Preclinical Evaluation of Vemurafenib as Therapy for BRAFV600E Mutated Sarcomas

Abstract
:1. Introduction
2. Results
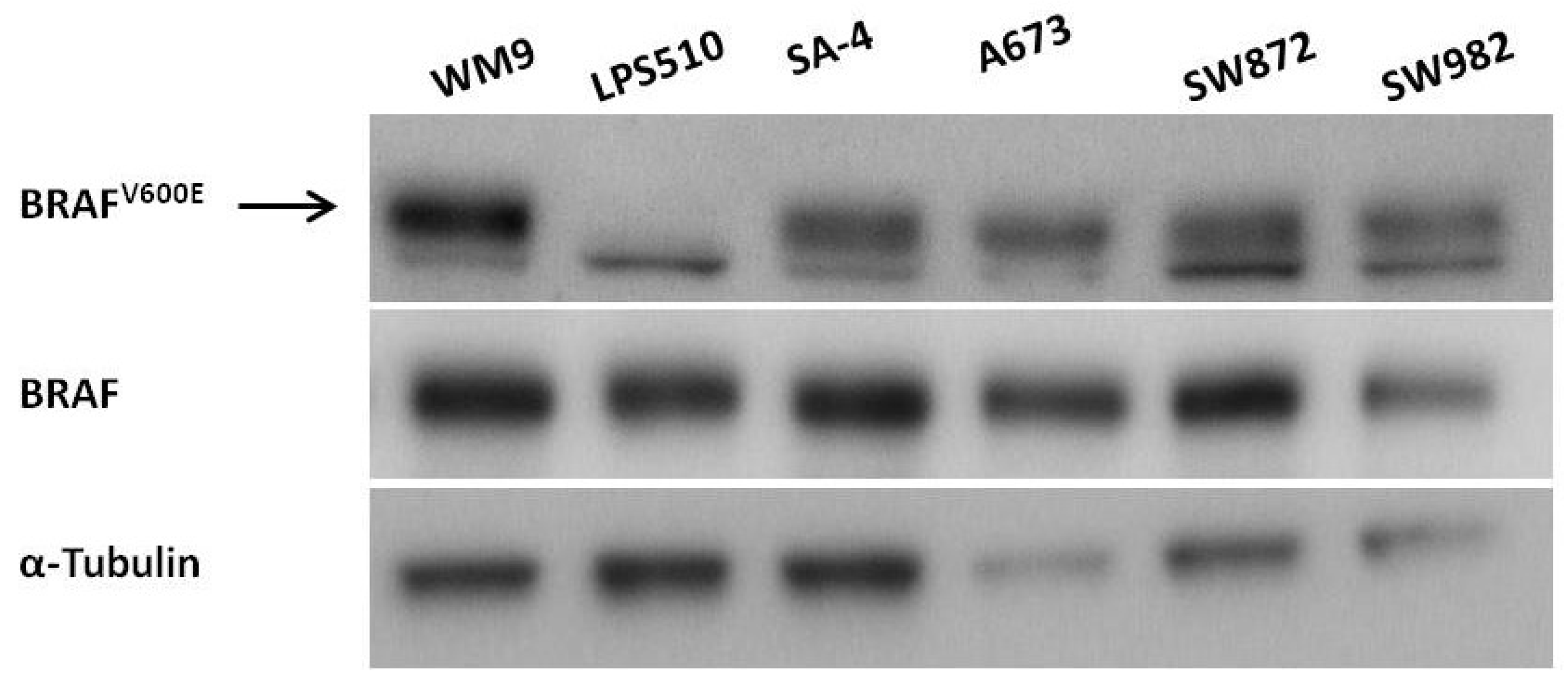
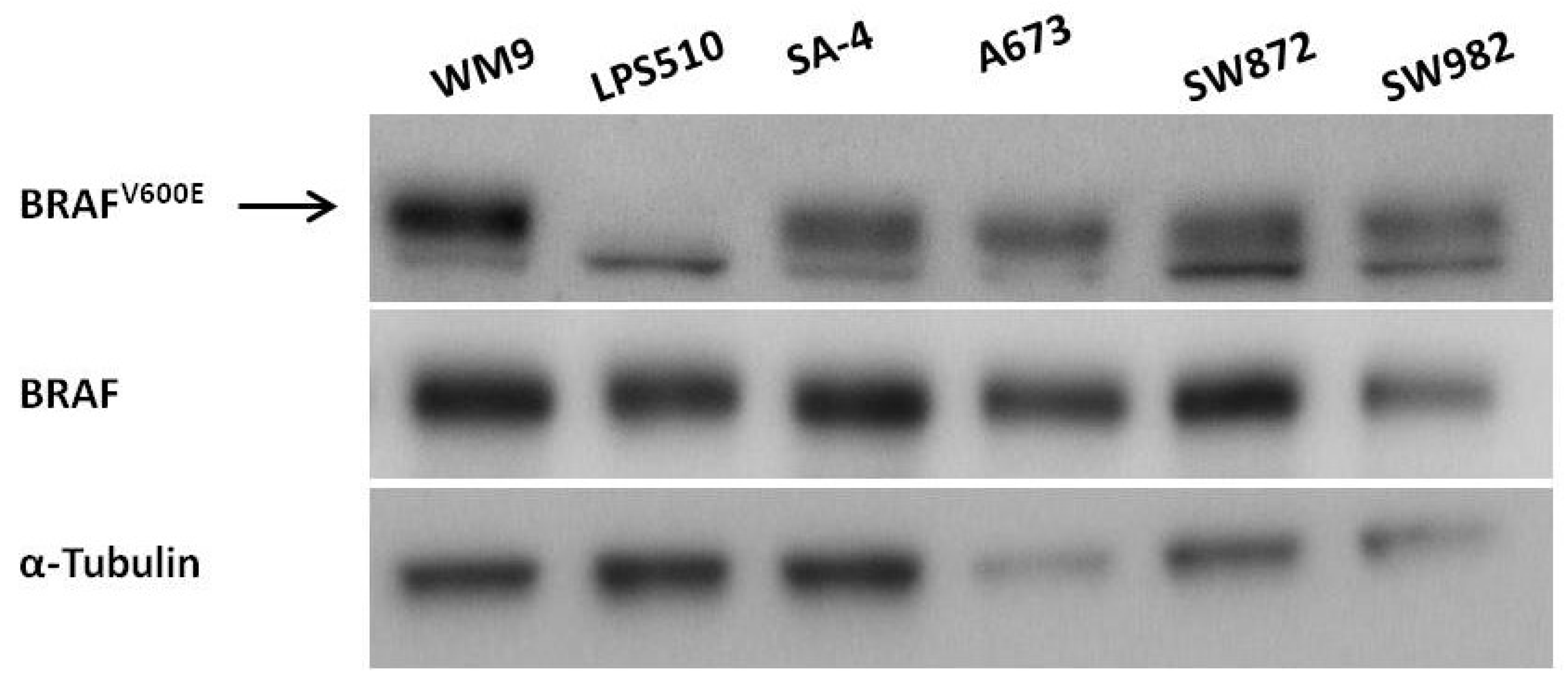
2.1. Expression of BRAFV600E in Sarcoma Cell Lines
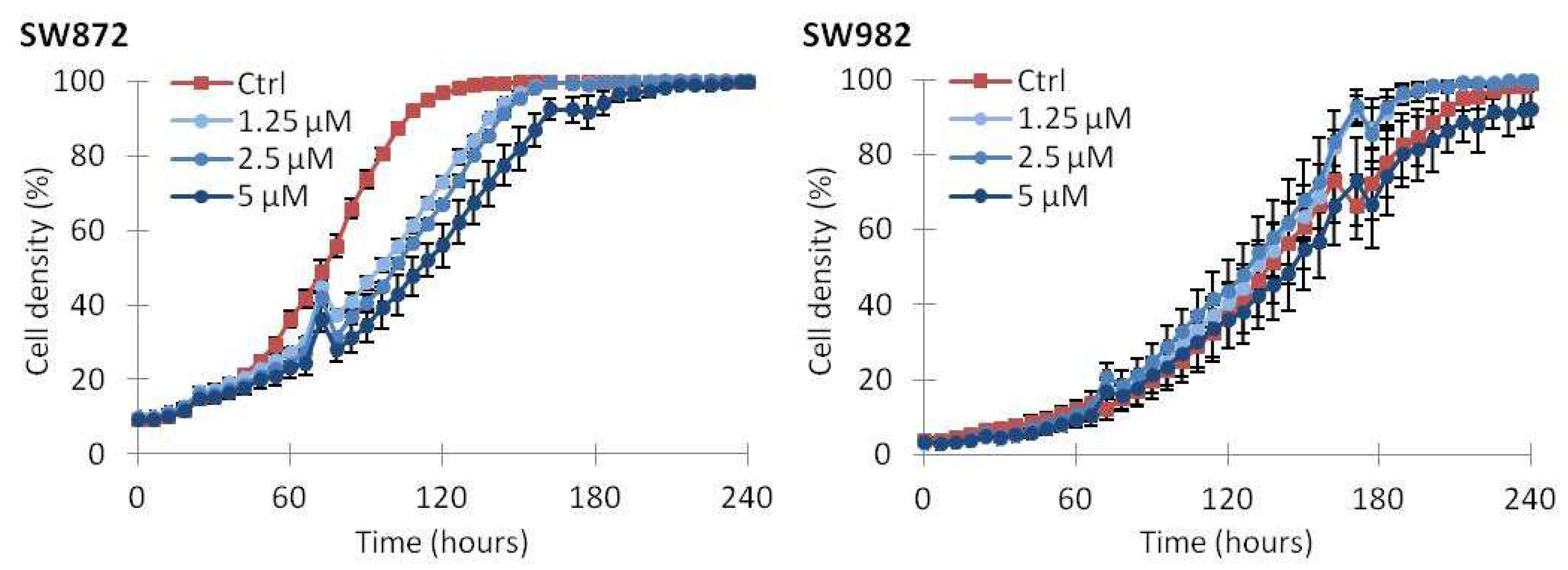
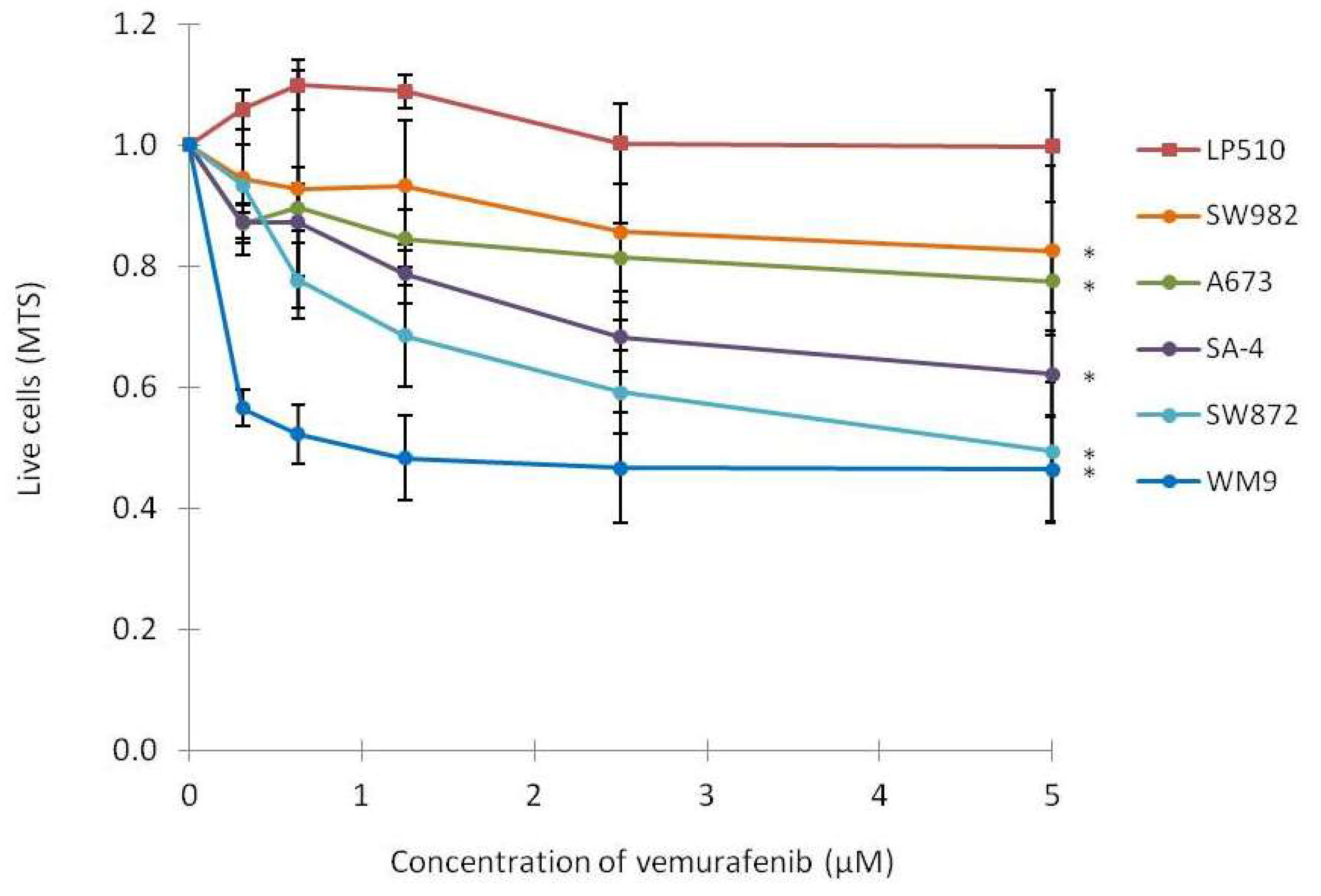
2.2. Vemurafenib Inhibits Cell Growth in BRAF Mutated Cell Lines
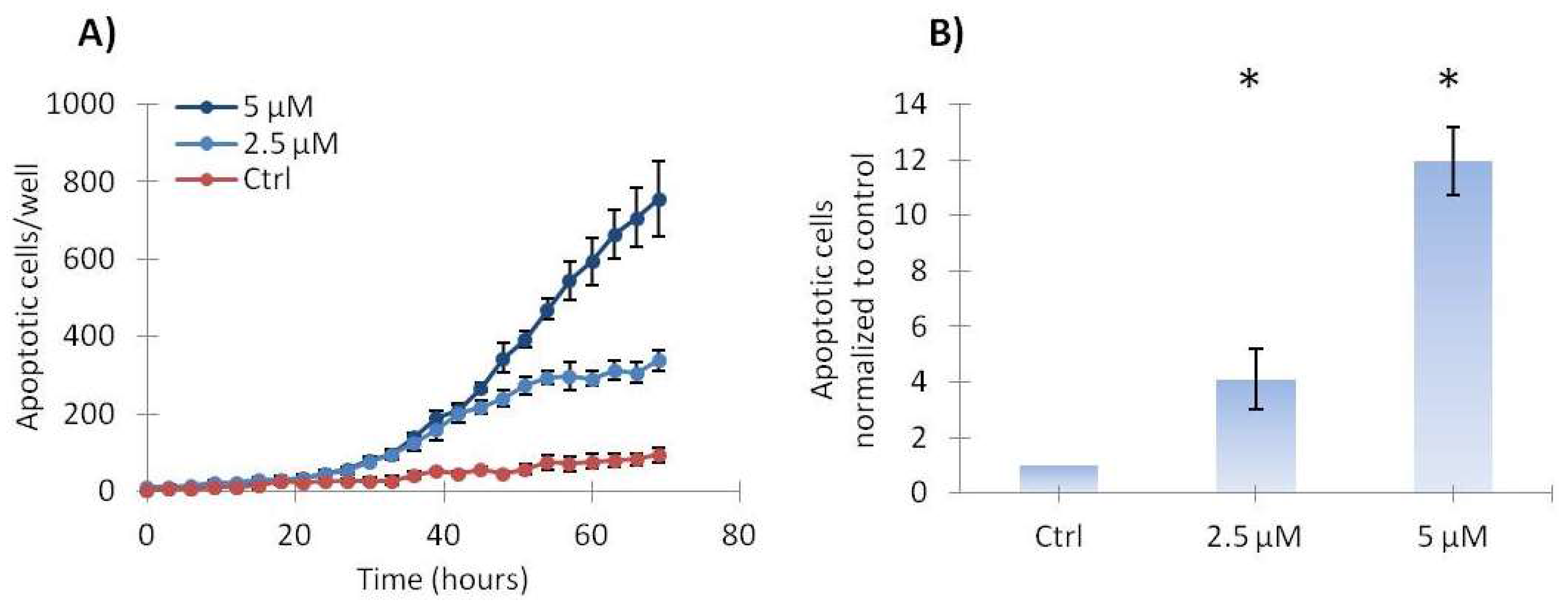
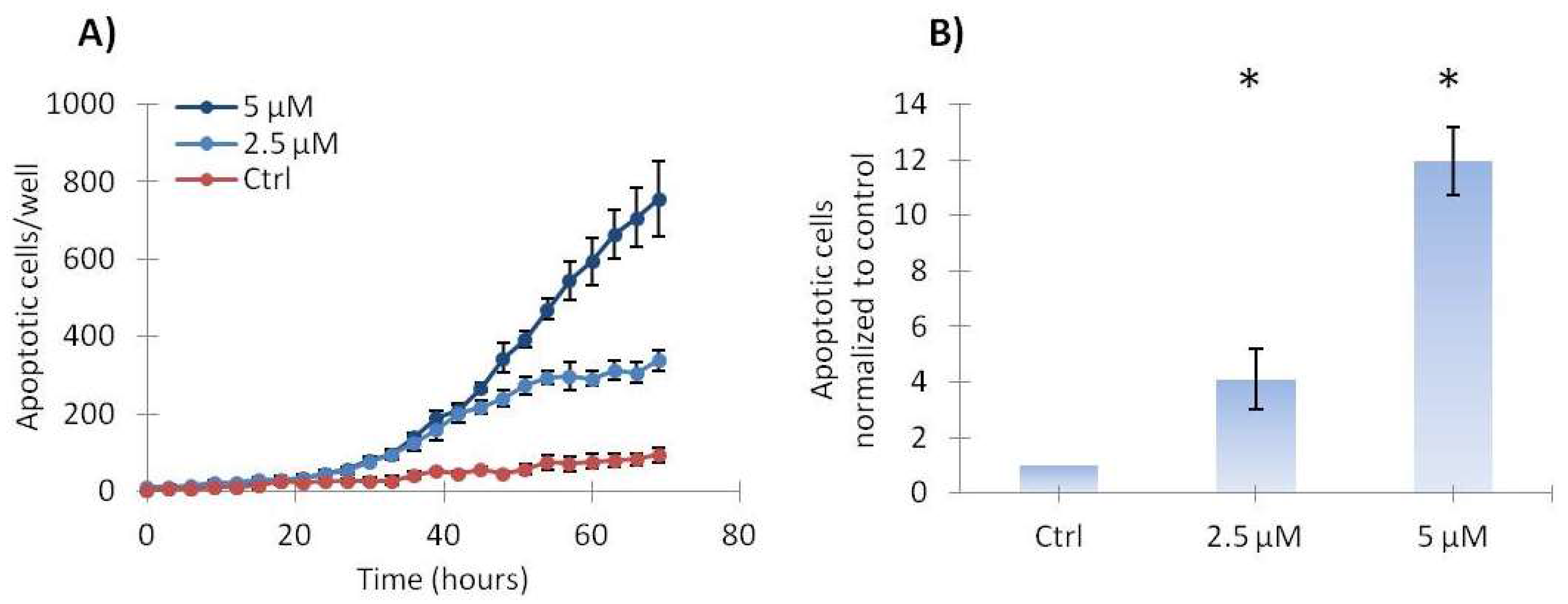
2.3. Vemurafenib Induces a Low Level of Apoptosis in SA-4 Cells
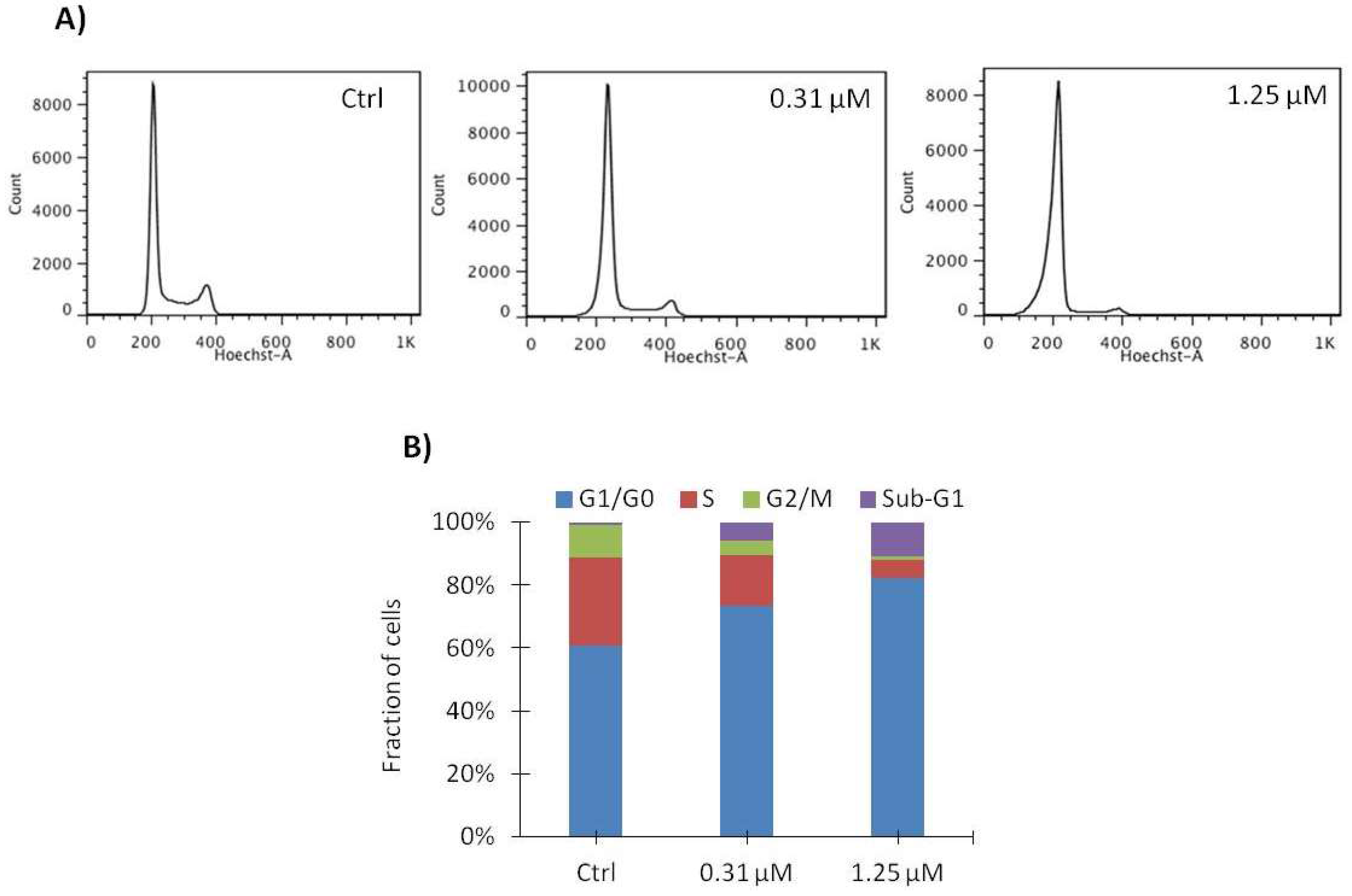
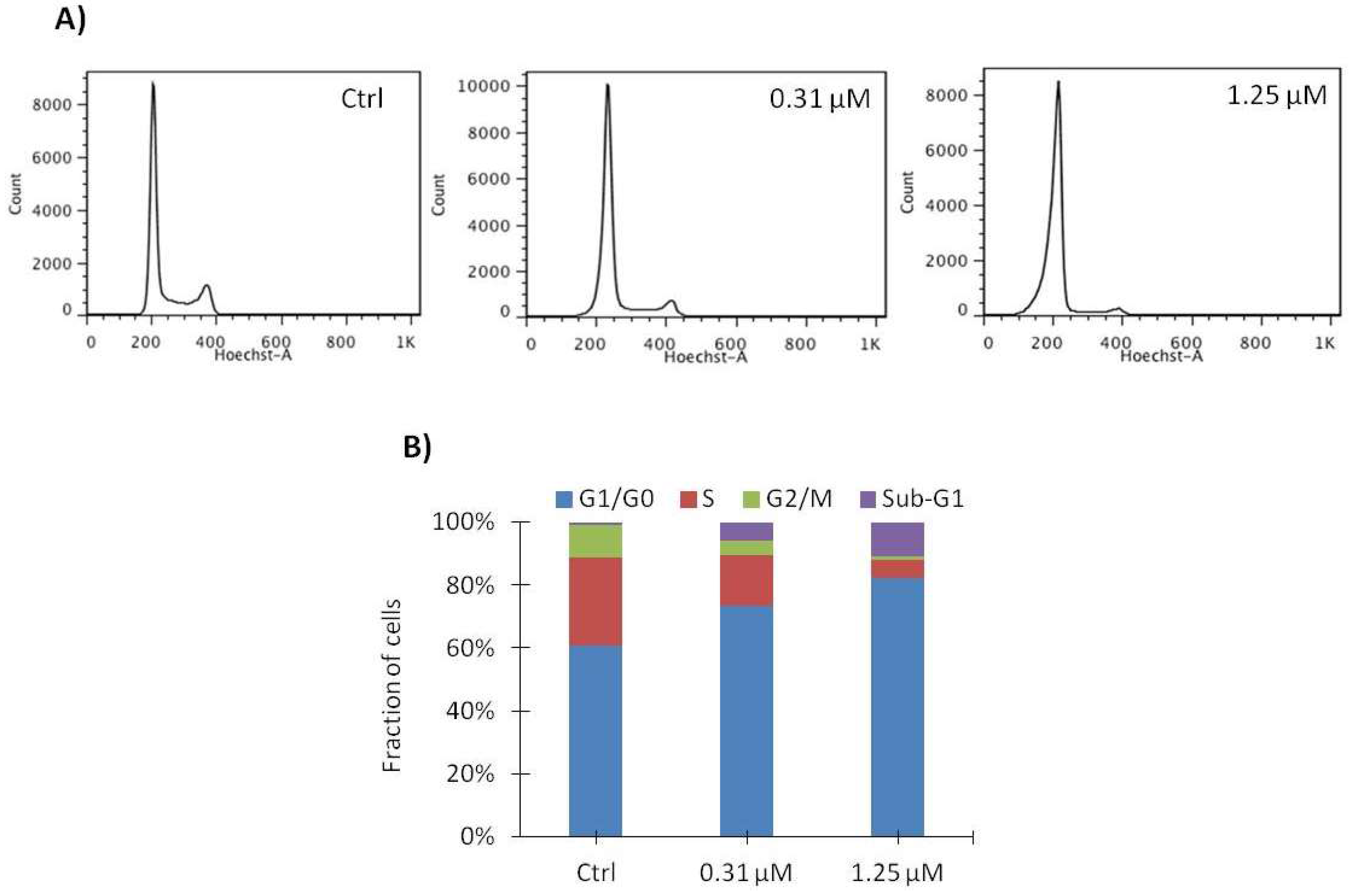
2.4. Vemurafenib Induces G1 Arrest in SA-4 Cells
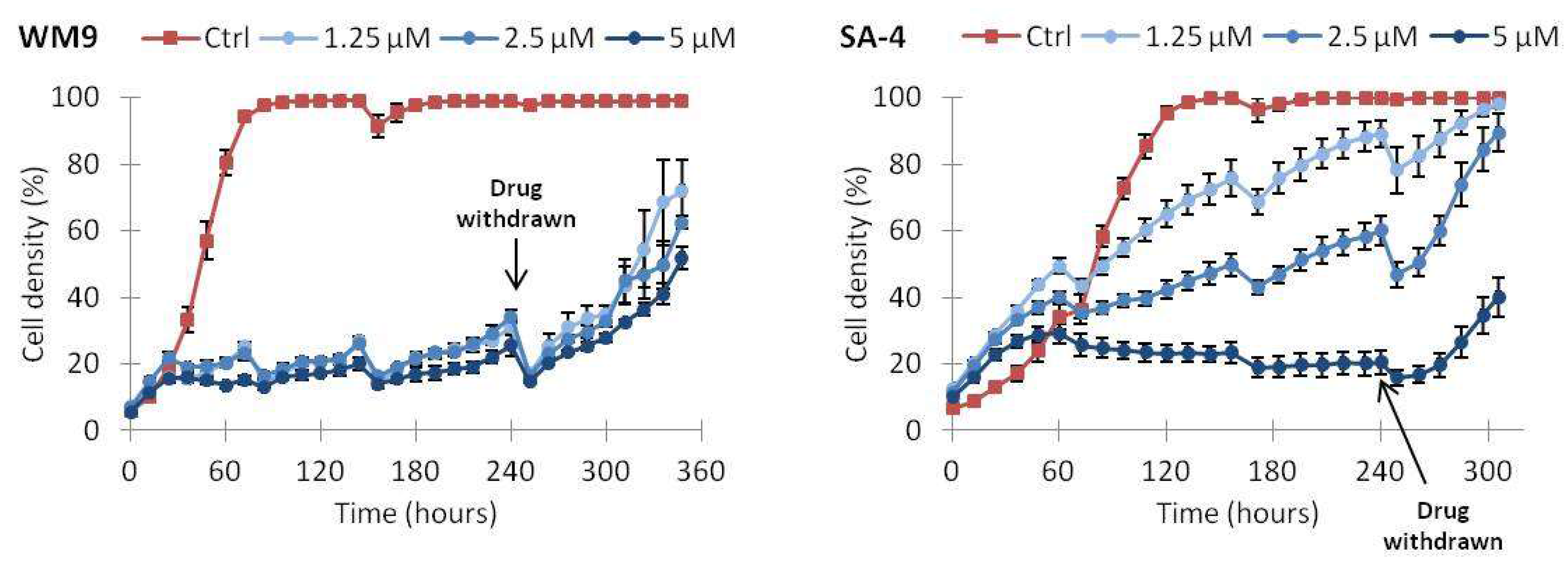
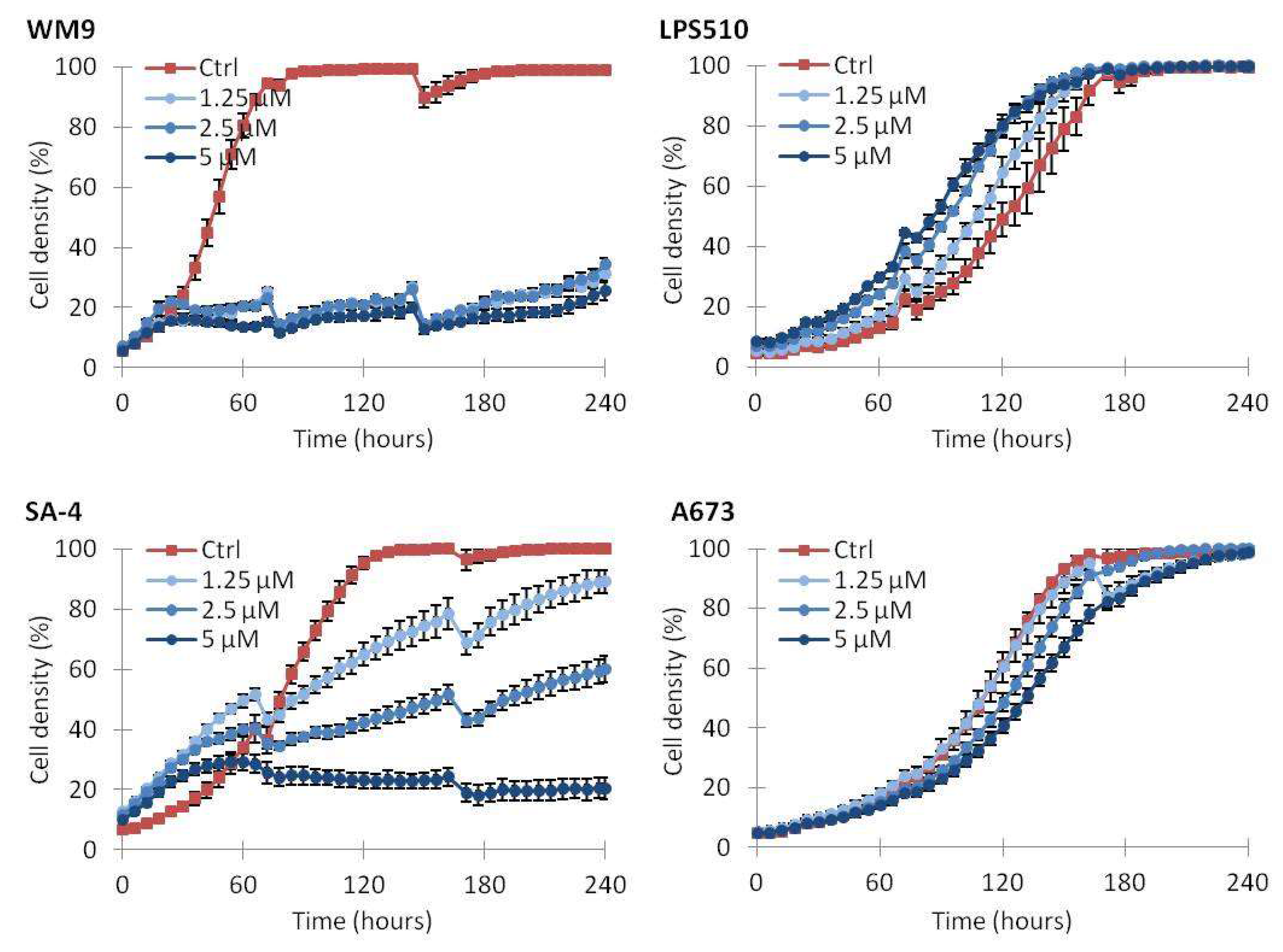
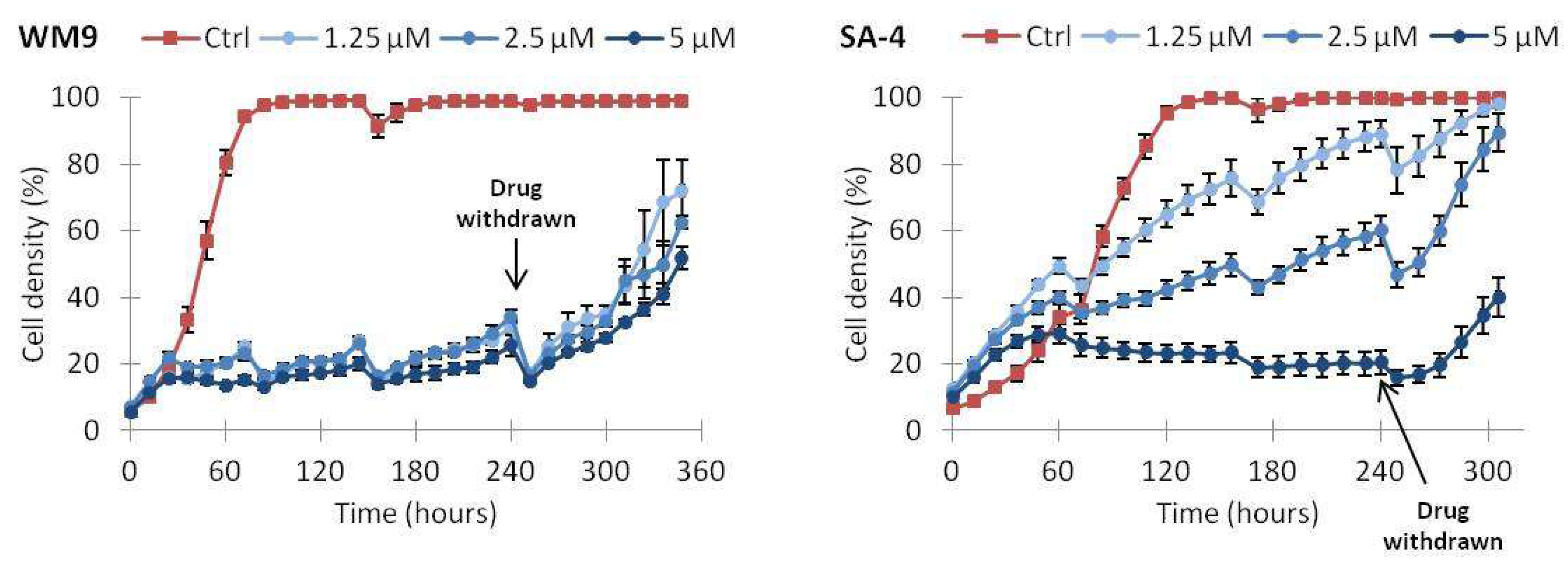
2.5. Continuous Treatment with Vemurafenib Is Necessary to Sustain Growth Inhibition
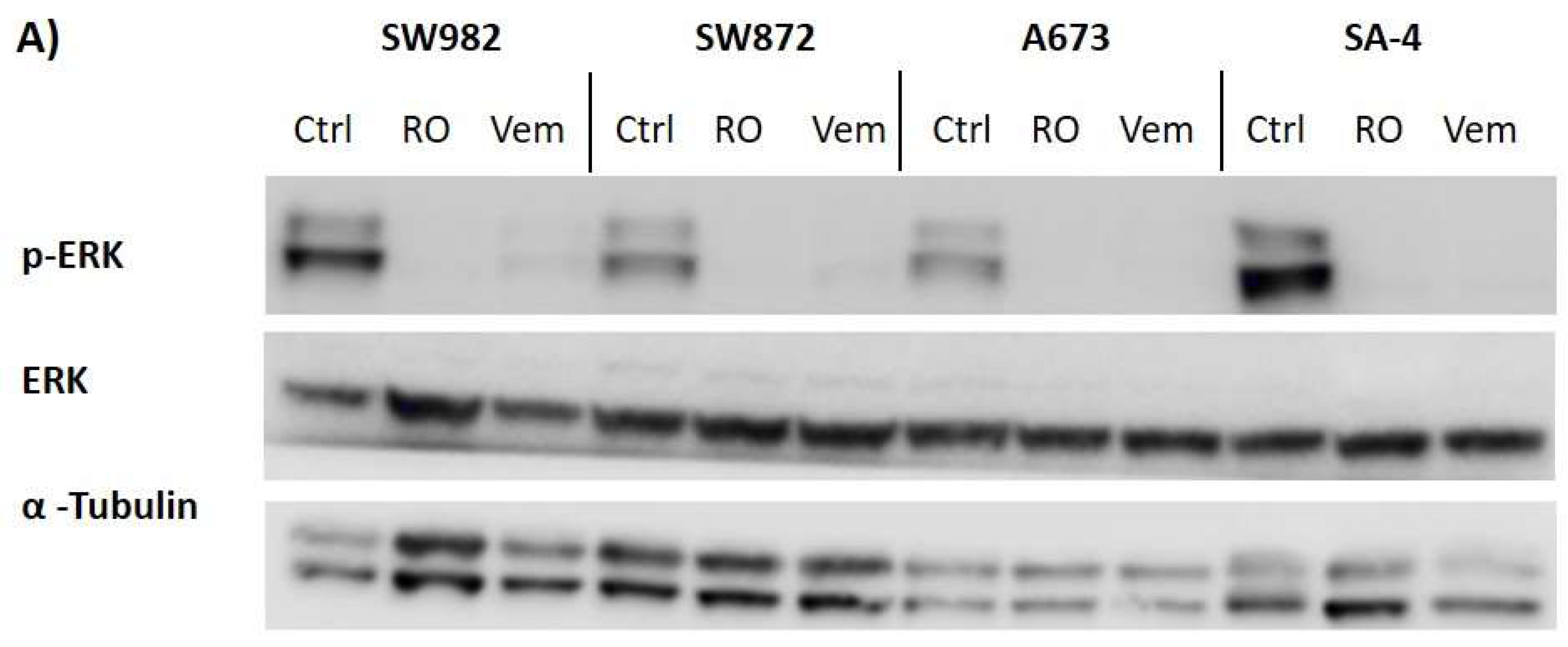
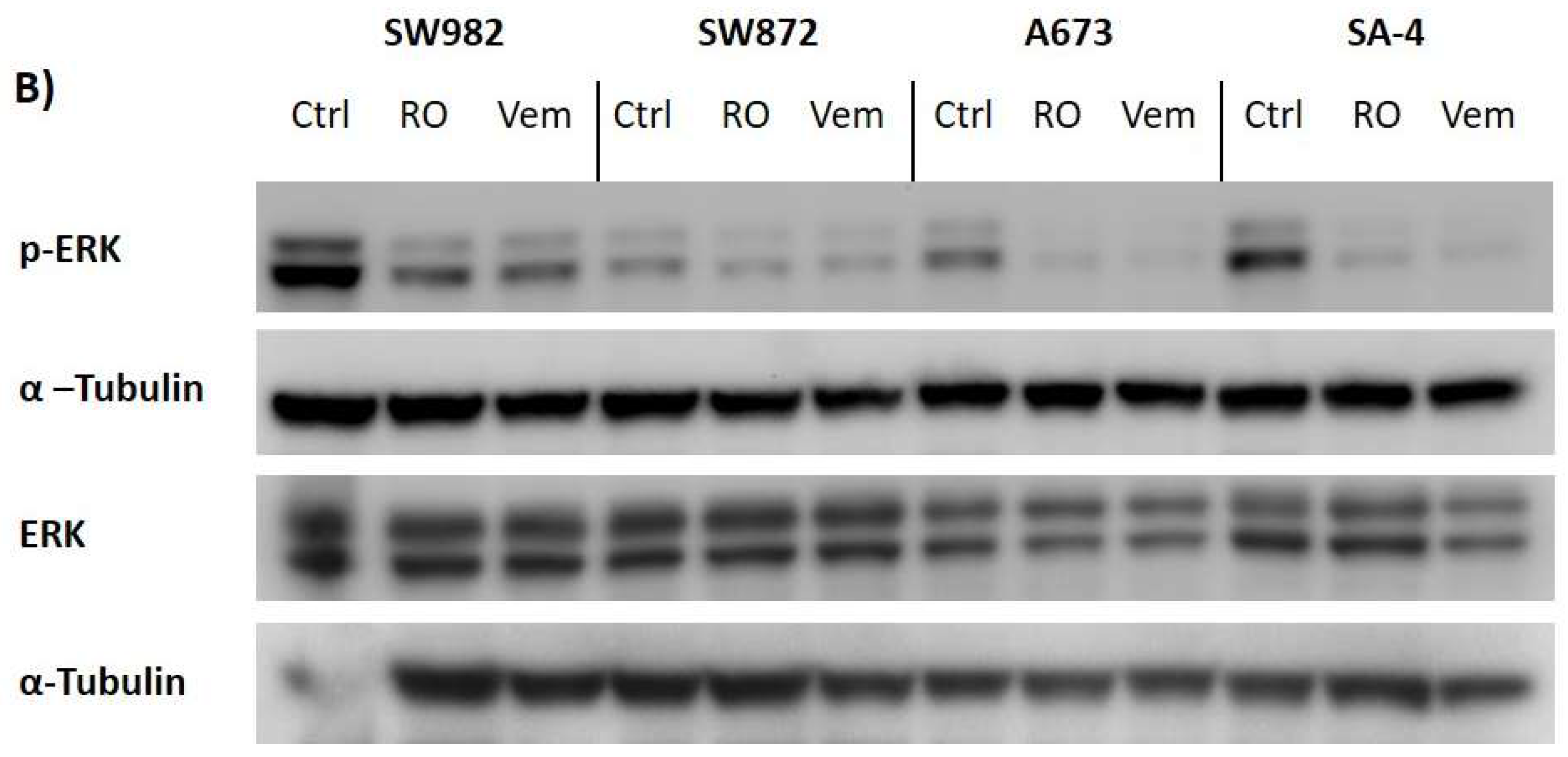
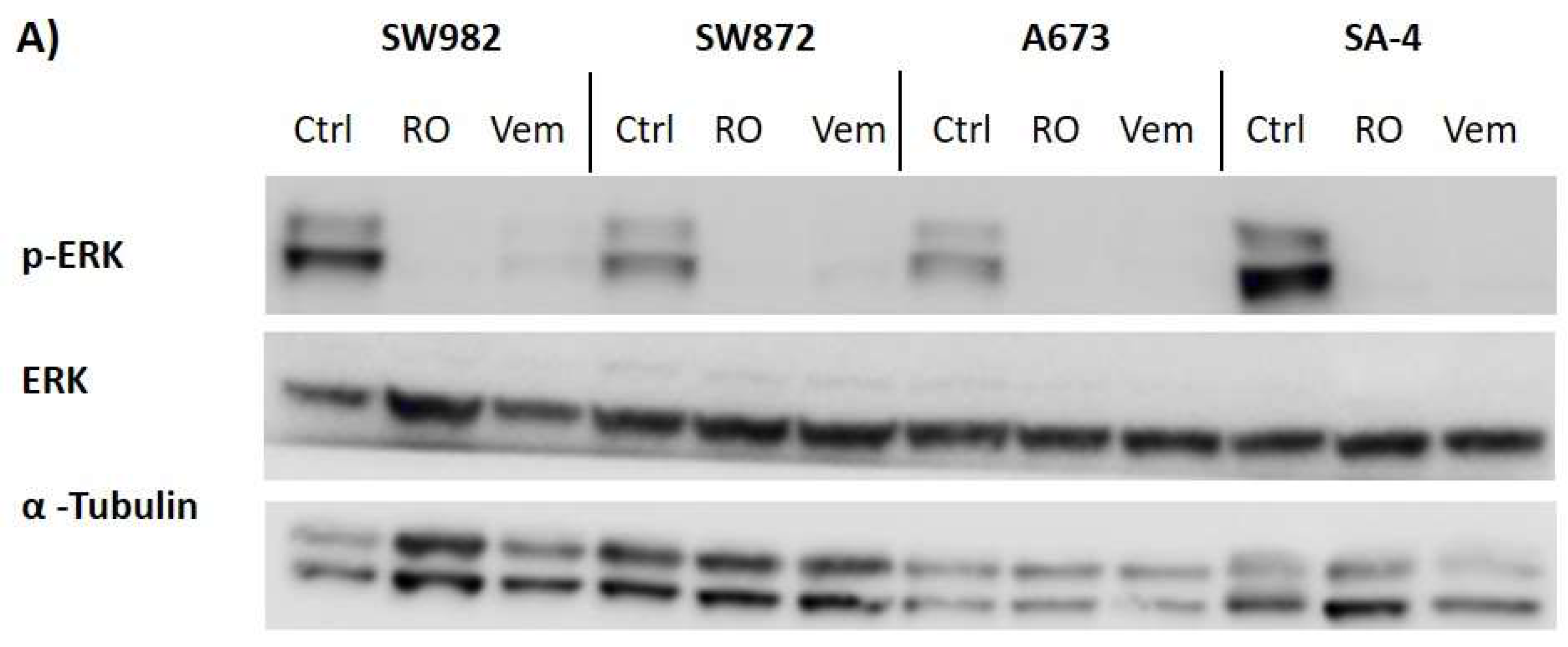
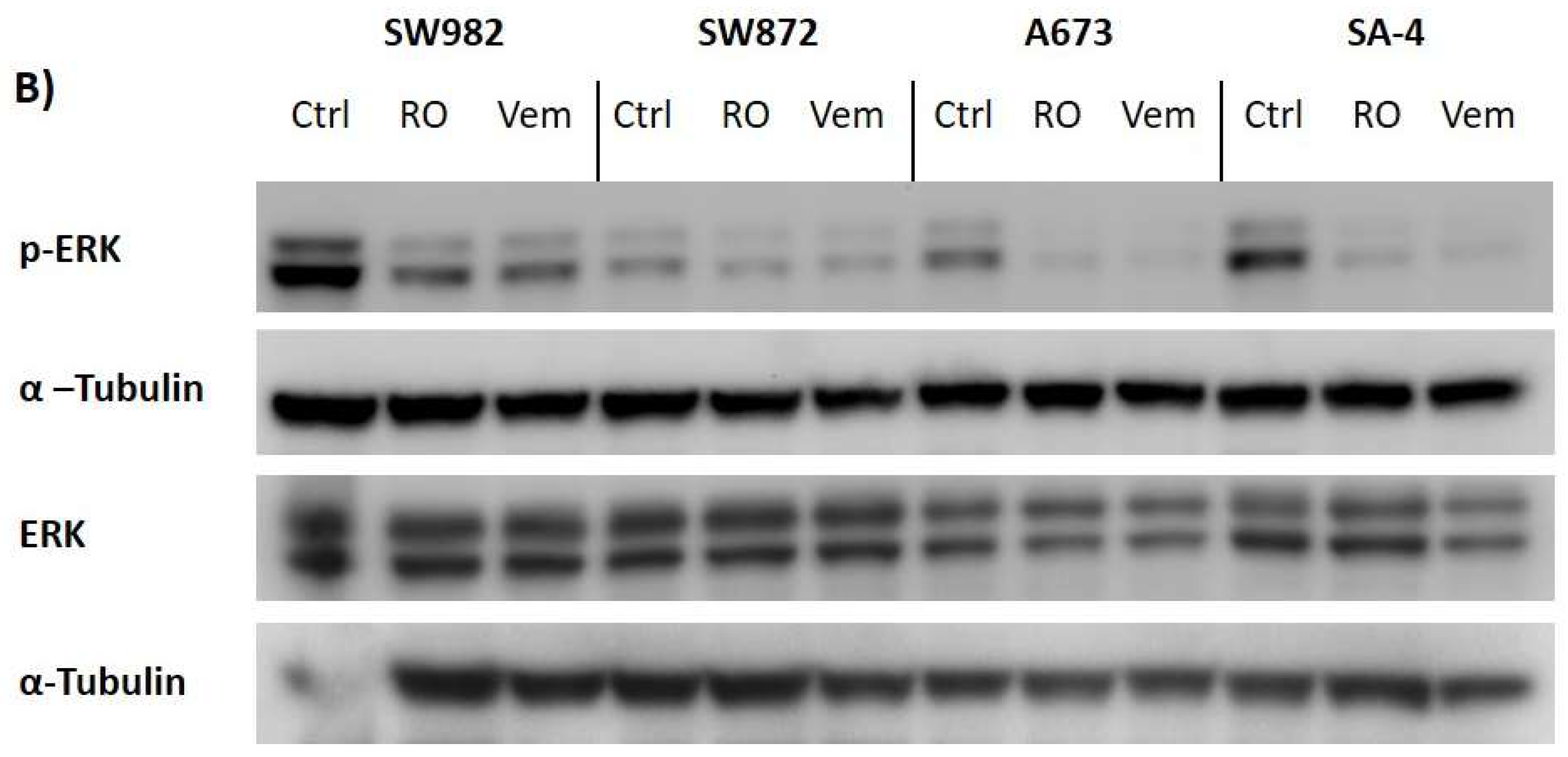
2.6. Vemurafenib De-Phosphorylates ERK Protein in All BRAF Mutated Sarcoma Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cancer Cell Lines and Cell Culturing
4.2. Evaluation of Mutational Status in Cell Lines
4.3. Western Blotting
4.4. Drug Treatment
4.5. Cell Growth by MTS
4.6. Cell Cycle Analysis
4.7. Apoptotic Cell Death
4.8. Growth Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| µ | Micro |
| µg | Microgram |
| µL | Microliter |
| µM | Micro molar |
| ARAF | v-Raf murine sarcoma 3611 viral oncogene homolog 1 |
| ATCC | American type culture collection |
| BRAF | v-Raf murine sarcoma viral oncogene homolog B |
| BRAFV600E | V600E-mutated BRAF |
| BRAFWT | Wild-type BRAF |
| DMSO | Dimethyl sulfoxide |
| DNA | Deoxyribonucleic acid |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| ERK | Extracellular signal-regulated kinase |
| FBS | Fetal bovine serum |
| FDA | Food and drug administration |
| FPKM | Fragments per kilobase million |
| GIST | Gastrointestinal stromal tumor |
| MAPK | Mitogen-activated protein kinase |
| MEK | Mitogen activated protein kinase |
| MITF | Microphthalmia-associated transcription factor |
| mL | Milliliter |
| MLANA | Melanoma antigen recognized by T-cells 1 |
| mM | Milli molar |
| mRNA | Messenger RNA |
| n | Number of biological replicates |
| nm | Nanometer |
| nM | Nano molar |
| p | Probability value |
| PAGE | Polyacrylamide gel electrophoresis |
| PBS | Phosphate-buffered saline |
| PCR | Polymerase chain reaction |
| PDGF | Platelet-derived growth factor |
| p-ERK | Phosphorylated ERK |
| PVDF | Polyvinylidene difluoride |
| RAF | Rapidly accelerated fibrosarcoma |
| RAF1 | v-Raf-1 murine leukemia viral oncogene homolog 1 |
| RAS | Ras sarcoma |
| RNA | Ribonucleic acid |
| RPKM | Reads per kilobase million |
| RT | Room temperature |
| RTK | Receptor tyrosine kinase |
| SD | Standard deviation |
| SDS | Sodium dodecyl sulfate |
| SEM | Standard error of mean |
| STR | Short tandem repeat |
| STS | Soft tissue sarcoma |
| TBS | Tris-buffered saline |
| α | Alpha |
| β | Beta |
References
- Williams, R.F.; Fernandez-Pineda, I.; Gosain, A. Pediatric Sarcomas. Surg. Clin. N. Am. 2016, 96, 1107–1125. [Google Scholar] [CrossRef] [PubMed]
- Burningham, Z.; Hashibe, M.; Spector, L.; Schiffman, J.D. The Epidemiology of Sarcoma. Clin. Sarcoma Res. 2012, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.D.M.; Bridge, J.A.; Hogendoorn, P.C.W.; Mertens, F. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone, 4th ed.; IARC Press: Lyon, France, 2013. [Google Scholar]
- Din, O.S.; Woll, P.J. Treatment of gastrointestinal stromal tumor: Focus on imatinib mesylate. Ther. Clin. Risk Manag. 2008, 4, 149–162. [Google Scholar] [PubMed]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.-J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the RAF/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873. [Google Scholar] [CrossRef] [PubMed]
- Satyamoorthy, K.; Li, G.; Gerrero, M.R.; Brose, M.S.; Volpe, P.; Weber, B.L.; van Belle, P.; Elder, D.E.; Herlyn, M. Constitutive Mitogen-activated Protein Kinase Activation in Melanoma Is Mediated by Both BRAF Mutations and Autocrine Growth Factor Stimulation. Cancer Res. 2003, 63, 756–759. [Google Scholar] [PubMed]
- Shelledy, L.; Roman, D. Vemurafenib: First-in-Class BRAF-Mutated Inhibitor for the Treatment of Unresectable or Metastatic Melanoma. J. Adv. Pract. Oncol. 2015, 6, 361–365. [Google Scholar] [PubMed]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Pelle, P.D.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Oddo, D.; Sennott, E.M.; Barault, L.; Valtorta, E.; Arena, S.; Cassingena, A.; Filiciotto, G.; Marzolla, G.; Elez, E.; van Geel, R.M.J.M.; et al. Molecular Landscape of Acquired Resistance to Targeted Therapy Combinations in BRAF-Mutant Colorectal Cancer. Cancer Res. 2016, 76, 4504. [Google Scholar] [CrossRef] [PubMed]
- Notarangelo, T.; Sisinni, L.; Condelli, V.; Landriscina, M. Dual EGFR and BRAF blockade overcomes resistance to vemurafenib in BRAF mutated thyroid carcinoma cells. Cancer Cell Int. 2017, 17, 86. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Kim, H.; Jang, K.W.; Kim, M.H.; Sohn, J.; Yun, M.R.; Kang, H.N.; Kang, C.W.; Kim, H.R.; Lim, S.M.; et al. EGFR-Mediated Reactivation of MAPK Signaling Induces Acquired Resistance to GSK2118436 in BRAF V600E–Mutant NSCLC Cell Lines. Mol. Cancer Ther. 2016, 15, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Park, B.M.; Jin, S.A.; Choi, Y.D.; Shin, S.H.; Jung, S.T.; Lee, J.B.; Lee, S.C.; Yun, S.J. Two cases of clear cell sarcoma with different clinical and genetic features: Cutaneous type with BRAF mutation and subcutaneous type with KIT mutation. Br. J. Dermatol. 2013, 169, 1346–1352. [Google Scholar] [CrossRef] [PubMed]
- Hocar, O.; Le Cesne, A.; Berissi, S.; Terrier, P.; Bonvalot, S.; Vanel, D.; Auperin, A.; Le Pechoux, C.; Bui, B.; Coindre, J.M.; et al. Clear Cell Sarcoma (Malignant Melanoma) of Soft Parts: A Clinicopathologic Study of 52 Cases. Dermatol. Res. Pract. 2012, 2012, 984096. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Trent, J.C.; Heinrich, M.C.; Beadling, C.; Patterson, J.; Bastida, C.C.; Blackman, S.C.; Kurzrock, R. BRAF Mutant Gastrointestinal Stromal Tumor: First report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget 2013, 4, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Kanojia, D.; Nagata, Y.; Garg, M.; Lee, D.H.; Sato, A.; Yoshida, K.; Sato, Y.; Sanada, M.; Mayakonda, A.; Bartenhagen, C.; et al. Genomic landscape of liposarcoma. Oncotarget 2015, 6, 42429–42444. [Google Scholar] [CrossRef] [PubMed]
- Becerikli, M.; Jacobsen, F.; Rittig, A.; Kohne, W.; Nambiar, S.; Mirmohammadsadegh, A.; Stricker, I.; Tannapfel, A.; Wieczorek, S.; Epplen, J.T.; et al. Growth rate of late passage sarcoma cells is independent of epigenetic events but dependent on the amount of chromosomal aberrations. Exp. Cell Res. 2013, 319, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Vultur, A.; Herlyn, M. Resistance to BRAF inhibitors: Unraveling mechanisms and future treatment options. Cancer Res. 2011, 71, 7137–7140. [Google Scholar] [CrossRef] [PubMed]
- Protsenko, S.A.; Semionova, A.I.; Komarov, Y.I.; Aleksakhina, S.N.; Ivantsov, A.O.; Iyevleva, A.G.; Imyanitov, E.N. BRAF-mutated clear cell sarcoma is sensitive to vemurafenib treatment. Investig. New Drugs 2015, 33, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600–Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Subbiah, V.; Janku, F.; Ludwig, J.A.; Naing, A.; Benjamin, R.S.; Brown, R.E.; Anderson, P.; Kurzrock, R. Novel Secondary Somatic Mutations in Ewing’s Sarcoma and Desmoplastic Small Round Cell Tumors. PLoS ONE 2014, 9, e93676. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Peng, W.; Calvo, E. Rational Approaches for Combination Therapy Strategies Targeting the MAP Kinase Pathway in Solid Tumors. Mol. Cancer Ther. 2018, 17, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Zhang, X.; Liu, J.; Estrem, S.; Li, S.; Gong, X.-Q.; Buchanan, S.; Henry, J.R.; Starling, J.J.; Peng, S.-B. Reactivation of Mitogen-activated Protein Kinase (MAPK) Pathway by FGF Receptor 3 (FGFR3)/Ras Mediates Resistance to Vemurafenib in Human B-RAF V600E Mutant Melanoma. J. Biol. Chem. 2012, 287, 28087–28098. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, F.; Ghiorzo, P.; Orgiano, L.; Pastorino, L.; Picasso, V.; Tornari, E.; Ottaviano, V.; Queirolo, P. BRAF-mutant melanoma: Treatment approaches, resistance mechanisms, and diagnostic strategies. OncoTargets Ther. 2015, 8, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Flaherty, K.T. Resistance to BRAF-targeted therapy in melanoma. Eur. J. Cancer 2013, 49, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, L.K.; Hindley, A.D.; O’Neill, E.; Kolch, W. Regulation and Role of Raf-1/B-Raf Heterodimerization. Mol. Cell. Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Joseph, E.W.; Pratilas, C.A.; Poulikakos, P.I.; Tadi, M.; Wang, W.; Taylor, B.S.; Halilovic, E.; Persaud, Y.; Xing, F.; Viale, A.; et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc. Natl. Acad. Sci. USA 2010, 107, 14903–14908. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Cohen, M.S. The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma. Expert Opin. Drug Discov. 2016, 11, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Li, L.; Brafford, P.A.; van den Eijnden, M.; Halloran, M.B.; Sproesser, K.; Haass, N.K.; Smalley, K.S.M.; Tsai, J.; Bollag, G.; et al. PLX4032, a Potent Inhibitor of the B-Raf V600E Oncogene, Selectively Inhibits V600E-positive Melanomas. Pigment Cell Melanoma Res. 2010, 23, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Hartsough, E.J.; Aplin, A.E. A STATement on Vemurafenib-Resistant Melanoma. J. Investig. Dermatol. 2013, 133, 1928–1929. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; MacConaill, L.E.; Hahn, W.C.; et al. Dissecting Therapeutic Resistance to RAF Inhibition in Melanoma by Tumor Genomic Profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Stratford, E.W.; Castro, R.; Daffinrud, J.; Skårn, M.; Lauvrak, S.; Munthe, E.; Myklebost, O. Characterization of Liposarcoma Cell Lines for Preclinical and Biological Studies. Sarcoma 2012, 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Asmann, Y.W.; Erickson-Johnson, M.R.; Oliveira, J.L.; Zhang, H.; Moura, R.D.; Lazar, A.J.; Lev, D.; Bill, K.; Lloyd, R.V.; et al. High-resolution genomic mapping reveals consistent amplification of the fibroblast growth factor receptor substrate 2 gene in well-differentiated and dedifferentiated liposarcoma. Genes Chromosomes Cancer 2011, 50, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Crozat, A.; Aman, P.; Mandahl, N.; Ron, D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993, 363, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ramirez, A.; Rodriguez-Perales, S.; Melendez, B.; Martinez-Delgado, B.; Urioste, M.; Cigudosa, J.C.; Benitez, J. Characterization of the A673 cell line (Ewing tumor) by molecular cytogenetic techniques. Cancer Genet. Cytogenet. 2003, 141, 138–142. [Google Scholar] [CrossRef]
- Teicher, B.A.; Polley, E.; Kunkel, M.; Evans, D.; Silvers, T.; Delosh, R.; Laudeman, J.; Ogle, C.; Reinhart, R.; Selby, M.; et al. Sarcoma Cell Line Screen of Oncology Drugs and Investigational Agents Identifies Patterns Associated with Gene and microRNA Expression. Mol. Cancer Ther. 2015, 14, 2452–2462. [Google Scholar] [CrossRef] [PubMed]
- Namløs, H.M.; Zaikova, O.; Bjerkehagen, B.; Vodák, D.; Hovig, E.; Myklebost, O.; Boye, K.; Meza-Zepeda, L.A. Use of liquid biopsies to monitor disease progression in a sarcoma patient: A case report. BMC Cancer 2017, 17, 29. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE SYMBOL | SA-4 | SW872 | LPS510 |
|---|---|---|---|
| ARAF | 9.0 | 24.1 | 21.7 |
| BRAFWT | 4.3 | 4.4 | 2.7 |
| BRAFV600E | 4.0 | 4.6 | 0 |
| RAF1 | 19.0 | 26.5 | 33.3 |
| G0/G1 | S | G2 | Sub-G1 | |
|---|---|---|---|---|
| Vehicle (Ctrl) | 55.2 ± 4.8 | 33.1 ± 4.9 | 11.3 ± 1.7 | 0.5 ± 0.7 |
| 0.31 μM | 72.4 ± 2.1 | 18.5 ± 3.8 | 5.5 ± 1.0 | 3.7 ± 2.7 |
| 1.25 μM | 81.6 ± 0.7 | 7.4 ± 2.0 | 2.3 ± 0.8 | 8.7 ± 2.4 |
| GENE SYMBOL | SA-4 | SW872 | LPS510 |
|---|---|---|---|
| S100B | 662 | 0.12 | 0 |
| MITF | 39.2 | 2.7 | 0.99 |
| TYR | 71.7 | 0.02 | 0 |
| MLANA | 278 | 0 | 0 |
| SOX10 | 262 | 0.05 | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gouravan, S.; Meza-Zepeda, L.A.; Myklebost, O.; Stratford, E.W.; Munthe, E. Preclinical Evaluation of Vemurafenib as Therapy for BRAFV600E Mutated Sarcomas. Int. J. Mol. Sci. 2018, 19, 969. https://doi.org/10.3390/ijms19040969
Gouravan S, Meza-Zepeda LA, Myklebost O, Stratford EW, Munthe E. Preclinical Evaluation of Vemurafenib as Therapy for BRAFV600E Mutated Sarcomas. International Journal of Molecular Sciences. 2018; 19(4):969. https://doi.org/10.3390/ijms19040969
Chicago/Turabian StyleGouravan, Sarina, Leonardo A. Meza-Zepeda, Ola Myklebost, Eva W. Stratford, and Else Munthe. 2018. "Preclinical Evaluation of Vemurafenib as Therapy for BRAFV600E Mutated Sarcomas" International Journal of Molecular Sciences 19, no. 4: 969. https://doi.org/10.3390/ijms19040969
APA StyleGouravan, S., Meza-Zepeda, L. A., Myklebost, O., Stratford, E. W., & Munthe, E. (2018). Preclinical Evaluation of Vemurafenib as Therapy for BRAFV600E Mutated Sarcomas. International Journal of Molecular Sciences, 19(4), 969. https://doi.org/10.3390/ijms19040969