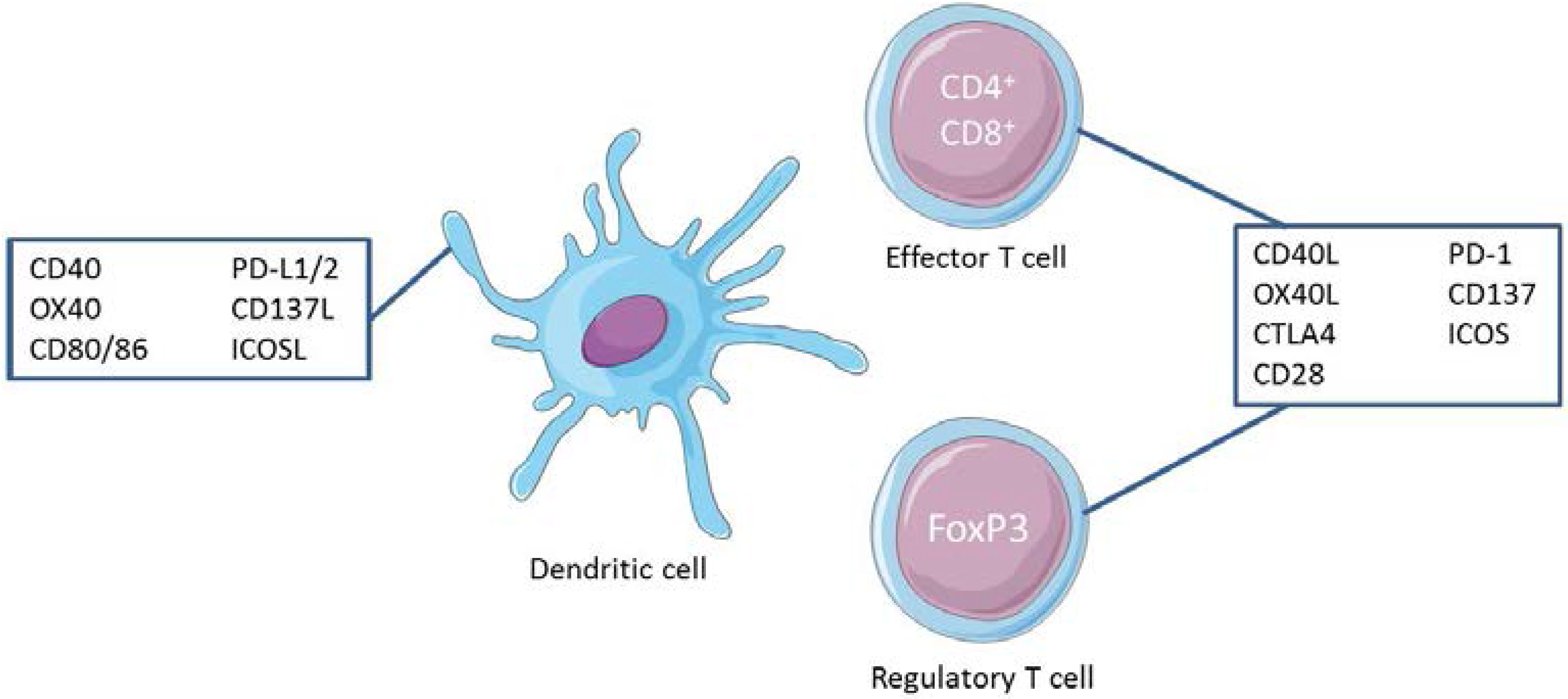{kind=link}
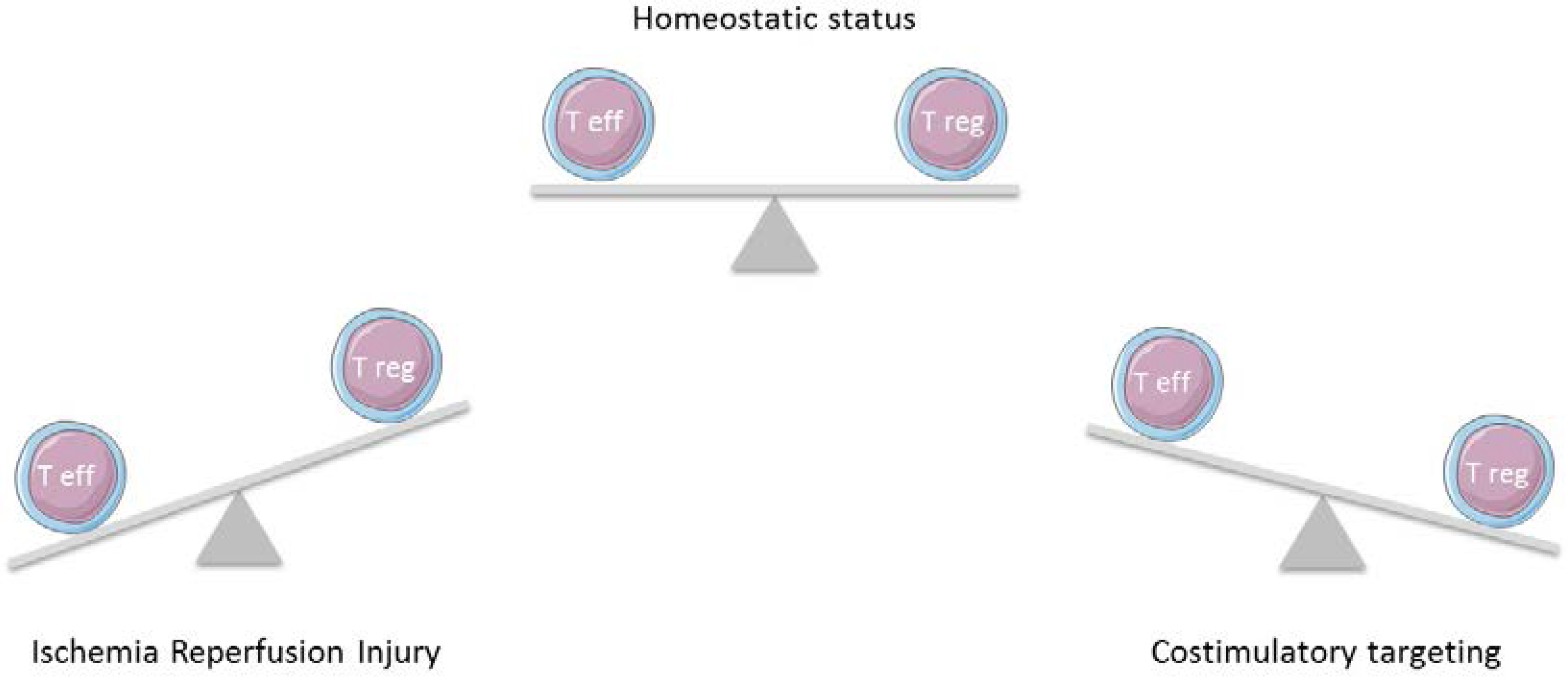{kind=link}
Abstract
:1. The Importance of Costimulatory Pathways in Ischemia-Reperfusion Injury
2. Diversity of Costimulatory Molecules
2.1. The Ig Superfamily (IgSF)
2.2. The TNF (Tumor Necrosis Factor) Superfamily
2.3. Integrins or Cell Adhesion Molecules
2.4. TIM Molecules
3. Costimulatory Blockade in IRI (Ischemia-Reperfusion Injury)
4. Role of Tregs in IRI
5. Treg Kinetics Targeting Costimulatory Pathways
5.1. Tregs and CD28/CTLA4–B7
5.2. Tregs and CD40–CD40L
5.3. Tregs and OX40–OX40L
5.4. Tregs and PD-1–PD-L1⁄PD-L2
5.5. Tregs and TIM-1 and TIM-4
6. Summary and Future Directions
Acknowledgments
Conflicts of Interest
References
- Ysebaert, D.K.; de Greef, K.E.; de Beuf, A.; van Rompay, A.R.; Vercauteren, S.; Persy, V.P.; de brOE, M.E. T cells as mediators in renal ischemia/reperfusion injury. Kidney Int. 2004, 66, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, J.M. Vascular and circulating microRNAs in renal ischaemia-reperfusion injury. J. Physiol. 2015, 593, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- De Ramon, L.; Ripoll, E.; Merino, A.; Lúcia, M.; Aran, J.M.; Pérez-Rentero, S.; Lloberas, N.; Cruzado, J.M.; Grinyó, J.M.; Torras, J. CD154-CD40 T-cell co-stimulation pathway is a key mechanism in kidney ischemia-reperfusion injury. Kidney Int. 2015, 88, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Friedewald, J.J.; Rabb, H. Inflammatory cells in ischemic acute renal failure. Kidney Int. 2004, 66, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Pilat, N.; Sayegh, M.H.; Wekerle, T. Costimulatory pathways in transplantation. Semin. Immunol. 2011, 23, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Zumerle, S.; Molon, B.; Viola, A. Membrane rafts in T cell activation: A spotlight on CD28 costimulation. Front. Immunol. 2017, 8, 1467. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, D.J.; Walunas, T.L.; Jeffrey, A. CD28/B7 System of T Cell costimulation. Annu. Rev. Immunol. 1996, 14, 233–258. [Google Scholar] [CrossRef] [PubMed]
- Salomon, B.; Lenschow, D.J.; Rhee, L.; Ashourian, N.; Singh, B.; Sharpe, A.; Bluestone, J.A. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 2000, 12, 431–440. [Google Scholar] [CrossRef]
- Jaworska, K.; Ratajczak, J.; Huang, L.; Whalen, K.; Yang, M.; Stevens, B.K.; Kinsey, G.R. Both PD-1 ligands protect the kidney from ischemia reperfusion injury. J. Immunol. 2015, 194, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, F.; Knoblock, D.M.; Liu, A.; McAllister, F.; Beatty, G.L. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol. Immunother. 2017, 66, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Ward-Kavanagh, L.K.; Lin, W.W.; Šedý, J.R.; Ware, C.F. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity 2016, 44, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, E.; Pluvinet, R.; Torras, J.; Olivar, R.; Vidal, A.; Franquesa, M.; Cassis, L.; Cruzado, J.M.; Bestard, O.; Grinyó, J.M.; et al. In vivo therapeutic efficacy of intra-renal CD40 silencing in a model of humoral acute rejection. Gene Ther. 2011, 18, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Vu, M.D.; Xiao, X.; Gao, W.; Degauque, N.; Chen, M.; Kroemer, A.; Killeen, N.; Ishii, N.; Li, X.C. OX40 costimulation turns off Foxp3 + Tregs. Blood 2007, 110, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- Lippert, U.; Zachmann, K.; Ferrari, D.M.; Schwarz, H.; Brunner, E.; Latif, A.H.; Neumann, C.; Soruri, A. CD137 ligand reverse signaling has multiple functions in human dendritic cells during an adaptive immune response. Eur. J. Immunol. 2008, 38, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Beiske, K.; Clark, E.A.; Holte, H.; Ledbetter, J.A.; Smeland, E.B.; Godal, T. Triggering of neoplastic B cells via surface IgM and the cell surface antigens CD20 and CDw40. Responses differ from normal blood B cells and are restricted to certain morphologic subsets. Int. J. Cancer 1988, 42, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Nicolls, M.R.; Gill, R.G. LFA-1 (CD11a) as a therapeutic target. Am. J. Transplant. 2006, 6, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Nicolls, M.R.; Coulombe, M.; Yang, H.; Bolwerk, A.; Gill, R.G. Anti-LFA-1 therapy induces long-term islet allograft acceptance in the absence of IFN- or IL-4. J. Immunol. 2000, 164, 3627–3634. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Haines, G.K.; Harlow, L.A.; Koch, A.E. Adhesion molecule expression in human synovial tissue. Arthritis Rheum. 1993, 36, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Varon, D.; Jackson, D.E.; Shenkman, B.; Dardik, R.; Tamarin, I.; Savion, N.; Newman, P.J. Platelet/endothelial cell adhesion molecule-1 serves as a costimulatory agonist receptor that modulates integrin-dependent adhesion and aggregation of human platelets. Blood 1998, 91, 500–507. [Google Scholar] [PubMed]
- Rival, Y.; del Maschio, A.; Rabiet, M.J.; Dejana, E.; Duperray, A. Inhibition of platelet endothelial cell adhesion molecule-1 synthesis and leukocyte transmigration in endothelial cells by the combined action of TNF-α and IFN-γ. J. Immunol. 1996, 157, 1233–1241. [Google Scholar] [PubMed]
- Rodriguez-Manzanet, R.; Dekruyff, R.; Kuchroo, V.K.; Umetsu, D.T. The costimulatory role of TIM molecules. Immunol. Rev. 2009, 229, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Umetsu, S.E.; Lee, W.-L.; McIntire, J.J.; Downey, L.; Sanjanwala, B.; Akbari, O.; Berry, G.J.; Nagumo, H.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat. Immunol. 2005, 6, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Degauque, N.; Mariat, C.; Kenny, J.; Zhang, D.; Gao, W.; Minh, D.V.; Alexopoulos, S.; Oukka, M.; Umetsu, D.T.; DeKruyff, R.H.; et al. Immunostimulatory Tim-1-specific antibody deprograms Tregs and prevents transplant tolerance in mice. J. Clin. Investig. 2008, 118, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.D.; Irving, B.A.; Crabtree, G.R.; Weiss, A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science 1991, 251, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Boise, L.H.; Minn, A.J.; Noel, P.J.; June, C.H.; Accavitti, M.A.; Lindsten, T.; Thompson, C.B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-xL. Immunity 1995, 3, 87–98. [Google Scholar] [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; St. Clair, E.W.; Turka, L.A. CTLA4Ig: Bridging the basic immunology with clinical application. Immunity 2006, 24, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Bour-Jordan, H.; Esensten, J.H.; Martinez-Llordella, M.; Penaranda, C.; Stumpf, M.; Bluestone, J.A. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol. Rev. 2011, 241, 180–205. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.J.; Ledbetter, J.A.; Morishita, Y.; June, C.H.; Beatty, P.G.; Hansen, J.A. A 44 kilodalton cell surface homodimer regulates interleukin 2 production by activated human T lymphocytes. J. Immunol. 1986, 136, 3282–3287. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Manger, B.; Imboden, J. Synergy between the T3/antigen receptor complex and Tp44 in the activation of human T cells. J. Immunol. 1986, 137, 819–825. [Google Scholar] [PubMed]
- Engelhardt, J.J.; Sullivan, T.J.; Allison, J.P. CTLA-4 overexpression inhibits T cell responses through a CD28-B7-dependent mechanism. J. Immunol. 2006, 177, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Takada, M.; Chandraker, A.; Nadeau, K.C.; Sayegh, M.H.; Tilney, N.L. The role of the B7 costimulatory pathway in experimental cold ischemia/reperfusion injury. J. Clin. Investig. 1997, 100, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Chandraker, A.; Takada, M.; Nadeau, K.C.; Peach, R.; Tilney, N.L.; Sayegh, M.H. CD28-b7 blockade in organ dysfunction secondary to cold ischemia/reperfusion injury. Kidney Int. 1997, 52, 1678–1684. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, M.A.; Summers, S.A.; Gan, P.Y.; Chan, A.J.; Khouri, M.B.; Ooi, J.D.; Ghali, J.R.; Odobasic, D.; Hickey, M.J.; Kitching, A.R.; et al. Endogenous Toll-like receptor 9 regulates AKI by promoting regulatory T cell recruitment. J. Am. Soc. Nephrol. 2016, 27, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Hathcock, K.S. Comparative analysis of B7-1 and B7-2 costimulatory ligands: Expression and function. J. Exp. Med. 1994, 180, 631–640. [Google Scholar] [CrossRef] [PubMed]
- De Greef, K.E.; Ysebaert, D.K.; Dauwe, S.; Persy, V.; Vercauteren, S.R.; Mey, D.; De Broe, M.E. Anti-B7-1 blocks mononuclear cell adherence in vasa recta after ischemia. Kidney Int. 2001, 60, 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Rabb, H.; Mendiola, C.C.; Saba, S.R.; Dietz, J.R.; Wayne Smith, C.; Bonventre, J.V.; Ramirez, G. Antibodies to ICAM-1 protect kidneys in severe ischemic reperfusion injury. Biochem. Biophys. Res. Commun. 1995, 211, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.J.; Williams, W.W.; Colvin, R.B.; Bonventre, J.V. Antibody to intercellular adhesion molecule 1 protects the kidney against ischemic injury. Proc. Natl. Acad. Sci. USA 1994, 91, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Solez, K.; Kramer, E.C.; Fox, J.A.; Heptinstall, R.H. Medullary plasma flow and intravascular leukocyte accumulation in acute renal failure. Kidney Int. 1974, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Rabb, H.; Daniels, F.; O’Donnell, M.; Haq, M.; Saba, S.R.; Keane, W.; Tang, W.W. Pathophysiological role of T lymphocytes in renal ischemia-reperfusion injury in mice. Am. J. Physiol. Ren. Physiol. 2000, 279, F525–F531. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Juedes, A.E.; Temann, U.A.; Shresta, S.; Allison, J.P.; Ruddle, N.H.; Flavell, R.A. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 2001, 409, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Tafuri, A.; Shahinian, A.; Bladt, F.; Yoshinaga, S.K.; Jordana, M.; Wakeham, A.; Boucher, L.M.; Bouchard, D.; Chan, V.S.; Duncan, G.; et al. ICOS is essential for effective T-helper-cell responses. Nature 2001, 409, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T. Co-stimulatory molecules as targets for treatment of lupus. Clin. Immunol. 2013, 148, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Podojil, J.R.; Miller, S.D. Targeting the B7 family of co-stimulatory molecules: Successes and challenges. BioDrugs 2013, 27, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bauquet, A.T.; Jin, H.; Paterson, A.M.; Mitsdoerffer, M.; Ho, I.C.; Sharpe, A.H.; Kuchroo, V.K. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH -17 cells. Nat. Immunol. 2009, 10, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, Y.; Zhang, H.; Zhang, T.; Wang, Y.; Tan, S.; Xu, Y.; Li, D.; Ye, L.; Chen, P. Effect of Inducible Co-Stimulatory Molecule siRNA in Cerebral Infarction Rat Models. Med. Sci. Monit. 2015, 21, 3003–3007. [Google Scholar] [CrossRef] [PubMed]
- Kosuge, H.; Suzuki, J.I.; Kakuta, T.; Haraguchi, G.; Koga, N.; Futamatsu, H.; Gotoh, R.; Inobe, M.; Isobe, M.; Uede, T. Attenuation of graft arterial disease by manipulation of the LIGHT pathway. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1409–1415. [Google Scholar] [CrossRef] [PubMed]
- Koga, N.; Suzuki, J.I.; Kosuge, H.; Haraguchi, G.; Onai, Y.; Futamatsu, H.; Maejima, Y.; Gotoh, R.; Saiki, H.; Tsushima, F.; et al. Blockade of the interaction between PD-1 and PD-L1 accelerates graft arterial disease in cardiac allografts. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Kosuge, H.; Suzuki, J.I.; Haraguchi, G.; Koga, N.; Maejima, Y.; Inobe, M.; Isobe, M.; Uede, T. Critical role of inducible costimulator signaling in the development of arteriosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2660–2665. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B. CD137-CD137 Ligand Interactions in Inflammation. Immune Netw. 2009, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, H.; Lee, S.W.; Hong, H.; Potter, K.G.; Maeda-Yamamoto, M.; Kinoshita, T.; Kawakami, Y.; Mittler, R.S.; Kwon, B.S.; Ware, C.F.; et al. Costimulation of mast cells by 4-1BB, a member of the tumor necrosis factor receptor superfamily, with the high-affinity IgE receptor. Blood 2005, 106, 4241–4248. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Choi, B.K.; Bae, J.S.; Kim, W.Y.; Gebhardt, B.M.; Kwon, B.S. CD137-deficient mice have reduced NK/NKT cell numbers and function, are resistant to lipopolysaccharide-induced shock syndromes, and have lower IL-4 responses. J. Immunol. 2004, 173, 4218–4229. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Schwarz, H. CD137 ligand, a member of the tumor necrosis factor family, regulates immune responses via reverse signal transduction. J. Leukoc. Biol. 2011, 89, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, J.S.; Kim, J.D.; Cha, H.J.; Kim, A.; Lee, S.K.; Lee, S.C.; Kwon, B.S.; Mittler, R.S.; Cho, H.R.; et al. PNAS Plus: Reverse signaling through the costimulatory ligand CD137L in epithelial cells is essential for natural killer cell-mediated acute tissue inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, E13–E22. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, E.N.; Kim, E.Y.; Park, H.J.; Chang, C.Y.; Jung, D.Y.; Choi, S.Y.; Lee, S.K.; Lee, K.W.; Kwon, G.Y.; et al. Administration of agonistic anti-4-1BB monoclonal antibody leads to the amelioration of inflammatory bowel disease. Immunol. Lett. 2005, 101, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.J.; Park, K.; Kim, J.; Choi, H.J.; Yagita, H.; Nam, S.H.; Cho, H.R.; Kwon, B. Costimulatory molecule-targeted immunotherapy of cutaneous graft-versus-host disease. Blood 2007, 110, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; de Paoli, P.; Valle, A.; Garcia, E.; Rousset, F. Long-term human B cell lines dependent on interleukin-4 and antibody to CD40. Science 1991, 251, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Van Kooten, C.; Gaillard, C.; Galizzi, J.-P.; Hermannv, P.; Fossiez, F.; Banchereau, J.; Blanchard, D. B cells regulate expression of CD40 ligand on activated T cells by lowering the mRNA level and through the release of soluble CD40. Eur. J. Immunol. 1994, 24, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Freudenthal, P.S.; Steinman, R.M. The distinct surface of human blood dendritic cells, as observed after an improved isolation method. Proc. Natl. Acad. Sci. USA 1990, 87, 7698–7702. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Hakamada, R.; Yamane, H.; Nariuchi, H. Induction of IL-12 p40 messenger RNA expression and IL-12 production of macrophages via CD40-CD40 ligand interaction. J. Immunol. 1996, 156, 3932–3938. [Google Scholar] [PubMed]
- Gauchat, J.F.; Henchoz, S.; Mazzei, G.; Aubry, J.P.; Brunner, T.; Blasey, H.; Life, P.; Talabot, D.; Flores-Romo, L.; Thompson, J.; et al. Induction of human IgE synthesis in B cells by mast cells and basophils. Nature 1993, 365, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, L.M.; Klaus, S.J.; Magaletti, D.M.; Pinchuk, G.V.; Norsen, J.P.; Clark, E.A. Functional CD40 ligand expressed by human blood dendritic cells is up-regulated by CD40 ligation. J. Immunol. 1996, 157, 4363–4370. [Google Scholar] [PubMed]
- Klaus, S.J.; Berberich, I.; Shu, G.; Clark, E.A. Cd40 and its ligand in the regulation of humoral immunity. Semin. Immunol. 1994, 6, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Defrance, T.; Vanbervliet, B.; Durand, I.; Briolay, J.; Banchereau, J. Proliferation and differentiation of human CD5+ and CD5− B cell subsets activated through their antigen receptors or CD40 antigens. Eur. J. Immunol. 1992, 22, 2831–2839. [Google Scholar] [CrossRef] [PubMed]
- Armitage, R.J.; Macduff, B.M.; Spriggs, M.K.; Fanslow, W.C. Human B cell proliferation and Ig secretion induced by recombinant CD40 ligand are modulated by soluble cytokines. J. Immunol. 1993, 150, 3671–3680. [Google Scholar] [PubMed]
- Arpin, C.; Dechanet, J.; Van Kooten, C.; Merville, P.; Grouard, G.; Briere, F.; Banchereau, J.; Liu, Y.J. Generation of memory B cells and plasma cells in vitro. Science 1995, 268, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-D.; Ke, B.; Zhai, Y.; Amersi, F.; Gao, F.; Anselmo, D.M.; Busuttil, R.W.; Kupiec-Weglinski, J.W. CD154-CD40 T-cell costimulation pathway is required in the mechanism of hepatic ischemia/reperfusion injury, and its blockade facilitates and depends on heme oxygenase-1 mediated cytoprotection. Transplantation 2002, 74, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Kirk, A.D.; Burkly, L.C.; Batty, D.S.; Baumgartner, R.E.; Berning, J.D.; Buchanan, K.; Fechner, J.H., Jr.; Germond, R.L.; Kampen, R.L.; Patterson, N.B.; et al. Treatment with humanized monoclonal antibody against CD154 prevents acute renal allograft rejection in nonhuman primates. Nat. Med. 1999, 5, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Buhler, L.; Alwayn, I.P.; Appel, J.Z., 3rd; Robson, S.C.; Cooper, D.K. Anti-CD154 monoclonal antibody and thromboembolism. Transplantation 2001, 71, 491. [Google Scholar] [CrossRef] [PubMed]
- Ko, G.J.; Linfert, D.; Jang, H.R.; Higbee, E.; Watkins, T.; Cheadle, C.; Liu, M.; Racusen, L.; Grigoryev, D.N.; Rabb, H. Transcriptional analysis of infiltrating T cells in kidney ischemia-reperfusion injury reveals a pathophysiological role for CCR5. Am. J. Physiol. Ren. Physiol. 2012, 302, F762–F773. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, È.; Merino, A.; Goma, M.; Aran, J.M.; Bolaños, N.; de Ramon, L.; Herrero-Fresneda, I.; Bestard, O.; Cruzado, J.M.; Grinyó, J.M.; et al. CD40 gene silencing reduces the progression of experimental lupus nephritis modulating local milieu and systemic mechanisms. PLoS ONE 2013, 8, e65068. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Gao, W.; Demirci, G.; Strom, T.B.; Li, X.C. Stimulating PD-1-negative signals concurrent with blocking CD154 co-stimulation induces long-term islet allograft survival. Transplantation 2003, 76, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Ozkaynak, E.; Wang, L.; Goodearl, A.; McDonald, K.; Qin, S.; O’Keefe, T.; Duong, T.; Smith, T.; Gutierrez-Ramos, J.C.; Rottman, J.B.; et al. Programmed Death-1 Targeting Can Promote Allograft Survival. J. Immunol. 2002, 169, 6546–6553. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Shen, X.; Gao, F.; Ke, B.; Freitas, M.C.S.; Uchida, Y.; Busuttil, R.W.; Zhai, Y.; Kupiec-Weglinski, J.W. Programmed death-1/B7-H1 negative costimulation protects mouse liver against ischemia and reperfusion injury. Hepatology 2010, 52, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 pathway in the immune response. Am. J. Transplant. 2012, 12, 2575–2587. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kinsey, G.R. Regulatory T cells in acute and chronic kidney diseases. Am. J. Physiol. Ren. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, G.R.; Sharma, R.; Okusa, M.D. Regulatory T cells in AKI. J. Am. Soc. Nephrol. 2013, 24, 1720–1726. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Wang, Y.; Zheng, G.; Tan, T.K.; Lee, S.; Zhang, J.; Zhang, G.Y.; Hu, M.; Wang, C.; et al. Regulatory T cells require renal antigen recognition through the TCR to protect against injury in nephritis. Int. J. Clin. Exp. Pathol. 2014, 7, 38–47. [Google Scholar] [PubMed]
- Demirci, G.; Li, X.C. Novel roles of OX40 in the allograft response. Curr. Opin. Organ Transplant. 2008, 13, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M. From vanilla to 28 flavors: Multiple varieties of T regulatory cells. Immunity 2006, 25, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. J. Immunol. 2017, 198, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T cell lineage specification by the forkhead transcription factor Foxp3. Immunity 2005, 22, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Khattri, R.; Cox, T.; Yasayko, S.A.; Ramsdell, F. An essential role for Scurfin in CD4+CD25+T regulatory cells. J. Immunol. 2017, 198, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, A.; Singbartl, K. A review of the role of immune cells in acute kidney injury. Pediatr. Nephrol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tao, Y. Research progress on regulatory T cells in acute kidney injury. J. Immunol. Res. 2015, 2015, 174164. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, G.R.; Sharma, R.; Huang, L.; Li, L.; Vergis, A.L.; Ye, H.; Ju, S.T.; Okusa, M.D. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2009, 20, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Benoist, C.; Bluestone, J.A.; Campbell, D.J.; Ghosh, S.; Hori, S.; Kuchroo, V.K.; Mathis, D.; Roncarolo, M.G.; Rudensky, A. Regulatory T cells: Recommendations to simplify the nomenclature. Nat. Immunol. 2013, 14, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M.; Thornton, A.M. tTregs, pTregs, and iTregs: Similarities and differences. Immunol. Rev. 2014, 259, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kryczek, I.; Zou, W. Regulatory T-cell compartmentalization and trafficking. Blood 2006, 108, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J.; Koch, M.A. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat. Rev. Immunol. 2011, 11, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Bodor, J.; Bopp, T.; Vaeth, M.; Klein, M.; Serfling, E.; Hünig, T.; Becker, C.; Schild, H.; Schmitt, E. Cyclic AMP underpins suppression by regulatory T cells. Eur. J. Immunol. 2012, 42, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, G.R.; Okusa, M.D. Expanding role of T cells in acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2014, 23, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.L.; Teijaro, J.R.; Njau, M.N.; Chandran, S.S.; Azimzadeh, A.; Nadler, S.G.; Rothstein, D.M.; Farber, D.L. CTLA4 Expression is an indicator and regulator of steady-state CD4+FoxP3+ T cell homeostasis. J. Immunol. 2008, 181, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Henriksen, K.J.; Boden, E.K.; Tooley, A.J.; Ye, J.; Subudhi, S.K.; Zheng, X.X.; Strom, T.B.; Bluestone, J.A. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T Cells. J. Immunol. 2003, 171, 3348–3352. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.-W.; Yong, K.-C.; Lien, Y.-H.H. Pharmacologic recruitment of regulatory T cells as a therapy for ischemic acute kidney injury. Kidney Int. 2012, 81, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, L.Z.; Blazar, B.R.; Adeyi, O.A.; Strom, T.B.; Noelle, R.J. CD154 on the surface of CD4+CD25+regulatory T cells contributes to skin transplant tolerance. Transplantation 2003, 76, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Wu, Z.; Wang, Y.; Lassman, C.; Busuttil, R.W.; Zhai, Y.; Kupiec-Weglinski, J.W. Differential impact of CD154 costimulation blockade on alloreactive effector and regulatory T cells in murine renal transplant recipients. Transplantation 2008, 85, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Vogel, I.; Verbinnen, B.; van Gool, S.; Ceuppens, J.L. Regulatory T cell–dependent and –independent mechanisms of immune suppression by CD28/B7 and CD40/CD40L costimulation blockade. J. Immunol. 2016, 197, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Zouggari, Y.; Ait-Oufella, H.; Waeckel, L.; Vilar, J.; Loinard, C.; Cochain, C.; Récalde, A.; Duriez, M.; Levy, B.I.; Lutgens, E.; et al. Regulatory T cells modulate postischemic neovascularization. Circulation 2009, 120, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Wang, Y.-H.; Duramad, O.; Hanabuchi, S.; Perng, O.A.; Gilliet, M.; Qin, F.X.; Liu, Y.J. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13138–13143. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, G.R.; Huang, L.; Vergis, A.L.; Li, L.; Okusa, M.D. Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney. Kidney Int. 2010, 77, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Munger, M.E.; Highfill, S.L.; Tolar, J.; Weigel, B.J.; Riddle, M.; Sharpe, A.H.; Vallera, D.A.; Azuma, M.; Levine, B.L.; et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood 2010, 116, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lau, R.; Yu, D.; Zhu, W.; Korman, A.; Weber, J. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+CD25Hi regulatory T cells. Int. Immunol. 2009, 21, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.Y.; McGrath, M.M.; Nakayama, M.; Shimizu, T.; Boenisch, O.; Magee, C.N.; Abdoli, R.; Akiba, H.; Ueno, T.; Turka, L.A.; et al. Interruption of dendritic cell-mediated TIM-4 signaling induces regulatory T Cells and promotes skin allograft survival. J. Immunol. 2013, 191, 4447–4455. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Rothstein, D.M.; Sayegh, M.H. Costimulatory pathways in transplantation: Challenges and new developments. Immunol. Rev. 2009, 229, 271–293. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Ramon, L.; Guiteras, J.; Guiteras, R.; Cruzado, J.M.; Grinyó, J.M.; Torras, J. The Costimulatory Pathways and T Regulatory Cells in Ischemia-Reperfusion Injury: A Strong Arm in the Inflammatory Response? Int. J. Mol. Sci. 2018, 19, 1283. https://doi.org/10.3390/ijms19051283
De Ramon L, Guiteras J, Guiteras R, Cruzado JM, Grinyó JM, Torras J. The Costimulatory Pathways and T Regulatory Cells in Ischemia-Reperfusion Injury: A Strong Arm in the Inflammatory Response? International Journal of Molecular Sciences. 2018; 19(5):1283. https://doi.org/10.3390/ijms19051283
Chicago/Turabian StyleDe Ramon, Laura, Jordi Guiteras, Roser Guiteras, Josep M. Cruzado, Josep M. Grinyó, and Juan Torras. 2018. "The Costimulatory Pathways and T Regulatory Cells in Ischemia-Reperfusion Injury: A Strong Arm in the Inflammatory Response?" International Journal of Molecular Sciences 19, no. 5: 1283. https://doi.org/10.3390/ijms19051283