Genetic and Epigenetic Control of CDKN1C Expression: Importance in Cell Commitment and Differentiation, Tissue Homeostasis and Human Diseases

 ,
, 

Abstract
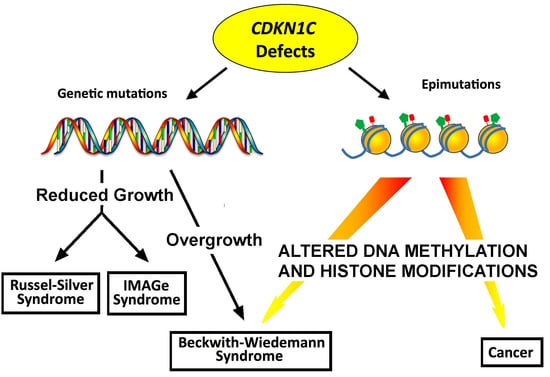
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. p57Kip2 Protein
1.2. p57Kip2 in Embryonic and Adult Tissues
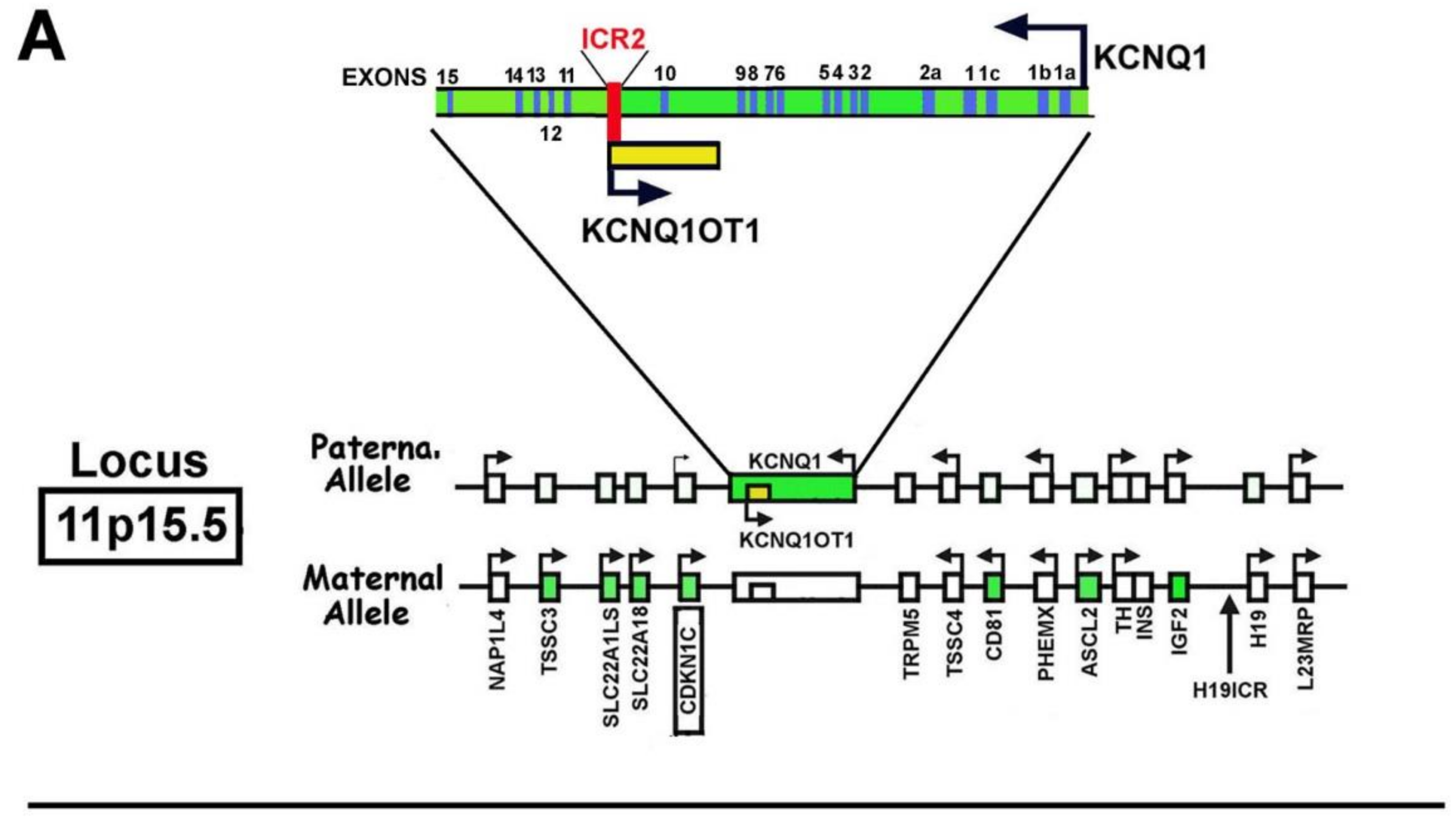
1.3. CDKN1C Mapping and Structure
2. Control of CDKN1C Transcription
2.1. DNA CpG Island Methylation
2.2. Histone Marks
2.3. LncRNA Involvement in Epigenetic Regulation
3. CDKN1C Expression and Human Diseases
3.1. Human Developmental Disorders
3.2. Human Cancers
3.3. Other CDKN1C-Related Human Diseases
4. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BWS | Beckwith-Wiedmann syndrome |
| CSC | Cardia stem cell |
| CDK | Cyclin-dependent kinase |
| CKI | CDK inhibitor |
| DNMT | DNA methyltransferase |
| HSC | Hematopoietic stem cell |
| ICR | Imprinted control region |
| IGF2 | Insulin-like gorwth factor |
| HDAC | histone deacetylase |
| IUGR | Intra-uterine growth restriction |
| IUP | Intrinsically unstructurated protein |
| KID | CDK binding/inhibitory domain |
| LSH | lymphoid-specific helicase |
| NLS | Nuclear localization signal |
| NSC | Neural stem cell |
| PCNA | Proliferating cell nuclear antigen |
| PMD | Placental mesenchymal dysplasia |
| PRC2 | Polycomb repressive complex 2 |
| p57KO | CDKN1C knockout |
| p57KOp27KI | CDKN1C knockout + CDKN1B knockin |
| RSS | Russell-Silver syndrome |
| Skp2 | S-phase kinase-associated protein 2 |
| SCF | Skp1/Cul1/F-box |
| TUG | Taurine upregulated gene 1 |
| T310 | Threonin 310 |
References
- Borriello, A.; Caldarelli, I.; Bencivenga, D.; Criscuolo, M.; Cucciolla, V.; Tramontano, A.; Oliva, A.; Perrotta, S.; Della Ragione, F. p57(Kip2) and cancer: Time for a critical appraisal. Mol. Cancer Res. 2011, 9, 1269–1284. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Reynisdottir, I.; Massague, J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 1995, 9, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Edwards, M.C.; Bai, C.; Parker, S.; Zhang, P.; Baldini, A.; Harper, J.W.; Elledge, S.J. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995, 9, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Fotedar, R.; Fitzgerald, P.; Rousselle, T.; Cannella, D.; Dorée, M.; Messier, H.; Fotedar, A. p21 contains independent binding sites for cyclin and cdk2: Both sites are required to inhibit cdk2 kinase activity. Oncogene 1996, 12, 2155–2164. [Google Scholar] [PubMed]
- Hashimoto, Y.; Kohri, K.; Kaneko, Y.; Morisaki, H.; Kato, T.; Ikeda, K.; Nakanishi, M. Critical role for the 310 helix region of p57(Kip2) in cyclin-dependent kinase 2 inhibition and growth suppression. J. Biol. Chem. 1998, 273, 16544–16550. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, E.G.; Guillier, M.; Leibovitch, M.P.; Leibovitch, S.A. Dimerization of the amino terminal domain of p57Kip2 inhibits cyclin D1-cdk4 kinase activity. Oncogene 2000, 19, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Vaccarello, G.; Figliola, R.; Cramerotti, S.; Novelli, F.; Maione, R. p57Kip2 is induced by MyoD through a p73-dependent pathway. J. Mol. Biol. 2006, 356, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.; Andersson, E.R.; Vlachos, P.; Södersten, E.; Liu, L.; Teixeira, A.I.; Hermanson, O. p57Kip2 is a repressor of Mash1 activity and neuronal differentiation in neural stem cells. Cell Death Differ. 2009, 16, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Joaquin, M.; Watson, R.J. The cell cycle-regulated B-Myb transcription factor overcomes cyclin-dependent kinase inhibitory activity of p57(KIP2) by interacting with its cyclin-binding domain. J. Biol. Chem. 2003, 278, 44255–44264. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Kim, M.J.; Ryoo, K.; Park, J.; Eom, S.J.; Shim, J.; Nakayama, K.I.; Nakayama, K.; Tomita, M.; Takahashi, K.; et al. p57KIP2 modulates stress-activated signaling by inhibiting c-Jun NH2-terminal kinase/stress-activated protein Kinase. J. Biol. Chem. 2003, 278, 48092–48098. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Digumarthi, H.; Aranbayeva, Z.; Wataha, J.; Lewis, J.; Messer, R.; Qin, H.; Dickinson, D.; Osaki, T.; Schuster, G.S.; et al. EGCG-targeted p57/KIP2 reduces tumorigenicity of oral carcinoma cells: Role of c-Jun N-terminal kinase. Toxicol. Appl. Pharmacol. 2007, 224, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Pan, Z.Q.; Schreiber-Agus, N.; DePinho, R.A.; Hurwitz, J.; Xiong, Y. Suppression of cell transformation by the cyclin-dependent kinase inhibitor p57KIP2 requires binding to proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA 1998, 95, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Borriello, A.; Cucciolla, V.; Criscuolo, M.; Indaco, S.; Oliva, A.; Giovane, A.; Bencivenga, D.; Iolascon, A.; Zappia, V.; Della Ragione, F. Retinoic acid induces p27Kip1 nuclear accumulation by modulating its phosphorylation. Cancer Res. 2006, 66, 4240–4248. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Nakamoto, T.; Nishimori, S.; Tanaka, K.; Chiba, T. A new ubiquitin ligase involved in p57KIP2 proteolysis regulates osteoblast cell differentiation. EMBO Rep. 2008, 9, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, H.; Hatakeyama, S.; Nakayama, K.; Nagata, M.; Tomita, K.; Nakayama, K. Spatial and temporal expression patterns of the cyclin-dependent kinase (CDK) inhibitors p27Kip1 and p57Kip2 during mouse development. Anat. Embryol. 2001, 203, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Macleod, K.F.; Sherry, N.; Hannon, G.; Beach, D.; Tokino, T.; Kinzler, K.; Vogelstein, B.; Jacks, T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 1995, 9, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.B.; Eichele, G.; Zhang, P.; Rawls, A.; Sands, A.T.; Bradley, A.; Olson, E.N.; Harper, J.W.; Elledge, S.J. p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science 1995, 267, 1024–1027. [Google Scholar] [CrossRef] [PubMed]
- Halevy, O.; Novitch, B.G.; Spicer, D.B.; Skapek, S.X.; Rhee, J.; Hannon, G.J.; Beach, D.; Lassar, A.B. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science 1995, 267, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Fredersdorf, S.; Milne, A.W.; Hall, P.A.; Lu, X. Characterization of a panel of novel anti-p21Waf1/Cip1 monoclonal antibodies and immunochemical analysis of p21Waf1/Cip1 expression in normal human tissues. Am. J. Pathol. 1996, 148, 825–835. [Google Scholar] [PubMed]
- Sood, R.; Zehnder, J.L.; Druzin, M.L.; Brown, P.O. Gene expression patterns in human placenta. Proc. Natl. Acad. Sci. USA 2006, 103, 5478–5483. [Google Scholar] [CrossRef] [PubMed]
- Tunster, S.J.; Van de Pette, M.; John, R.M. Fetal overgrowth in the Cdkn1c mouse model of Beckwith-Wiedemann syndrome. Dis. Models Mech. 2011, 4, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Tunster, S.J.; van de Pette, M.; John, R.M. Impact of genetic background on placental glycogen storage in mice. Placenta 2012, 33, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Enders, A.C.; Carter, A.M. What can comparative studies of placental structure tell us?—A review. Placenta 2004, 25 (Suppl. A), S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Frisén, J.; Lee, M.H.; Massagué, J.; Barbacid, M. Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 1997, 11, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liégeois, N.J.; Wong, C.; Finegold, M.; Hou, H.; Thompson, J.C.; Silverman, A.; Harper, J.W.; DePinho, R.A.; Elledge, S.J. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 1997, 387, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wong, C.; DePinho, R.A.; Harper, J.W.; Elledge, S.J. Cooperation between the Cdk inhibitors p27(KIP1) and p57(KIP2) in the control of tissue growth and development. Genes Dev. 1998, 12, 3162–3167. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nakayama, K.; Nakayama, K. Mice lacking a CDK inhibitor, p57Kip2, exhibit skeletal abnormalities and growth retardation. J. Biochem. 2000, 127, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Zhang, P.; Harper, J.W.; Elledge, S.J.; Leder, P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995, 82, 675–684. [Google Scholar] [CrossRef]
- Brugarolas, J.; Chandrasekaran, C.; Gordon, J.I.; Beach, D.; Jacks, T.; Hannon, G.J. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 1995, 377, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Fero, M.L.; Rivkin, M.; Tasch, M.; Porter, P.; Carow, C.E.; Firpo, E.; Polyak, K.; Tsai, L.H.; Broudy, V.; Perlmutter, R.M.; et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 1996, 85, 733–744. [Google Scholar] [CrossRef]
- Kiyokawa, H.; Kineman, R.D.; Manova-Todorova, K.O.; Soares, V.C.; Hoffman, E.S.; Ono, M.; Khanam, D.; Hayday, A.C.; Frohman, L.A.; Koff, A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 1996, 85, 721–732. [Google Scholar] [CrossRef]
- Nakayama, K.; Ishida, N.; Shirane, M.; Inomata, A.; Inoue, T.; Shishido, N.; Horii, I.; Loh, D.Y.; Nakayama, K. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 1996, 85, 707–720. [Google Scholar] [CrossRef]
- Susaki, E.; Nakayama, K.I. Functional similarities and uniqueness of p27 and p57: Insight from a knock-in mouse model. Cell Cycle 2009, 8, 2497–2501. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Takeishi, S.; Kanie, T.; Susaki, E.; Onoyama, I.; Tateishi, Y.; Nakayama, K.; Nakayama, K.I. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell 2011, 9, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Furutachi, S.; Matsumoto, A.; Nakayama, K.I.; Gotoh, Y. p57 controls adult neural stem cell quiescence and modulates the pace of lifelong neurogenesis. EMBO J. 2013, 32, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Iwama, A.; Takayanagi, S.; Morita, Y.; Eto, K.; Ema, H.; Nakauchi, H. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J. 2006, 25, 3515–3523. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Rodrigues, N.; Shen, H.; Yang, Y.; Dombkowski, D.; Sykes, M.; Scadden, D.T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 2000, 287, 1804–1808. [Google Scholar] [CrossRef] [PubMed]
- van Os, R.; Kamminga, L.M.; Ausema, A.; Bystrykh, L.V.; Draijer, D.P.; van Pelt, K.; Dontje, B.; de Haan, G. A Limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells 2007, 25, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, T.; Yamato, M.; Nishida, K.; Yang, J.; Tano, Y.; Okano, T. p57Kip2 is expressed in quiescent mouse bone marrow side population cells. Biochem. Biophys. Res. Commun. 2005, 337, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Stehling-Sun, S.; Lezon-Geyda, K.; Juneja, S.C.; Coillard, L.; Chatterjee, G.; Wuertzer, C.A.; Camargo, F.; Perkins, A.S. PR-domain-containing Mds1-Evi1 is critical for long-term hematopoietic stem cell function. Blood 2011, 118, 3853–3861. [Google Scholar] [CrossRef] [PubMed]
- Pechnick, R.N.; Zonis, S.; Wawrowsky, K.; Pourmorady, J.; Chesnokova, V. p21Cip1 restricts neuronal proliferation in the subgranular zone of the dentate gyrus of the hippocampus. Proc. Natl. Acad. Sci. USA 2008, 105, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Takagi, Y.; Harada, J.; Topalkara, K.; Wang, Y.; Sims, J.R.; Zheng, G.; Huang, P.; Ling, Y.; Scadden, D.T.; et al. p27Kip1 constrains proliferation of neural progenitor cells in adult brain under homeostatic and ischemic conditions. Stem Cells 2009, 27, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Encinas, J.M.; Michurina, T.V.; Peunova, N.; Park, J.H.; Tordo, J.; Peterson, D.A.; Fishell, G.; Koulakov, A.; Enikolopov, G. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell 2011, 8, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Tury, A.; Mairet-Coello, G.; DiCicco-Bloom, E. The cyclin-dependent kinase inhibitor p57Kip2 regulates cell cycle exit, differentiation, and migration of embryonic cerebral cortical precursors. Cereb. Cortex 2011, 21, 1840–1856. [Google Scholar] [CrossRef] [PubMed]
- Dugas, J.C.; Ibrahim, A.; Barres, B.A. A crucial role for p57(Kip2) in the intracellular timer that controls oligodendrocyte differentiation. J. Neurosci. 2007, 27, 6185–6196. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.A.; Chen, Y.; Fischer, S.M.; Conti, C.J. Coordinated changes in cell cycle machinery occur during keratinocyte terminal differentiation. Oncogene 1999, 18, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Gosselet, F.P.; Magnaldo, T.; Culerrier, R.M.; Sarasin, A.; Ehrhart, J.C. BMP2 and BMP6 control p57(Kip2) expression and cell growth arrest/terminal differentiation in normal primary human epidermal keratinocytes. Cell Signal. 2007, 19, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Hiromura, K.; Haseley, L.A.; Zhang, P.; Monkawa, T.; Durvasula, R.; Petermann, A.T.; Alpers, C.E.; Mundel, P.; Shankland, S.J. Podocyte expression of the CDK-inhibitor p57 during development and disease. Kidney Int. 2001, 60, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Figliola, R.; Maione, R. MyoD induces the expression of p57Kip2 in cells lacking p21Cip1/Waf1: Overlapping and distinct functions of the two cdk inhibitors. J. Cell. Physiol. 2004, 200, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, E.G.; Pelpel, K.; Guillier, M.; Leibovitch, M.P.; Leibovitch, S.A. p57(Kip2) stabilizes the MyoD protein by inhibiting cyclin E-Cdk2 kinase activity in growing myoblasts. Mol. Cell. Biol. 1999, 19, 7621–7629. [Google Scholar] [CrossRef] [PubMed]
- Figliola, R.; Busanello, A.; Vaccarello, G.; Maione, R. Regulation of p57(KIP2) during muscle differentiation: Role of Egr1, Sp1 and DNA hypomethylation. J. Mol. Biol. 2008, 380, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Zullo, A.; Mancini, F.P.; Schleip, R.; Wearing, S.; Yahia, L.; Klingler, W. The interplay between fascia, skeletl muscle, nerves, adipose tissue, inflammation and mechanical stress in musculo-fascial regeneration. J. Gerontol. Geriatr. 2017, 65, 271–283. [Google Scholar]
- Zalc, A.; Hayashi, S.; Auradé, F.; Bröhl, D.; Chang, T.; Mademtzoglou, D.; Mourikis, P.; Yao, Z.; Cao, Y.; Birchmeier, C. Antagonistic regulation of p57kip2 by Hes/Hey downstream of Notch signaling and muscle regulatory factors regulates skeletal muscle growth arrest. Development 2014, 141, 2780–2790. [Google Scholar] [CrossRef] [PubMed]
- Fukada, S.; Uezumi, A.; Ikemoto, M.; Masuda, S.; Segawa, M.; Tanimura, N.; Yamamoto, H.; Miyagoe-Suzuki, Y.; Takeda, S. Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells 2007, 25, 2448–2459. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Mori, M.; Inagawa, M.; Miyata, K.; Hashimoto, N.; Tanaka, S.; Asahara, H. Dnmt3a Regulates Proliferation of Muscle Satellite Cells via p57Kip2. PLoS Genet. 2016, 12, e1006167. [Google Scholar] [CrossRef] [PubMed]
- Sommese, L.; Zullo, A.; Schiano, C.; Mancini, F.P.; Napoli, C. Possible Muscle Repair in the Human Cardiovascular System. Stem Cell Rev 2017, 13, 170–191. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.M.; Kartha, C.C. Proliferation of murine c-kit(pos) cardiac stem cells stimulated with IGF-1 is associated with Akt-1 mediated phosphorylation and nuclear export of FoxO3a and its effect on downstream cell cycle regulators. Growth Factors 2014, 32, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mignone, J.; MacLellan, W.R. Cardiac Regeneration and Stem Cells. Physiol. Rev. 2015, 95, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Evans-Anderson, H.J.; Alfieri, C.M.; Yutzey, K.E. Regulation of cardiomyocyte proliferation and myocardial growth during development by FOXO transcription factors. Circ. Res. 2008, 102, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, V.; Martelli, F. Removing the brakes to cardiomyocyte cell cycle. Cell Cycle 2011, 10, 1176–1177. [Google Scholar] [CrossRef] [PubMed]
- Haley, S.A.; Zhao, T.; Zou, L.; Klysik, J.E.; Padbury, J.F.; Kochilas, L.K. Forced expression of the cell cycle inhibitor p57Kip2 in cardiomyocytes attenuates ischemia-reperfusion injury in the mouse heart. BMC Physiol. 2008, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Gao, L.; Hou, Y.; Xu, C.; Chang, N.; Wang, F.; Hu, K.; He, A.; Luo, Y.; Wang, J.; et al. Chromatin-remodelling factor Brg1 regulates myocardial proliferation and regeneration in zebrafish. Nat. Commun. 2016, 7, 13787. [Google Scholar] [CrossRef] [PubMed]
- Borriello, A.; Caldarelli, I.; Bencivenga, D.; Cucciolla, V.; Oliva, A.; Usala, E.; Danise, P.; Ronzoni, L.; Perrotta, S.; Della Ragione, F. p57Kip2 is a downstream effector of BCR-ABL kinase inhibitors in chronic myelogenous leukemia cells. Carcinogenesis 2011, 32, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Scandura, J.M.; Boccuni, P.; Massagué, J.; Nimer, S.D. Transforming growth factor beta-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc. Natl. Acad. Sci. USA 2004, 101, 15231–15236. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Branco, G.; Wagner, J.; Rodriguez, F.J.; Kele, J.; Sousa, K.; Rawal, N.; Pasolli, H.A.; Fuchs, E.; Kitajewski, J.; Arenas, E. Differential regulation of midbrain dopaminergic neuron development by Wnt-1, Wnt-3a, and Wnt-5a. Proc. Natl. Acad. Sci. USA 2003, 100, 12747–12752. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Lin, M.; Zhang, L.; York, J.P.; Zhang, P. The Notch signaling pathway controls the size of the ocular lens by directly suppressing p57Kip2 expression. Mol. Cell. Biol. 2007, 27, 7236–7247. [Google Scholar] [CrossRef] [PubMed]
- Georgia, S.; Soliz, R.; Li, M.; Zhang, P.; Bhushan, A. p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev. Biol. 2006, 298, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Hatada, I.; Mukai, T. Genomic imprinting of p57KIP2, a cyclin-dependent kinase inhibitor, in mouse. Nat. Genet. 1995, 11, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Potikha, T.; Kassem, S.; Haber, E.P.; Ariel, I.; Glaser, B. p57Kip2 (cdkn1c): Sequence, splice variants and unique temporal and spatial expression pattern in the rat pancreas. Lab. Investig. 2005, 85, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, L.; Mar, L.; Bogutz, A.; Oh-McGinnis, R.; Mandegar, M.A.; Paderova, J.; Gertsenstein, M.; Squire, JA.; Nagy, A. The interval between Ins2 and Ascl2 is dispensable for imprinting centre function in the murine Beckwith-Wiedemann region. Hum. Mol. Genet. 2009, 18, 4255–4267. [Google Scholar] [CrossRef] [PubMed]
- Mancini-DiNardo, D.; Steele, S.J.; Ingram, R.S.; Tilghman, S.M. A differentially methylated region within the gene Kcnq1 functions as an imprinted promoter and silencer. Hum. Mol. Genet. 2003, 12, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Choufani, S.; Ferreira, J.C.; Weksberg, R. Growth regulation, imprinted genes, and chromosome 11p15.5. Pediatr. Res. 2007, 61 Pt 2, 43r–47r. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.P.; DeBaun, M.R.; Mitsuya, K.; Galonek, H.L.; Brandenburg, S.; Oshimura, M.; Feinberg, A.P. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc. Natl. Acad. Sci. USA 1999, 96, 5203–5208. [Google Scholar] [CrossRef] [PubMed]
- Hark, A.T.; Schoenherr, C.J.; Katz, D.J.; Ingram, R.S.; Levorse, J.M.; Tilghman, S.M. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000, 405, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Cucciolla, V.; Borriello, A.; Criscuolo, M.; Sinisi, A.A.; Bencivenga, D.; Tramontano, A.; Scudieri, A.C.; Oliva, A.; Zappia, V.; Della Ragione, F. Histone deacetylase inhibitors upregulate p57Kip2 level by enhancing its expression through Sp1 transcription factor. Carcinogenesis 2008, 29, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Goufman, E.; Najmabadi, F.; Tyner, A.L. Sp1 and Sp3 activate p21 (WAF1/CIP1) gene transcription in the Caco-2 colon adenocarcinoma cell line. Oncogene 2000, 19, 5182–5188. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Matsuzaki, Y.; Miyazawa, K.; Zindy, F.; Roussel, M.F.; Sakai, T. Histone deacetylase inhibitors activate INK4d gene through Sp1 site in its promoter. Oncogene 2004, 23, 5340–5349. [Google Scholar] [CrossRef] [PubMed]
- Billing, M.; Rörby, E.; May, G.; Tipping, A.J.; Soneji, S.; Brown, J.; Salminen, M.; Karlsson, G.; Enver, T.; Karlsson, S. A network including TGFbeta/Smad4, Gata2, and p57 regulates proliferation of mouse hematopoietic progenitor cells. Exp. Hematol. 2016, 44, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ruan, X.; Zhang, P.; Yu, Y.; Gao, M.; Yuan, S.; Zhao, Z.; Yang, J.; Zhao, L. TBX3 promotes proliferation of papillary thyroid carcinoma cells through facilitating PRC2-mediated p57(KIP2) repression. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Toyota, M.; Itoh, F.; Suzuki, H.; Obata, T.; Yamamoto, H.; Kakiuchi, H.; Kusano, M.; Issa, J.P.; Tokino, T.; et al. Inactivation of p57KIP2 by regional promoter hypermethylation and histone deacetylation in human tumors. Oncogene 2002, 21, 2741–2749. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Edelkraut, U.; Daniel, G.; Hoffmann, A.; Spengler, D. Zac1 regulates cell cycle arrest in neuronal progenitors via Tcf4. Mol. Cell. Biol. 2014, 34, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Pfurr, S.; Chu, Y.H.; Bohrer, C.; Greulich, F.; Beattie, R.; Mammadzada, K.; Hils, M.; Arnold, S.J.; Taylor, V.; Schachtrup, K.; et al. The E2A splice variant E47 regulates the differentiation of projection neurons via p57(KIP2) during cortical development. Development 2017, 144, 3917–3931. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Cong, Q.; Chua, J.F.; Liu, H.; Xia, X.; Zhang, X.; Lin, J.; Habib, S.L.; Ao, J.; Zuo, Q.; et al. p57Kip2 is an unrecognized DNA damage response effector molecule that functions in tumor suppression and chemoresistance. Oncogene 2015, 34, 3568–3581. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Shivdasani, R.A. Genetic evidence that intestinal Notch functions vary regionally and operate through a common mechanism of Math1 repression. J. Biol. Chem. 2011, 286, 11427–11433. [Google Scholar] [CrossRef] [PubMed]
- Alheim, K.; Corness, J.; Samuelsson, M.K.; Bladh, L.G.; Murata, T.; Nilsson, T.; Okret, S. Identification of a functional glucocorticoid response element in the promoter of the cyclin-dependent kinase inhibitor p57Kip2. J. Mol. Endocrinol. 2003, 30, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [PubMed]
- John, R.M.; Ainscough, J.F.; Barton, S.C.; Surani, M.A. Distant cis-elements regulate imprinted expression of the mouse p57(Kip2) (Cdkn1c) gene: Implications for the human disorder, Beckwith-Wiedemann syndrome. Hum. Mol. Genet. 2001, 10, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Gurrieri, F.; Zollino, M.; Oliva, A.; Pascali, V.; Orteschi, D.; Pietrobono, R.; Camporeale, A.; Coll Vidal, M.; Partemi, S.; Brugada, R.; et al. Mild Beckwith-Wiedemann and severe long-QT syndrome due to deletion of the imprinting center 2 on chromosome 11p. Eur. J. Hum. Genet. 2013, 21, 965–969. [Google Scholar] [CrossRef] [PubMed]
- John, R.M.; Surani, M.A. Genomic imprinting, mammalian evolution, and the mystery of egg-laying mammals. Cell 2000, 101, 585–588. [Google Scholar] [CrossRef]
- Surani, M.A.; Barton, S.C.; Norris, M.L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984, 308, 548–550. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984, 37, 179–183. [Google Scholar] [CrossRef]
- Monk, D.; Sanches, R.; Arnaud, P.; Apostolidou, S.; Hills, F.A.; Abu-Amero, S.; Murrell, A.; Friess, H.; Reik, W.; Stanier, P.; et al. Imprinting of IGF2 P0 transcript and novel alternatively spliced INS-IGF2 isoforms show differences between mouse and human. Hum. Mol. Genet. 2006, 15, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Surani, M.A. Imprinting and the initiation of gene silencing in the germ line. Cell 1998, 93, 309–312. [Google Scholar] [CrossRef]
- Hatada, I.; Inazawa, J.; Abe, T.; Nakayama, M.; Kaneko, Y.; Jinno, Y.; Niikawa, N.; Ohashi, H.; Fukushima, Y.; Iida, K.; et al. Genomic imprinting of human p57KIP2 and its reduced expression in Wilms’ tumors. Hum. Mol. Genet. 1996, 5, 783–788. [Google Scholar] [CrossRef] [PubMed]
- John, R.M.; Lefebvre, L. Developmental regulation of somatic imprints. Differentiation 2011, 81, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, B.; Arnaudo, A.; Dymkowski, A.; Best, A.; Davis, T.L. Methylation at mouse Cdkn1c is acquired during postimplantation development and functions to maintain imprinted expression. Genomics 2004, 84, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.N.; Andresini, O.; Matteini, F.; Maione, R. Transcriptional regulation of p57(kip2) expression during development, differentiation and disease. Front. Biosci. 2018, 23, 83–108. [Google Scholar]
- Fan, T.; Hagan, J.P.; Kozlov, S.V.; Stewart, C.L.; Muegge, K. Lsh controls silencing of the imprinted Cdkn1c gene. Development 2005, 132, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, M.J.; Reifenberger, J.; Reifenberger, G. Frequent biallelic inactivation and transcriptional silencing of the DIRAS3 gene at 1p31 in oligodendroglial tumors with 1p loss. Int. J. Cancer 2008, 122, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Neumann, L.C.; Weinhäusel, A.; Thomas, S.; Horsthemke, B.; Lohmann, D.R.; Zeschnigk, M. EFS shows biallelic methylation in uveal melanoma with poor prognosis as well as tissue-specific methylation. BMC Cancer 2011, 11, 380. [Google Scholar] [CrossRef] [PubMed]
- Andresini, O.; Ciotti, A.; Rossi, M.N.; Battistelli, C.; Carbone, M.; Maione, R. A cross-talk between DNA methylation and H3 lysine 9 dimethylation at the KvDMR1 region controls the induction of Cdkn1c in muscle cells. Epigenetics 2016, 11, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, F.; Mondal, T.; Guseva, N.; Pandey, G.K.; Kanduri, C. Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 2010, 137, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Nagai, H.; Ohno, T.; Yuge, M.; Hatano, S.; Ito, E.; Mori, N.; Saito, H.; Kinoshita, T. Aberrant DNA methylation of p57(KIP2) gene in the promoter region in lymphoid malignancies of B-cell phenotype. Blood 2002, 100, 2572–2577. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Choi, H.P.; Wang, X.; Wu, F.; Chen, X.; Lü, X.; Jing, R.; Ryu, H.; Wang, X.; Azadzoi, K.M.; et al. Post-Translational Modification of Human Histone by Wide Tolerance of Acetylation. Cells 2017, 6, E34. [Google Scholar] [CrossRef] [PubMed]
- Faundes, V.; Newman, W.G.; Bernardini, L.; Canham, N.; Clayton-Smith, J.; Dallapiccola, B.; Davies, S.J.; Demos, M.K.; Goldman, A.; Gill, H.; et al. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am. J. Hum. Genet. 2018, 102, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Umlauf, D.; Goto, Y.; Cao, R.; Cerqueira, F.; Wagscha, A.; Zhang, Y.; Feil, R. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat. Genet. 2004, 36, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Mitsuya, K.; Umlauf, D.; Smith, P.; Dean, W.; Walter, J.; Higgins, M.; Feil, R.; Reik, W. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat. Genet. 2004, 36, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Algar, E.M.; Muscat, A.; Dagar, V.; Rickert, C.; Chow, C.W.; Biegel, J.A.; Ekert, P.G.; Saffery, R.; Craig, J.; Johnstone, R.W.; et al. Imprinted CDKN1C is a tumor suppressor in rhabdoid tumor and activated by restoration of SMARCB1 and histone deacetylase inhibitors. PLoS ONE 2009, 4, e4482. [Google Scholar] [CrossRef] [PubMed]
- Attia, M.; Rachez, C.; De Pauw, A.; Avner, P.; Rogner, U.C. Nap1l2 promotes histone acetylation activity during neuronal differentiation. Mol. Cell. Biol. 2007, 27, 6093–6102. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.L.; Yue, Z.; Liu, L.; Pei, L.; Yin, Y.; Qin, L.; Zhao, J.; Liu, H.; Wang, H.; Jia, M. The expression of histone deacetylase HDAC1 correlates with the progression and prognosis of gastrointestinal malignancy. Oncotarget 2017, 8, 39241–39253. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.K.; Park, N.Y.; Shin, J.H.; Jo, D.S.; Bae, J.E.; Choi, E.S.; Maeng, S.; Jeon, H.B.; Roh, S.A.; Chang, J.W.; et al. Up-regulation of UVRAG by HDAC1 Inhibition Attenuates 5FU-induced Cell Death in HCT116 Colorectal Cancer Cells. Anticancer Res. 2018, 38, 271–277. [Google Scholar] [PubMed]
- Zhou, H.; Cai, Y.; Liu, D.; Li, M.; Sha, Y.; Zhang, W.; Wang, K.; Gong, J.; Tang, N.; Huang, A.; et al. Pharmacological or transcriptional inhibition of both HDAC1 and 2 leads to cell cycle blockage and apoptosis via p21(Waf1/Cip1) and p19(INK4d) upregulation in hepatocellular carcinoma. Cell Prolif. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wakahara, M.; Sakabe, T.; Kubouchi, Y.; Hosoya, K.; Hirooka, Y.; Yurugi, Y.; Nosaka, K.; Shiomi, T.; Nakamura, H.; Umekita, Y. Subcellular Localization of maspin correlates with histone deacetylase 1 expression in human breast cancer. Anticancer Res. 2017, 37, 5071–5077. [Google Scholar] [PubMed]
- Cai, M.H.; Xu, X.G.; Yan, S.L.; Sun, Z.; Ying, Y.; Wang, B.K.; Tu, Y.X. Depletion of HDAC1, 7 and 8 by histone deacetylase inhibition confers elimination of pancreatic cancer stem cells in combination with gemcitabine. Sci. Rep. 2018, 8, 1621. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Ballestar, E.; Alaminos, M.; Arango, D.; Schwartz, S., Jr.; Esteller, M. Transforming pathways unleashed by a HDAC2 mutation in human cancer. Oncogene 2008, 27, 4008–4012. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, F.; Qu, Y.; Chen, X.; Gao, M.; Yang, J.; Zhang, D.; Zhang, N.; Li, W.; Liu, H. HDAC2 regulates cell proliferation, cell cycle progression and cell apoptosis in esophageal squamous cell carcinoma EC9706 cells. Oncol. Lett. 2017, 13, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Hulsurkar, M.; Li, Z.; Zhang, Y.; Li, X.; Zheng, D.; Li, W. Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1. Oncogene 2017, 36, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Noh, J.H.; Lee, J.H.; Eun, J.W.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; Yoo, N.J.; Lee, J.Y.; et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 2005, 113, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Iida, A.; Iwagawa, T.; Baba, Y.; Satoh, S.; Mochizuki, Y.; Nakauchi, H.; Furukawa, T.; Koseki, H.; Murakami, A.; Watanabe, S. Roles of histone H3K27 trimethylase Ezh2 in retinal proliferation and differentiation. Dev. Neurobiol. 2015, 75, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Heinen, A.; Tzekova, N.; Graffmann, N.; Torres, K.J.; Uhrberg, M.; Hartung, H.P.; Küry, P. Histone methyltransferase enhancer of zeste homolog 2 regulates Schwann cell differentiation. Glia 2012, 60, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Mitsuya, K.; Meguro, M.; Lee, M.P.; Katoh, M.; Schulz, T.C.; Kugoh, H.; Yoshida, M.A.; Niikawa, N.; Feinberg, A.P.; Oshimura, M. LIT1, an imprinted antisense RNA in the human KvLQT1 locus identified by screening for differentially expressed transcripts using monochromosomal hybrids. Hum. Mol. Genet. 1999, 8, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Zhou, W.; Beatty, L.G.; Weksberg, R.; Sadowski, P.D. The KCNQ1OT1 promoter, a key regulator of genomic imprinting in human chromosome 11p15.5. Genomics 2004, 84, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; He, X.; Yin, D.; Han, L.; Qiu, M.; Xu, T.; Xia, R.; Xu, L.; Yin, R.; De, W. Increased expression of long noncoding RNA TUG1 predicts a poor prognosis of gastric cancer and regulates cell proliferation by epigenetically silencing of p57. Cell Death Dis. 2016, 7, e2109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Yin, D.; Han, L.; He, X.; Si, X.; Chen, W.; Xia, R.; Xu, T.; Gu, D.; De, W.; et al. E2F1-induced upregulation of long noncoding RNA LINC00668 predicts a poor prognosis of gastric cancer and promotes cell proliferation through epigenetically silencing of CKIs. Oncotarget 2016, 7, 23212–23226. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, L.; Huo, X.S.; Yuan, J.H.; Xu, D.; Yuan, S.X.; Zhu, N.; Zhou, W.P.; Yang, G.S.; Wang, Y.Z.; et al. Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology 2011, 54, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: An international consensus statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Russo, S.; De Crescenzo, A.; Freschi, A.; Calzari, L.; Maitz, S.; Macchiaiolo, M.; Molinatto, C.; Baldassarre, G.; Mariani, M.; et al. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2016, 24, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Heide, S.; Chantot-Bastaraud, S.; Keren, B.; Harbison, M.D.; Azzi, S.; Rossignol, S.; Michot, C.; Lackmy-Port Lys, M.; Demeer, B.; Heinrichs, C.; et al. Chromosomal rearrangements in the 11p15 imprinted region: 17 new 11p15.5 duplications with associated phenotypes and putative functional consequences. J. Med. Genet. 2018, 55, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Mussa, A.; Russo, S.; de Crescenzo, A.; Freschi, A.; Calzari, L.; Maitz, S.; Macchiaiolo, M.; Molinatto, C.; Baldassarre, G.; Mariani, M.; et al. Fetal growth patterns in Beckwith-Wiedemann syndrome. Clin. Genet. 2016, 90, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.N.; Schofield, P.N.; Reik, W.; Macdonald, F.; Maher, E.R. Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2005, 13, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Brioude, F.; Luharia, A.; Evans, G.A.; Raza, H.; Haire, A.C.; Grundy, R.; Bowdin, S.C.; Riccio, A.; Sebastio, G.; Bliek, J.; et al. Mutations of the Imprinted CDKN1C Gene as a Cause of the Overgrowth Beckwith-Wiedemann Syndrome: Clinical Spectrum and Functional Characterization. Hum. Mutat. 2015, 36, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Bourcigaux, N.; Gaston, V.; Logié, A.; Bertagna, X.; Le Bouc, Y.; Gicquel, C. High expression of cyclin E and G1 CDK and loss of function of p57KIP2 are involved in proliferation of malignant sporadic adrenocortical tumors. J. Clin. Endocrinol. Metab. 2000, 85, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Duquesnes, N.; Callot, C.; Jeannot, P.; Daburon, V.; Nakayama, K.I.; Manenti, S.; Davy, A.; Besson, A. p57(Kip2) knock-in mouse reveals CDK-independent contribution in the development of Beckwith-Wiedemann syndrome. J. Pathol. 2016, 239, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Arboleda, V.A.; Lee, H.; Parnaik, R.; Fleming, A.; Banerjee, A.; Ferraz-de-Souza, B.; Délot, E.C.; Rodriguez-Fernandez, I.A.; Braslavsky, D.; Bergadá, I.; et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat. Genet. 2012, 44, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Dias, R.P.; Maher, E.R. An imprinted IMAGe: Insights into growth regulation through genomic analysis of a rare disease. Genome Med. 2012, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- Begemann, M.; Spengler, S.; Kanber, D.; Haake, A.; Baudis, M.; Leisten, I.; Binder, G.; Markus, S.; Rupprecht, T.; Segerer, H.; et al. Silver-Russell patients showing a broad range of ICR1 and ICR2 hypomethylation in different tissues. Clin. Genet. 2011, 80, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Eggermann, K.; Bliek, J.; Brioude, F.; Algar, E.; Buiting, K.; Russo, S.; Tümer, Z.; Monk, D.; Moore, G.; Antoniadi, T.; et al. EMQN best practice guidelines for the molecular genetic testing and reporting of chromosome 11p15 imprinting disorders: Silver-Russell and Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2016, 24, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Monk, D.; Wakeling, E.L.; Proud, V.; Hitchins, M.; Abu-Amero, S.N.; Stanier, P.; Preece, M.A.; Moore, G.E. Duplication of 7p11.2-p13, including GRB10, in Silver-Russell syndrome. Am. J. Hum. Genet. 2000, 66, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Brioude, F.; Oliver-Petit, I.; Blaise, A.; Praz, F.; Rossignol, S.; Le Jule, M.; Thibaud, N.; Faussat, A.M.; Tauber, M.; Le Bouc, Y.; et al. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J. Med. Genet. 2013, 50, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Giordani, L.; Moretti, A.; Basso, G.; Borriello, A.; Della Ragione, F. Analysis of CDKN2A, CDKN2B, CDKN2C, and cyclin Ds gene status in hepatoblastoma. Hepatology 1998, 27, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Giordani, L.; Moretti, A.; Tonini, G.P.; Lo Cunsolo, C.; Mastropietro, S.; Borriello, A.; Della Ragione, F. Structural and functional analysis of cyclin-dependent kinase inhibitor genes (CDKN2A, CDKN2B, and CDKN2C) in neuroblastoma. Pediatr. Res. 1998, 43, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Pateras, I.S.; Apostolopoulou, K.; Niforou, K.; Kotsinas, A.; Gorgoulis, V.G. p57KIP2: “Kip”ing the cell under control. Mol. Cancer Res. 2009, 7, 1902–1919. [Google Scholar] [CrossRef] [PubMed]
- Bencivenga, D.; Caldarelli, I.; Stampone, E.; Mancini, F.P.; Balestrieri, M.L.; Della Ragione, F.; Borriello, A. p27(Kip1) and human cancers: A reappraisal of a still enigmatic protein. Cancer Lett. 2017, 403, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, F.; Orlow, I.; Cordon-Cardo, C. Mutational study of p16CDKN2/MTS1/INK4A and p57KIP2 genes in hepatocellular carcinoma. Int. J. Oncol. 1998, 12, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Kim, H.S.; Lee, K.S.; Kim, J.; Park, J.B.; Won, M.H.; Chae, S.W.; Choi, Y.H.; Choi, K.C.; Park, Y.E.; et al. Mutation and expression of the p27KIP1 and p57KIP2 genes in human gastric cancer. Exp. Mol. Med. 2000, 32, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Oya, M.; Schulz, W.A. Decreased expression of p57(KIP2)mRNA in human bladder cancer. Br. J. Cancer 2000, 83, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.J.; Flor, A.R.; Seifert, H.H.; Schulz, W.A. Multiple mechanisms downregulate CDKN1C in human bladder cancer. Int. J. Cancer 2005, 114, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Matsubayashi, H.; Abe, T.; Fukushima, N.; Goggins, M. Epigenetic down-regulation of CDKN1C/p57KIP2 in pancreatic ductal neoplasms identified by gene expression profiling. Clin. Cancer Res. 2005, 11, 4681–4688. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, I.; Boldrini, R.; Benedetti, M.C.; Inserra, A.; De Pasquale, M.D.; Francalanci, P. Pediatric adrenocortical neoplasms: Immunohistochemical expression of p57 identifies loss of heterozygosity and abnormal imprinting of the 11p15.5. Pediatr. Res. 2017, 81, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jin, S.D.; Zhu, Q.; Han, L.; Feng, J.; Lu, X.Y.; Wang, W.; Wang, F.; Guo, R.H. Long non-coding RNA LUCAT1 is associated with poor prognosis in human non-small lung cancer and regulates cell proliferation via epigenetically repressing p21 and p57 expression. Oncotarget 2017, 8, 28297–28311. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Li, Y.; Zeng, B.; Guan, X.; Li, H. Downregulated CDKN1C/p57(kip2) drives tumorigenesis and associates with poor overall survival in breast cancer. Biochem. Biophys. Res. Commun. 2018, 497, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Radujkovic, A.; Dietrich, S.; Andrulis, M.; Benner, A.; Longerich, T.; Pellagatti, A.; Nanda, K.; Giese, T.; Germing, U.; Baldus, S.; et al. Expression of CDKN1C in the bone marrow of patients with myelodysplastic syndrome and secondary acute myeloid leukemia is associated with poor survival after conventional chemotherapy. Int. J. Cancer 2016, 139, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Nan, K.; Hu, T.; Meng, J.; Hui, W.; Zhang, X.; Qin, H.; Sui, C. Prognostic significance of co-expression of nm23 and p57 protein in hepatocellular carcinoma. Hepatol. Res. 2010, 40, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Kobatake, T.; Yano, M.; Toyooka, S.; Tsukuda, K.; Dote, H.; Kikuchi, T.; Toyota, M.; Ouchida, M.; Aoe, M.; Date, H.; et al. Aberrant methylation of p57KIP2 gene in lung and breast cancers and malignant mesotheliomas. Oncol. Rep. 2004, 12, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Toyota, M.; Kondo, Y.; Obata, T.; Daniel, S.; Pierce, S.; Imai, K.; Kantarjian, H.M.; Issa, J.P.; Garcia-Manero, G. Aberrant DNA methylation of p57KIP2 identifies a cell-cycle regulatory pathway with prognostic impact in adult acute lymphocytic leukemia. Blood 2003, 101, 4131–4136. [Google Scholar] [CrossRef] [PubMed]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Agirre, X.; Prosper, F.; Heiniger, A.; Torres, A. Lack of CpG island methylator phenotype defines a clinical subtype of T-cell acute lymphoblastic leukemia associated with good prognosis. J. Clin. Oncol. 2005, 23, 7043–7049. [Google Scholar] [CrossRef] [PubMed]
- Zohny, S.F.; Baothman, O.A.; El-Shinawi, M.; Al-Malki, A.L.; Zamzami, M.A.; Choudhry, H. The KIP/CIP family members p21^{Waf1/Cip1} and p57^{Kip2} as diagnostic markers for breast cancer. Cancer Biomark. 2017, 18, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Lin, C.L.; Huang, T.H.; Bouamar, H.; Sun, L.Z. MicroRNA-21 inhibits p57Kip2 expression in prostate cancer. Mol. Cancer 2014, 13, 212. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Yu, J.; Han, T.S.; Park, S.Y.; Namkoong, B.; Kim, D.H.; Hur, K.; Yoo, M.W.; Lee, H.J.; Yang, H.K.; et al. Functional links between clustered microRNAs: Suppression of cell-cycle inhibitors by microRNA clusters in gastric cancer. Nucleic Acids Res. 2009, 37, 1672–1681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gong, X.; Tian, K.; Chen, D.; Sun, J.; Wang, G.; Guo, M. miR-25 promotes glioma cell proliferation by targeting CDKN1C. Biomed. Pharmacother. 2015, 71, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Nass, D.; Rosenwald, S.; Meiri, E.; Gilad, S.; Tabibian-Keissar, H.; Schlosberg, A.; Kuker, H.; Sion-Vardy, N.; Tobar, A.; Kharenko, O.; et al. MiR-92b and miR-9/9* are specifically expressed in brain primary tumors and can be used to differentiate primary from metastatic brain tumors. Brain Pathol. 2009, 19, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Gramantieri, L.; Ferracin, M.; Veronese, A.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Giovannini, C.; Croce, C.M.; Bolondi, L.; et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene 2008, 27, 5651–5661. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wang, Q.; Chen, J.; Huang, X.; Chen, X.; Cao, L.; Tan, H.; Li, W.; Zhang, L.; Bi, J.; et al. Clinical significance of miR-221 and its inverse correlation with p27Kip1 in hepatocellular carcinoma. Mol. Biol. Rep. 2011, 38, 3029–3035. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.J.; Shen, W.G.; Liu, C.J.; Chen, Y.W.; Lu, H.H.; Tsai, M.M.; Lin, S.C. miR-221 and miR-222 expression increased the growth and tumorigenesis of oral carcinoma cells. J. Oral Pathol. Med. 2011, 40, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Wang, W.; Zeng, J.J.; Wu, C.T.; Lei, S.T.; Li, G.X. MicroRNA-221 inhibits CDKN1C/p57 expression in human colorectal carcinoma. Acta Pharmacol. Sin. 2011, 32, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Bazot, Q.; Paschos, K.; Skalska, L.; Kalchschmidt, J.S.; Parker, G.A.; Allday, M.J. Epstein-Barr Virus Proteins EBNA3A and EBNA3C Together Induce Expression of the Oncogenic MicroRNA Cluster miR-221/miR-222 and Ablate Expression of Its Target p57KIP2. PLoS Pathog. 2015, 11, e1005031. [Google Scholar] [CrossRef] [PubMed]
- Wurz, K.; Garcia, R.L.; Goff, B.A.; Mitchell, P.S.; Lee, J.H.; Tewari, M.; Swisher, E.M. MiR-221 and MiR-222 alterations in sporadic ovarian carcinoma: Relationship to CDKN1B, CDKNIC and overall survival. Genes Chromosomes Cancer 2010, 49, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Unek, G.; Ozmen, A.; Mendilcioglu, I.; Simsek, M.; Korgun, E.T. The expression of cell cycle related proteins PCNA, Ki67, p27 and p57 in normal and preeclamptic human placentas. Tissue Cell 2014, 46, 198–205. [Google Scholar] [CrossRef] [PubMed]
- McMinn, J.; Wei, M.; Schupf, N.; Cusmai, J.; Johnson, E.B.; Smith, A.C.; Weksberg, R.; Thaker, H.M.; Tycko, B. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta 2006, 27, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Allias, F.; Lebreton, F.; Collardeau-Frachon, S.; Azziza, J.; Pasquier, C.J.; Arcin-Thoury, F.; Patrier, S.; Devouassoux-Shisheboran, M. Immunohistochemical expression of p57 in placental vascular proliferative disorders of preterm and term placentas. Fetal Pediatr. Pathol. 2009, 28, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, M. Immunohistochemical characterization of p57(KIP2) expression in early hydatidiform moles. Hum. Pathol. 2002, 33, 1188–1192. [Google Scholar] [CrossRef] [PubMed]
- McCowan, L.M.; Becroft, D.M. Beckwith-Wiedemann syndrome, placental abnormalities, and gestational proteinuric hypertension. Obstet. Gynecol. 1994, 83 Pt 2, 813–817. [Google Scholar] [PubMed]
- Linn, R.L.; Minturn, L.; Yee, L.M.; Maniar, K.; Zhang, Y.; Fritsch, M.K.; Kashireddy, P.; Kapur, R.; Ernst, L.M. Placental mesenchymal dysplasia without fetal development in a twin gestation: A case report and review of the spectrum of androgenetic biparental mosaicism. Pediatr. Dev. Pathol. 2015, 18, 146–154. [Google Scholar] [CrossRef] [PubMed]
- H’Mida, D.; Gribaa, M.; Yacoubi, T.; Chaieb, A.; Adala, L.; Elghezal, H.; Saad, A. Placental mesenchymal dysplasia with beckwith-wiedemann syndrome fetus in the context of biparental and androgenic cell lines. Placenta 2008, 29, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Kaiser-Rogers, K.A.; McFadden, D.E.; Livasy, C.A.; Dansereau, J.; Jiang, R.; Knops, J.F.; Lefebvre, L.; Rao, K.W.; Robinson, W.P. Androgenetic/biparental mosaicism causes placental mesenchymal dysplasia. J. Med. Genet. 2006, 43, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Kalish, J.M.; Conlin, L.K.; Bhatti, T.R.; Dubbs, H.A.; Harris, M.C.; Izumi, K.; Mostoufi-Moab, S.; Mulchandani, S.; Saitta, S.; States, L.J.; et al. Clinical features of three girls with mosaic genome-wide paternal uniparental isodisomy. Am. J. Med. Genet. A 2013, 161a, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Samadder, A.; Kar, R. Utility of p57 immunohistochemistry in differentiating between complete mole, partial mole & non-molar or hydropic abortus. Indian J. Med. Res. 2017, 145, 133–137. [Google Scholar] [PubMed]
- Sasaki, S.; Sasaki, Y.; Kunimura, T.; Sekizawa, A.; Kojima, Y.; Iino, K. Clinical Usefulness of Immunohistochemical Staining of p57 kip2 for the Differential Diagnosis of Complete Mole. Biomed. Res. Int. 2015, 2015, 905648. [Google Scholar] [CrossRef] [PubMed]
- Castrillon, D.H.; Sun, D.; Weremowicz, S.; Fisher, R.A.; Crum, C.P.; Genest, D.R. Discrimination of complete hydatidiform mole from its mimics by immunohistochemistry of the paternally imprinted gene product p57KIP2. Am. J. Surg. Pathol. 2001, 25, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- De Nigris, F.; Balestrieri, M.L.; Napoli, C. Targeting c-Myc, Ras and IGF cascade to treat cancer and vascular disorders. Cell Cycle 2006, 5, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, M.L.; Fiorito, C.; Crimi, E.; Felice, F.; Schiano, C.; Milone, L.; Casamassimi, A.; Giovane, A.; Grimaldi, V.; del Giudice, V.; et al. Effect of red wine antioxidants and minor polyphenolic constituents on endothelial progenitor cells after physical training in mice. Int. J. Cardiol. 2008, 126, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, M.; Zullo, A.; Servillo, L.; Mancini, F.P.; Borriello, A.; Giovane, A.; Della Ragione, F.; D’Onofrio, N.; Balestrieri, M.L. Multiple pathways of SIRT6 at the crossroads in the control of longevity, cancer, and cardiovascular diseases. Ageing Res. Rev. 2017, 35, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Kassem, S.A.; Ariel, I.; Thornton, P.S.; Hussain, K.; Smith, V.; Lindley, K.J.; Aynsley-Green, A.; Glaser, B. p57(KIP2) expression in normal islet cells and in hyperinsulinism of infancy. Diabetes 2001, 50, 2763–2769. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, D.; Li, C.; Yu, M.; Jiao, Y.; Zhang, J.; Naji, A.; Ziaie, S.; Glaser, B.; Kaestner, K.H. Targeting the cell cycle inhibitor p57Kip2 promotes adult human beta cell replication. J. Clin. Investig. 2014, 124, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Kerns, S.L.; Guevara-Aguirre, J.; Andrew, S.; Geng, J.; Guevara, C.; Guevara-Aguirre, M.; Guo, M.; Oddoux, C.; Shen, Y.; Zurita, A. A novel variant in CDKN1C is associated with intrauterine growth restriction, short stature, and early-adulthood-onset diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E2117–E2122. [Google Scholar] [CrossRef] [PubMed]
- Van De Pette, M.; Tunster, S.J.; McNamara, G.I.; Shelkovnikova, T.; Millership, S.; Benson, L.; Peirson, S.; Christian, M.; Vidal-Puig, A.; John, R.M. Cdkn1c Boosts the Development of Brown Adipose Tissue in a Murine Model of Silver Russell Syndrome. PLoS Genet. 2016, 12, e1005916. [Google Scholar] [CrossRef] [PubMed]
- Asahara, S.; Etoh, H.; Inoue, H.; Teruyama, K.; Shibutani, Y.; Ihara, Y.; Kawada, Y.; Bartolome, A.; Hashimoto, N.; Matsuda, T. Paternal allelic mutation at the Kcnq1 locus reduces pancreatic beta-cell mass by epigenetic modification of Cdkn1c. Proc. Natl. Acad. Sci. USA 2015, 112, 8332–8337. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Choi, J.; Zhao, C.; Ma, Z.A. FTY720 normalizes hyperglycemia by stimulating beta-cell in vivo regeneration in db/db mice through regulation of cyclin D3 and p57(KIP2). J. Biol. Chem. 2012, 287, 5562–5573. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, V.; Schiano, C.; Casamassimi, A.; Zullo, A.; Soricelli, A.; Mancini, F.P.; Napoli, C. Imaging Techniques to Evaluate Cell Therapy in Peripheral Artery Disease: State of the Art and Clinical Trials. Clin. Physiol. Funct. Imaging 2016, 36, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Nödl, M.-T.; Fossati, S.M.; Domingues, P.; Sánchez, F.J.; Zullo, L. The Making of an Octopus Arm. Evodevo 2015, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Fossati, S.M.; Candiani, S.; Nödl, M.-T.; Maragliano, L.; Pennuto, M.; Domingues, P.; Benfenati, F.; Pestarino, M.; Zullo, L. Identification and Expression of Acetylcholinesterase in Octopus Vulgaris Arm Development and Regeneration: A Conserved Role for ACHE? Mol. Neurobiol. 2015, 52, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Yoshihara, H.; Hosokawa, K.; Tai, I.; Shinmyozu, K.; Tsukahara, F.; Maru, Y.; Nakayama, K.; Nakayama, K.I.; Suda, T. p57Kip2 and p27Kip1 Cooperate to Maintain Hematopoietic Stem Cell Quiescence through Interactions with Hsc70. Cell Stem Cell 2011, 9, 247–261. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stampone, E.; Caldarelli, I.; Zullo, A.; Bencivenga, D.; Mancini, F.P.; Della Ragione, F.; Borriello, A. Genetic and Epigenetic Control of CDKN1C Expression: Importance in Cell Commitment and Differentiation, Tissue Homeostasis and Human Diseases. Int. J. Mol. Sci. 2018, 19, 1055. https://doi.org/10.3390/ijms19041055
Stampone E, Caldarelli I, Zullo A, Bencivenga D, Mancini FP, Della Ragione F, Borriello A. Genetic and Epigenetic Control of CDKN1C Expression: Importance in Cell Commitment and Differentiation, Tissue Homeostasis and Human Diseases. International Journal of Molecular Sciences. 2018; 19(4):1055. https://doi.org/10.3390/ijms19041055
Chicago/Turabian StyleStampone, Emanuela, Ilaria Caldarelli, Alberto Zullo, Debora Bencivenga, Francesco Paolo Mancini, Fulvio Della Ragione, and Adriana Borriello. 2018. "Genetic and Epigenetic Control of CDKN1C Expression: Importance in Cell Commitment and Differentiation, Tissue Homeostasis and Human Diseases" International Journal of Molecular Sciences 19, no. 4: 1055. https://doi.org/10.3390/ijms19041055
APA StyleStampone, E., Caldarelli, I., Zullo, A., Bencivenga, D., Mancini, F. P., Della Ragione, F., & Borriello, A. (2018). Genetic and Epigenetic Control of CDKN1C Expression: Importance in Cell Commitment and Differentiation, Tissue Homeostasis and Human Diseases. International Journal of Molecular Sciences, 19(4), 1055. https://doi.org/10.3390/ijms19041055